Shellmycin A–D, Novel Bioactive Tetrahydroanthra-γ-Pyrone Antibiotics from Marine Streptomyces sp. Shell-016


Abstract
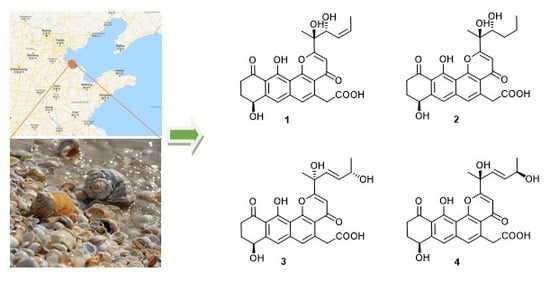
:
1. Introduction
2. Results and Discussion
2.1. Extraction and Purification
2.2. Structure Elucidation
2.3. Bioactivity Assay
2.4. Proposed Biosynthesis Pathway
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Biological Materials
3.3. Fermentation and Extraction
3.4. Purification of the Compounds
3.5. Spectral Data of the Compounds
3.6. Computational Chemistry
3.7. Cytotoxicity Assay
3.8. Antimicrobial Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [Green Version]
- Hutchings, M.; Truman, A.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Biotechnol. 2019, 51, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Bulaj, G.; Ahern, M.M.; Kuhn, A.; Judkins, Z.S.; Bowen, R.C.; Chen, Y. Incorporating Natural Products, Pharmaceutical Drugs, Self-Care and Digital/Mobile Health Technologies into Molecular-Behavioral Combination Therapies for Chronic Diseases. Curr. Clin. Pharmacol. 2016, 11, 128–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waclaw, B. Evolution of Drug Resistance in Bacteria. In Advances in Experimental Medicine and Biology; Springer Nature: New York, NY, USA, 2016; Volume 915, pp. 49–67. [Google Scholar]
- Namee, N.M.; O’Driscoll, L. Extracellular vesicles and anti-cancer drug resistance. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Olasehinde, T.A.; Olaniran, A.O.; Okoh, A.I. Macroalgae as a Valuable Source of Naturally Occurring Bioactive Compounds for the Treatment of Alzheimer’s Disease. Mar. Drugs 2019, 17, 609. [Google Scholar] [CrossRef] [Green Version]
- Nolsoe, J.M.J.; Aursnes, M.; Stenstrom, Y.H.; Hansen, T.V. Some Biogenetic Considerations Regarding the Marine Natural Product (-)-Mucosin. Molecules 2019, 24, 4147. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Li, Y.Y.; Shen, Y.; Li, J.; Li, W.J.; Shen, Y.M. Oxoprothracarcin, a novel pyrrolo[1,4]benzodiazepine antibiotic from marine Streptomyces sp. M10946. Drug. Discov. Ther. 2013, 7, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [Green Version]
- Nalini, S.; Sandy Richard, D.; Mohammed Riyaz, S.U.; Kavitha, G.; Inbakandan, D. Antibacterial macro molecules from marine organisms. Int. J. Biol. Macromol. 2018, 115, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Karpinski, T.M. Marine Macrolides with Antibacterial and/or Antifungal Activity. Mar. Drugs 2019, 17, 241. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Current Status of Marine-Derived Compounds as Warheads in Anti-Tumor Drug Candidates. Mar. Drugs 2017, 15, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, R.C.; Ng, T.B.; Wong, J.H.; Chen, Y.; Chan, W.Y. Marine natural products with anti-inflammatory activity. Appl. Microbiol. Biotechnol. 