2. Results and Discussion
The freeze-dried fronds of B. bifurcata were successively extracted with CH2Cl2 and MeOH. A solvent–solvent extraction of the combined organic extract by a modified Kupchan partition scheme afforded the n-hexanes, CHCl3 and aqueous subextracts. The n-hexanes and CHCl3 subextracts were subjected to automated flash chromatography and RP-HPLC to yield compounds 1–8.
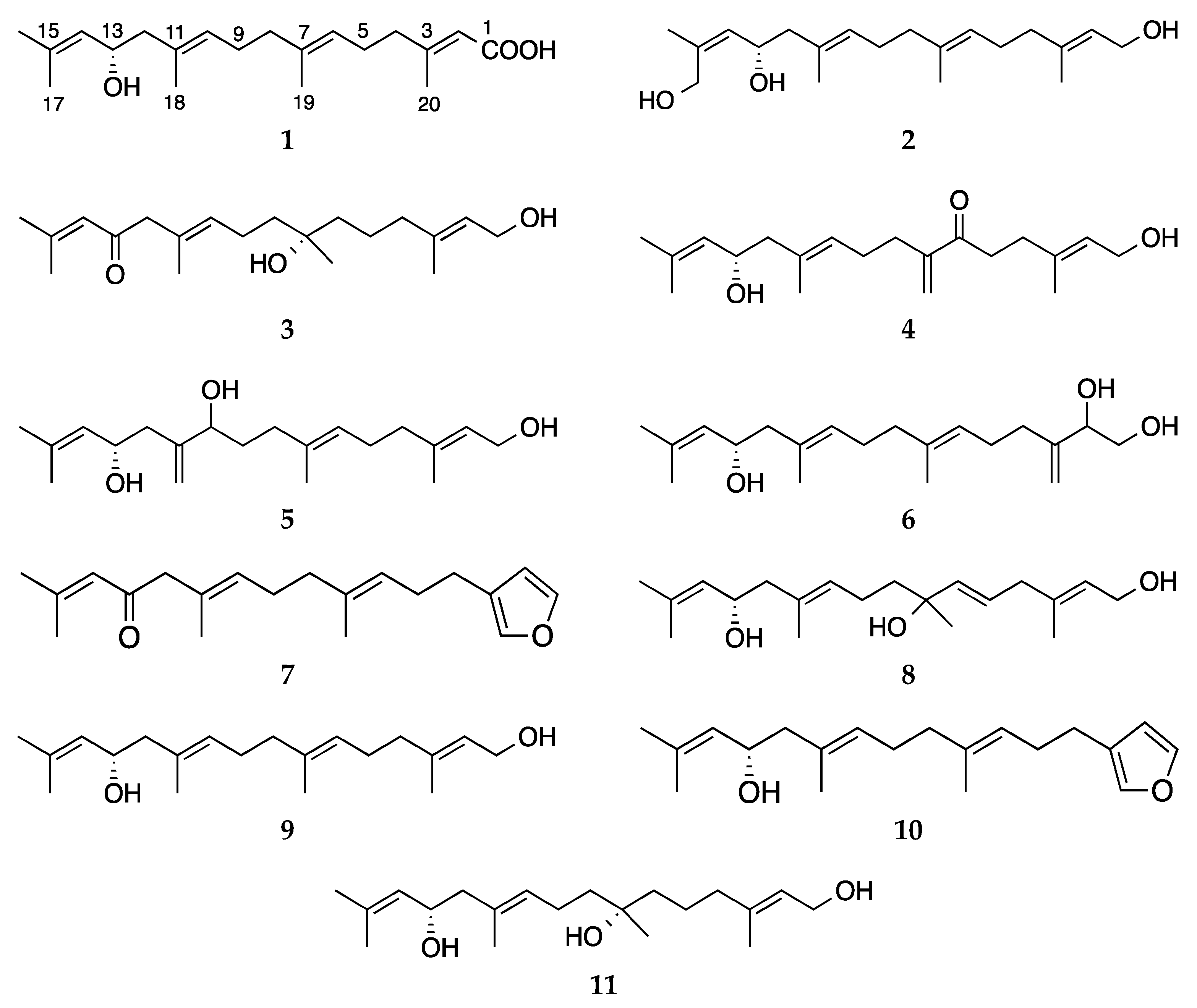
Compound
1 was isolated as a colorless oil. The molecular formula C
20H
32O
3 was assigned based on a sodium adduct
m/z 343.2247 [M + Na]
+ observed in its HRESIMS spectrum, indicating five double bond equivalents (DBEs). Its IR spectrum contained absorption bands typical for hydroxyl, conjugated carboxylic acid and alkene functions (
νmax 3369, 1690 and 1642 cm
−1, respectively). The
1H NMR spectrum of
1 (
Table 1) revealed the presence of four methine protons at δ
H 5.06 (t,
J = 5.5 Hz, H-6), δ
H 5.13 (br. d,
J = 8.4 Hz, H-14), δ
H 5.18 (t,
J = 6.7 Hz, H-10) and δ
H 5.66 (br. s, H-2) and five olefinic methyl singlets at δ
H 1.70 (H
3-16), δ
H 1.67 (H
3-17), δ
H 1.63 (H
3-18), δ
H 1.58 (H
3-19) and δ
H 2.15 (H
3-20), plus an oxymethine proton at δ
H 4.38 (td,
J = 8.4, 5.3 Hz, H-13). The
13C NMR spectrum (
Table 2) contained 20 carbon resonances, which, in conjunction with the
gHSQC spectrum, were sorted out into a carbonyl group (δ
C 169.1, C-1), four quaternary sp
2, four sp
2 methine, one sp
3 oxymethine, five sp
3 methylene and four tertiary methyl carbons. The comparison of these data with those of the known compound eleganediol (
9) [
14] indicated that
1 is a linear diterpene containing an OH substitution, four double bonds and a carboxylic acid group, fulfilling the required number (5) of DBEs within
1.