2016, 100, 1645–1666. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.S.; Kim, S.K. Marine-derived polysaccharides for regulation of allergic responses. In Advances in Food Nutrition Research; Academic Press: Cambridge, MA, USA, 2014; Volume 73, pp. 1–13. [Google Scholar]
- Santos, M.F.; Harper, P.M.; Williams, D.E.; Mesquita, J.T.; Pinto, E.G.; da Costa-Silva, T.A.; Hajdu, E.; Ferreira, A.G.; Santos, R.A.; Murphy, P.J.L.; et al. Anti-parasitic Guanidine and Pyrimidine Alkaloids from the Marine Sponge Monanchora arbuscula. J. Nat. Prod. 2015, 78, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, S.; Hodges, T.W.; Rajbhandari, I.; Gerwick, W.H.; Hamann, M.T.; Nagle, D.G. Marine natural products as novel antioxidant prototypes. J. Nat. Prod. 2003, 66, 605–608. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Liu, J.; Yang, B.; Lin, X.; Yang, X.W.; Liu, Y. Marine natural products with anti-HIV activities in the last decade. Curr. Med. Chem. 2013, 20, 953–973. [Google Scholar] [PubMed]
- Hara, M.; Takiguchi, T.; Ashizawa, T.; Gomi, K.; Nakano, H. Sapurimycin, new antitumor antibiotic produced by Streptomyces. Producing organism, fermentation, isolation and biological properties. J. Antibiot. 1991, 44, 33–39. [Google Scholar]
- Hou, X.F.; Song, Y.J.; Zhang, M.; Lan, W.; Meng, S.; Wang, C.; Pan, H.X.; Cao, C.; Tang, G.L. Enzymology of Anthraquinone-gamma-Pyrone Ring Formation in Complex Aromatic Polyketide Biosynthesis. Angew. Chem. Int. Ed. 2018, 57, 13475–13479. [Google Scholar] [CrossRef]
- Gonzales, G.B.; Smagghe, G.; Grootaert, C.; Zotti, M.; Raes, K.; Van Camp, J. Flavonoid interactions during digestion, absorption, distribution and metabolism: A sequential structure-activity/property relationship-based approach in the study of bioavailability and bioactivity. Drug Metab. Rev. 2015, 47, 175–190. [Google Scholar] [CrossRef]
- Lu, S.; Tian, J.; Sun, W.; Meng, J.; Wang, X.; Fu, X.; Wang, A.; Lai, D.; Liu, Y.; Zhou, L. Bis-naphtho-gamma-pyrones from fungi and their bioactivities. Molecules 2014, 19, 7169–7188. [Google Scholar] [CrossRef] [Green Version]
- Maskey, R.P.; Helmke, E.; Fiebig, H.H.; Laatsch, H. Parimycin: Isolation and structure elucidation of a novel cytotoxic 2,3-dihydroquinizarin analogue of gamma-indomycinone from a marine streptomycete isolate. J. Antibiot. 2002, 55, 1031–1035. [Google Scholar] [CrossRef] [Green Version]
- Murphy, B.T.; Narender, T.; Kauffman, C.A.; Woolery, M.; Jensen, P.R.; Fenical, W. Saliniquinones A-F, New Members of the Highly Cytotoxic Anthraquinone-gamma-Pyrones from the Marine Actinomycete Salinispora arenicola. Aust. J. Chem. 2010, 63. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.C.; Matsui, S.I.; Beerman, T.A. Hedamycin, a DNA alkylator, induces (gamma)H2AX and chromosome aberrations: Involvement of phosphatidylinositol 3-kinase-related kinases and DNA replication fork movement. Mol. Cancer Ther. 2005, 4, 1175–1185. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, R.W.; Davidson, B.S.; Montenegro, D.A.; Bernan, V.S. Gamma-indomycinone, a new pluramycin metabolite from a deep-sea derived actinomycete. J. Nat. Prod. 1995, 58, 613–617. [Google Scholar] [CrossRef]
- Qu, F.; Meng, L.; Yu, J.; Liu, J.; Sun, J.; Yang, H.; Dong, L. Influences of micro-geomorphology on the stoichiometry of C, N and P in Chenier Island soils and plants in the Yellow River Delta, China. PLoS ONE 2017, 12, e0189431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Zhang, G.; Zhang, S.; Sun, J.; Zhao, Y.; Shao, H.; Liu, J. Photosynthetic and water use characteristics in three natural secondary shrubs on Shell Islands, Shandong, China. Plant Biosyst. 2014, 148, 109–117. [Google Scholar] [CrossRef]
- Koutsaviti, A.; Daskalaki, M.G.; Agusti, S.; Kampranis, S.C.; Tsatsanis, C.; Duarte, C.M.; Roussis, V.; Ioannou, E. Thuwalallenes A-E and Thuwalenynes A-C: New C15 Acetogenins with Anti-Inflammatory Activity from a Saudi Arabian Red Sea Laurencia sp. Mar. Drugs 2019, 17, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javidpour, P.; Das, A.; Khosla, C.; Tsai, S.C. Structural and biochemical studies of the hedamycin type II polyketide ketoreductase (HedKR): Molecular basis of stereo- and regiospecificities. Biochemistry 2011, 50, 7426–7439. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Khosla, C. In vivo and in vitro analysis of the hedamycin polyketide synthase. Chem. Biol. 2009, 16, 1197–1207. [Google Scholar] [CrossRef] [Green Version]
- Bililign, T.; Hyun, C.G.; Williams, J.S.; Czisny, A.M.; Thorson, J.S. The hedamycin locus implicates a novel aromatic PKS priming mechanism. Chem. Biol. 2004, 11, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2017, e1327. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pos. | δH (mult., J = Hz) | δc | 1H-1H COSY | HMBC |
|---|---|---|---|---|
| 2 | 174.9s | |||
| 3 | 6.73 (s) | 111.6d | C-14, C-2, C-4, C-4a | |
| 4 | 182.1s | |||
| 4a | 120.6s | |||
| 5 | 138.0s | |||
| 6 | 7.47 (s) | 129.9d | C-13, C-12a, C-7, C-4a, C-6a | |
| 6a | 142.6s | |||
| 7 | 7.43 (s) | 117.4d | C-8, C-6, C-11a, C-6a, C-11 | |
| 7a | 147.8s | |||
| 8 | 4.95 (dd, 7.5, 3.8) | 69.0d | H-9 | C-10, C-7, C-7a |
| 9 | 2.18, 2.37 (m) | 32.7t | H-8, H-10 | C-10, C-8, C-7a, C-11 |