In order to confirm and the position of the carboxylic acid and the OH substitution, and thereby the planar structure of 1, we ran gCOSY and gHMBC experiments. Three short spin systems were visible in the COSY spectrum. The first two proton networks included two methylene and one olefinic methine protons each, i.e., H2-4/H2-5/H-6 (fragment a) and H2-8/H2-9/H-10 (fragment b). The third network consisted of H2-12, the oxymethine proton (H-13, δH 4.38) and the olefinic methine H-14, yielding the fragment c. The obtained COSY fragments a–c were readily connected through 1H-13C HMBC correlations between H-6/C-8 and H3-19/C-6; H-10/C-12 and H-10/CH3-18, and from terminal methyl groups H3-16 and H3-17 to both C-14 and C-15. The significantly downfield resonances of C-3 (δC 162.6), H3-20 (δH 2.15) and H-2 (δH 5.66), as well as the appearance of H-2 as an isolated broad singlet, suggested the presence of a terminal carboxylic acid at C-1. Further evidence for the location of the carboxylic acid at C-1 was provided by the diagnostic HMBC cross peaks between H-2/C-1, H3-20/C-1 and H3-20/C-2, plus a W coupling of H-2 with H2-4 in the COSY spectrum. This finalised the gross structure of compound 1.
The NOESY spectrum of
1 assisted in the identification of the
E geometry of all double bonds, on the basis of key NOE couplings observed between H-2/H
2-4 (for C-2), H-6/H
2-4, H-6/H
2-8 (for C-6), H-10/H
2-8, H-10/H
2-12 (for C-10) and finally between H-14/H
2-12 (for C-14). The assignment of H
3-16 as
pro-
E and H
3-17 as
pro-
Z was possible due to additional NOE correlations between H-14/H
3-16 and H-13/H
3-17, respectively. The absolute configuration of the only stereogenic center at C-13 was assigned by vibrational circular dichroism (VCD) spectroscopy. We have shown in a previous VCD study on eleganediol (
9) and bifurcane (
10) [
14] that the C-13 stereocenter gives rise to a distinct VCD spectroscopic signature which is independent of the substitution at C-1. The experimental VCD spectrum of
1 showed the same spectral pattern as observed for
9 (
Supplementary Materials Figure S42), which led us to conclude without further theoretical spectra analysis that the AC of
1 is also 13
S.The molecular formula C
20H
34O
3 was assigned to compound
2 by its HRESIMS data (
m/z 345.2398 [M + Na]
+). Compound
2 was identified as possessing a 13
S-hydroxygeranylgeraniol scaffold with high similarities to eleganediol (
9). However, 1D NMR and the
gHSQC spectra indicated that one of the vinylic methyl signals (CH
3-17) was replaced with a primary alcohol group (δ
H 3.99 d and δ
H 4.25 d,
J = 12.4 Hz; δ
c 62.3, t). These methylene protons only showed geminal coupling with each other on the
gCOSY spectrum. The HMBC spectrum that contained correlations between H
3-16/C-17; H
2-17/C-14 and H
2-17/C-16 clearly proved that the H
3-17 methyl group was converted to a hydroxymethylene function, completing the structure of
2. The all-
E geometry of the double bond carbons and
S configuration of C-13 were confirmed by the characteristic
13C and
1H resonances of
2 that were similar to those of compounds
1 and
9 [
14,
15].