| 10 | 2.81, 3.00 (dq, 17.6, 4.6) | 36.8t | H-9 | C-9, C-8, C-11 |
| 11 | 207.0s | |||
| 11a | 113.3s | |||
| 12 | 165.8s | |||
| 12a | 114.7s | |||
| 12b | 159.1s | |||
| 13 | 4.16,4.26 (m) | 42.9t | C-13-COOH, C-5, C-4a, C-6 | |
| 13-COOH | 175.7s | |||
| 14 | 78.1s | |||
| 15 | 1.55 (s) | 24.0q | C-3, C-2, C-16 | |
| 16 | 5.28 (d, 10.5) | 71.5d | H-17 | C-3, C-2, C-15 |
| 17 | 5.73 (ddd, 11.7, 10.5, 1.5) | 130.0d | H-16, H-18 | C-19, C-16, C-14 |
| 18 | 5.88 (dq, 11.7, 6.8) | 130.1d | H-17, H-19 | C-19, C-16 |
| 19 | 1.92 (dd, 6.9, 1.8) | 14.4q | H-18 | C-17, C-16 |
| Pos. | δH (mult., J = Hz) | δc | 1H-1H COSY | HMBC |
|---|---|---|---|---|
| 2 | 174.9s | |||
| 3 | 6.72 (s) | 111.6d | C-14, C-4a, C-2 | |
| 4 | 182.5s | |||
| 4a | 121.1s | |||
| 5 | 138.4s | |||
| 6 | 7.59 (s) | 130.3d | C-13, C-4a, C-7, C-12a | |
| 6a | 143.0s | |||
| 7 | 7.54 (s) | 117.7d | C-8, C-11a, C-6, C-6a | |
| 7a | 148.3s | |||
| 8 | 5.00 (dd, 8.6, 3.9) | 69.3d | H-9 | |
| 9 | 2.21, 2.40 (m) | 33.0t | H-8, H-10 | |
| 10 | 2.87, 3.04 (m) | 37.0t | H-9 | |
| 11 | 207.3 | |||
| 11a | 113.7s | |||
| 12 | 166.2s | |||
| 12a | 115.3s | |||
| 12b | 159.7s | |||
| 13 | 4.26-4.33 (m) | 43.3t | C-13-COOH, C-4a, C-6, C-5 | |
| 13-COOH | 176.2s | |||
| 14 | 78.5s | |||
| 15 | 1.65 (s) | 24.2q | C-3, C-2, C-16 | |
| 16 | 4.19 (dd, 10.2, 1.8) | 76.7d | H-17 | |
| 17 | 1.58, 1.69 (m) | 34.3t | H-16, H-18 | |
| 18 | 1.47, 1.68 (m) | 21.3t | H-17, H-19 | |
| 19 | 1.02 (t, 6.9, 1.8) | 15.1q | H-18 | C-17, C-16, C-14 |
| Pos. | δH (mult., J = Hz) | δc | 1H-1H COSY | HMBC |
|---|---|---|---|---|
| 2 | 174.8s | |||
| 3 | 6.67 (s) | 109.8d | C-14, C-4a, C-2 | |
| 4 | 182.6s | |||
| 4a | 121.0s | |||
| 5 | 138.3s | |||
| 6 | 7.58 (s) | 130.4d | C-13, C-4a, C-7, C-12a | |
| 6a | 143.0s | |||
| 7 | 7.54 (s) | 117.6d | C-8, C-11a, C-6, C-6a | |
| 7a | 148.4s | |||
| 8 | 5.00 (dd, 8.7, 4.0) | 69.4d | H-9 | C-7, C-7a |
| 9 | 2.21, 2.40 (m) | 33.1t | H-8, H-10 | |
| 10 | 2.87, 3.04 (m) | 37.1t | H-9 | C-8, C-9 |
| 11 | 207.2s | |||
| 11a | 113.8s | |||
| 12 | 166.2s | |||
| 12a | 115.1s | |||
| 12b | 159.7s | |||
| 13 | 4.28 (m) | 43.3t | C-13-COOH, C-4a, C-6, C-5 | |
| 13-COOH | 176.1s | |||
| 14 | 74.8s | |||
| 15 | 1.82 (s) | 28.2q | C-17, C-2, C-14 | |
| 16 | 6.25 (dd, 15.5, 1.3) | 133.9d | H-17 | C-14, C-18 |
| 17 | 6.05 (dd, 15.5, 5.8) | 136.3d | H-16, H-18 | C-14, C-18 |
| 18 | 4.32 (m) | 69.5d | H-17, H-19 | C-16 |
| 19 | 1.26 (t, 6.9, 1.8) | 24.4q | H-18 | C-17, C-18 |
| Pos. | δH (mult., J = Hz) | δc | 1H-1H COSY | HMBC |
|---|---|---|---|---|
| 2 | 174.9s | |||
| 3 | 6.67 (s) | 109.8d | C-14, C-4a, C-2 | |
| 4 | 182.6s | |||
| 4a | 121.0s | |||
| 5 | 138.3s | |||