The HRESIMS analysis of compound
3 revealed a molecular formula C
20H
34O
3 (m/z 345.2402 [M + Na]
+). The detailed inspection of the 1D and 2D NMR data of compound
3 indicated that it was an analogue of bifurcatriol (
11), which is 13
S,7
S-dihydroxygeranylgeraniol [
15]. The only difference between the two compounds was the conversion of the C-13-OH functionality in
11 to a ketone group (δ
C 197.7) in
3, which was also confirmed by the IR absorption band at
νmax 1703 cm
−1. The oxidation at C-13 led to deshielding of H-14 (δ
H 5.96), H
3-17 (δ
H 2.12) and H
2-12 (δ
H 2.97, 2H), with the latter resonating as an isolated singlet (
Table 1). The position of the C-13 ketone group was verified by the key HMBC correlations observed between H
2-12/C-13 and H-14/C-13. The inspection of its full 2D NMR data (DEPT-HSQC, COSY, HMBC and NOESY) supported the suggested structure of the ketoalcohol
3, as shown in
Figure 1. An
S configuration was assigned to C-7 based on comparison of the 1D NMR data of
3 with those of bifurcatriol (
11) [
15], the C-13-hydroxy derivative of
3.
Compound
4 was isolated as a colorless oil. The HRESIMS of
4 returned a sodium adduct ion at
m/z 343.2246 [M + Na]
+ in agreement with a molecular formula C
20H
32O
3 requiring five DBEs. A careful investigation of its 1D and 2D NMR spectra indicated that
4 lacked one of the olefinic methyl signals, but contained instead two broad singlets at δ
H 5.99 and δ
H 5.72, and a CH
2 signal at δ
C 124.2 (t). Compound
4 had identical NMR data to those of
1 and
9 regarding the three double bonds (∆
2,3, ∆
10,11 and ∆
14,15) and four olefinic methyl groups (CH
3-16, CH
3-17, CH
3-18 and CH
3-20), hence CH
3-19 must have converted into an exomethylene group, to fulfil the fourth DBE. On the other hand, the notably deshielded resonance of H
3-19 and H
2-5 (δ
H 2.79 dd,
J = 7.9, 7.5 Hz,
Table 1) suggested the presence of a strong electronegative group in their vicinity. The
13C signal at δ
C 201.2 (C-6) and the FT-IR absorption band at
νmax 1718 cm
−1 of
4 supported the presence of a ketone group either at C-6 or C-8. The latter option (C-8-oxo) was ruled out, as H
2-8 was part of a proton network composed of H-10 (δ
H 5.19), H
2-9 (δ
H 2.14) and H
2-8 (δ
H 2.30) in the
gCOSY spectrum of
4 (
Figure S26). Thus, the ketone group was assigned to C-6. An in-depth analysis of the cross peaks in the HMBC spectrum of
4, particularly those between H
2-8/C-19, H-9/C-8, H
2-19/C-7 and H
2-19/C-8 lent proof for the presence of the exomethylene function at C-19, while further HMBC correlations from H
2-19, H
2-4, H
2-5 and H
2-8 to C-6 corroborated the position of the oxo function at C-6. Identical NMR data around the only stereocenter C-13 was indicative of the same
S configuration at C-13.
Compound
5 had a molecular formula C
20H
34O
3 deduced by its HRMS data (
m/z 345.2405 [M + Na]
+). Analysis of the NMR spectra indicated that
5 is another 13
S-hydroxygeranylgeraniol derivative comprising four olefinic methyl groups. It was evident from two singlets at δ
H 4.95 and δ
H 5.08 (1H each) that correlated with the CH
2 signal at δ
C 114.3 on the
gHSQC spectrum that one of the methyl groups (CH
3-18) was converted into an exomethylene group in
5. Furthermore, NMR signals at δ
H 4.06 (dd,
J = 7.8, 5.5 Hz, H-10) and δ
c 74.9 (C-10) suggested the presence of an additional secondary alcohol group (
Table 1 and
Table 2). The HMBC spectrum contained key correlations from H-10 and H
2-12 to C-18; from H
2-18 to C-10, C-11 and C-12; and finally from H
2-8 and H
2-9 to C-10 assigned, respectively, the exocyclic double bond to C-18 and the second OH group to H-10. Due to rapid decomposition of the compound
5 during the NOESY experiment, the stereochemistry at C-10 could not be assigned. The all-
E geometry of the double bond carbons and
S configuration of C-13 were confirmed by their characteristic
13C and
1H NMR resonances [
14,
15].