| 6 | 7.57 (s) | 130.4d | C-13, C-4a, C-7, C-12a | |
| 6a | 143.0s | |||
| 7 | 7.53 (s) | 117.6d | C-8, C-11a, C-6, C-6a | |
| 7a | 148.4s | |||
| 8 | 5.00 (dd, 8.7, 4.0) | 69.4d | H-9 | C-7, C-7a |
| 9 | 2.21, 2.40 (m) | 33.1t | H-8, H-10 | |
| 10 | 2.87, 3.04 (m) | 37.1t | H-9 | C-8, C-9 |
| 11 | 207.2s | |||
| 11a | 113.8s | |||
| 12 | 166.2s | |||
| 12a | 115.1s | |||
| 12b | 159.6s | |||
| 13 | 4.24-4.31 (m) | 43.3t | C-13-COOH, C-4a, C-6, C-5 | |
| 13-COOH | 176.1s | |||
| 14 | 74.9s | |||
| 15 | 1.82 (s) | 28.3q | C-17, C-2, C-14 | |
| 16 | 6.26 (d, 15.5) | 133.8d | H-17 | C-14, C-18 |
| 17 | 6.05 (dd, 15.5, 5.6) | 136.2d | H-16, H-18 | C-14, C-18 |
| 18 | 4.33 (m) | 69.4d | H-17, H-19 | C-16 |
| 19 | 1.25 (t, 6.9, 1.8) | 24.4q | H-18 | C-17, C-18 |
| Pathogen Strains | MIC (µg/mL) | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Candida albicans ATCC 18804 | >100 | >100 | >100 | >100 |
| Staphylococcus aureus ATCC 25923 | 3 | 21 | 46 | 16 |
| Enterococcus faecalis ATCC 29212 | 6 | 13 | 36 | 21 |
| Bacillus subtilis ATCC 6633 | 11 | 26 | 32 | 21 |
| Pseudomonas aeruginosa ATCC 27853 | >100 | >100 | >100 | >100 |
| Mycobacterium smegmatis ATCC 700084 | >100 | >100 | >100 | >100 |
| Compound | IC50 (µM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A375 | H1299 | HepG2 | HT29 | HCC1937 | ||||||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |
| 1 | 5.22 | 0.69 | 8.63 | 1.32 | 4.22 | 0.89 | 4.69 | 0.85 | 9.68 | 2.62 |
| 2 | 7.18 | 0.95 | 10.3 | 2.62 | 5.67 | 1.35 | 6.12 | 1.12 | 11.6 | 3.11 |
| 3 | 40.3 | 11.3 | >50 | 26.3 | 11.3 | 5.03 | 13.0 | 4.33 | >50 | 12.6 |
| 4 | 6.04 | 0.78 | 10.1 | 2.15 | 5.16 | 1.11 | 5.37 | 1.02 | 10.3 | 2.89 |
| cis-platinum | 23.1 | 9.32 | 21.2 | 7.3 | >50 | 18.9 | >50 | >50 | >50 | >50 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Wang, Y.; Yang, Y.; Chen, H. Shellmycin A–D, Novel Bioactive Tetrahydroanthra-γ-Pyrone Antibiotics from Marine Streptomyces sp. Shell-016. Mar. Drugs 2020, 18, 58. https://doi.org/10.3390/md18010058
Han Y, Wang Y, Yang Y, Chen H. Shellmycin A–D, Novel Bioactive Tetrahydroanthra-γ-Pyrone Antibiotics from Marine Streptomyces sp. Shell-016. Marine Drugs. 2020; 18(1):58. https://doi.org/10.3390/md18010058
Chicago/Turabian StyleHan, Yong, Yan Wang, Yuehan Yang, and Haotong Chen. 2020. "Shellmycin A–D, Novel Bioactive Tetrahydroanthra-γ-Pyrone Antibiotics from Marine Streptomyces sp. Shell-016" Marine Drugs 18, no. 1: 58. https://doi.org/10.3390/md18010058
APA StyleHan, Y., Wang, Y., Yang, Y., & Chen, H. (2020). Shellmycin A–D, Novel Bioactive Tetrahydroanthra-γ-Pyrone Antibiotics from Marine Streptomyces sp. Shell-016. Marine Drugs, 18(1), 58. https://doi.org/10.3390/md18010058