As deduced by HREIMS (m/z 345.2396 [M + Na]+), the new compound 6 was determined as a positional isomer of compound 5 with the same molecular formula C20H34O3. The only difference between the two compounds was the position of both the exocyclic double bond (δH 4.95 s, δH 5.13 s; δc 111.0 t) and the additional secondary OH group (δH 4.16 dd, J = 7.2, 3.4 Hz, δc 74.9). Thanks to the HMBC correlations observed between (i) from H2-20 to C-2, C- 3 and C-4, (ii) from H-2 to C-20, and from H2-4 to C-20 and (iii) from H-2 to C-1 and C-3, the olefinic methylene group was assigned to C-20 and the secondary alcohol to C-2. Similar to 5, compound 6 had low stability and decomposed during the final 2D NMR experiment (NOESY), hence the stereochemistry of C-2 remains unassigned.
In vitro anticancer activity of the isolated compounds was evaluated against the breast cancer cell line MDA-MB-231. The only compounds that displayed activity were the new compound 1 (IC50 30.7 μg/mL) and the known compounds 9 (IC50 11.6 μg/mL) and 10 (IC50 32.0 μg/mL). Compounds 7, 8 and 11 were inactive even at the highest test concentrations (100 μg/mL), while the remaining new compounds 2–6 were not tested due to availability of minute amounts or stability issues.
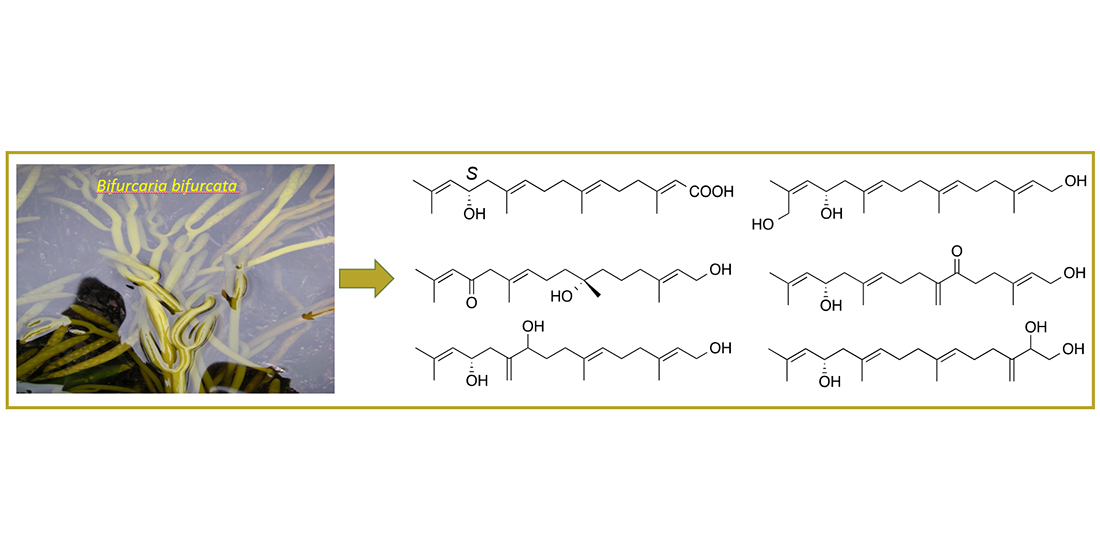
The present study has shown the Irish brown alga
B. bifurcata to contain a wide array of linear diterpenes. Oxygenation in multiple positions is common in many acyclic diterpenes obtained from this seaweed [
2,
3,
16,
17,
18]. Notably, all compounds isolated herein possessed oxygenation at C-13 accompanied by additional hydroxy, ketone, carboxylic acid substitutions or a terminal furan ring. We have previously applied VCD spectroscopy for identification of the absolute configuration (
S) of linear diterpenes from this seaweed, e.g., eleganediol (
9) and bifurcane (
10) [
14], with a single stereocenter (C-13) as well as for bifurcatriol (
11) that bears two stereocenters (C-7, C-13) [
15]. In the present study, it was again successfully applied to confirm the (S)-configuration of C-13 in compound
1. By structural analogy, NMR data comparison and biosynthetic considerations, we easily assigned 13
S stereochemistry to the other new compounds
2–
6.
Numerous algal metabolites belonging to different natural product classes have been shown to exert anticancer activity [
19,
20]. The halogenated monoterpene halomon or the highly oxygenated triterpene laurenmariannol represent well-known examples of seaweed-derived terpenes with low to submicromolar level cytotoxicity [
19], indicating the understudied potential of seaweeds. As for
B. bifurcata, several C-12 hydroxy-bearing linear diterpenes have been reported with such potential and their potency [
19,
20,
21,
22] is generally similar to C-13 OH-bearing acyclic diterpenes isolated in this study.
In summary, the current study implies the diversity of polyoxygenated acyclic diterpenes in Irish brown alga
B. bifurcata. Only three compounds showed modest anticancer activity against human breast cancer cells (MDA-MB-231). Several compounds could not be assessed for their bioactivity due to stability issues or low amounts available. Some compounds obtained in large amounts previously (e.g.,
9 and
10) [
14] can be submitted to semi-synthesis studies for enhancement of their anticancer potency and assessment of their further biological and ecological activities.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were determined with a Unipol L1000 Schmidt + Haensch polarimeter at the sodium D line (589.3 nm), at 20 °C, with a 10 cm cell. UV spectra were acquired in spectroscopic grade CHCl3 or MeOH on a Varian, Cary 100 UV-Vis spectrophotometer. IR spectra were recorded on a Perkin Elmer 400 or a Perkin Elmer Spectrum One ATR FT-IR spectrometer. NMR spectra were acquired using a Jeol 400 MHz, a Varian 500 MHz or an Agilent 600 MHz spectrometer. Chemical shifts are given on the δ (ppm) scale, referenced to the residual solvent signal (CDCl3: δH 7.24, δC 77.0 or C6D6: δH 7.16, δC 128.0), and J values in Hz. High-resolution mass spectrometric data were measured on an Agilent QTOF 6540 MS system, with electrospray ionisation (ESI) in the positive ion mode, coupled to an Agilent 1290 Infinity UPLC system, operating on the elution gradient: 50% B for 8 min, increasing to 100% B in 3 min, maintaining 100% B for 5 min (solvent A: H2O + 0.1% formic acid, solvent B: MeCN + 0.1% formic acid), on a Zorbax Eclipse Plus C18 RRHD (50 × 2.1 mm, 1.8 μm) column, at 0.5 mL/min, with UV detection at 200−600 nm. Thin layer chromatography (TLC) was performed with Kieselgel 60 F254 aluminium support plates (Merck) and spots were detected after spraying with 6% vanillin and 15% H2SO4 in MeOH reagent. All solvents were of HPLC or LCMS grade and were purchased from Sigma-Aldrich.
3.2. Algal Material
Bifurcaria bifurcata was collected from the intertidal rock pool at Kilkee, Co. Clare of Ireland, in May 2009. A voucher specimen is kept at the Herbarium of the Biodiscovery Laboratory in the Irish Marine Institute (BDV0015).
3.3. Extraction and Isolation
The algal thalli were cleaned with sea water. CH2Cl2 and MeOH were used for exhaustive extraction of the freeze-dried material (132.4 g dry weight) at room temperature. The organic extracts were combined and evaporated to dryness on a rotary evaporator. The resulting dark green paste (12.0 g) was subjected to a modified Kupchan liquid–liquid partition. Briefly, the crude extract was dissolved in 90% MeOH (200 mL) and partitioned against n-hexanes (3 × 200 mL). The water concentration was increased to 35%, before partitioning against CHCl3. Evaporation of the solvents under reduced pressure and temperature afforded n-hexanes (3.6 g) and CHCl3 subextracts (7.6 g).
An aliquot of the n-hexanes subextract (2.3 g) was chromatographed by flash column chromatography over silica gel using a 10% gradient of EtOAc in hexane to afford nine combined fractions (H1–H9). Fr. H2 (547 mg) which, eluted with 20% EtOAc, was subjected to fractionation on an Agilent 971FP system loaded with a pre-packed silica column SF15-12g (Agilent), operating at the following gradient: 0% B for 10 min, increasing to 6% B in 50 min, increasing to 10% B in 20 min, increasing to 100% B in 10 min, maintaining 100% B for 15 min (solvent A: n-hexanes, solvent B: EtOAc), at a flow of 15 mL/min, to afford 7 (1.2 mg).
The CHCl3 subextract (6.8 g) was subjected to gradient flash CC fractionation (Agilent 971FP system, pre-packed Agilent silica column SF25-80g, operating with the following gradient: 0% B for 5 min, to 5% B in 15 min, at 5% B for 10 min, to 10% B in 10 min, at 10% B for 10 min, to 40% B in 40 min, to 100% B in 10 min, at 100% B for 10 min, solvent A: n-hexanes, solvent B: EtOAc, flow of 25 mL/min, affording 21 fractions (C1–C21). Fr. C4 (43.9 mg) was subjected to RP-HPLC. The separation was conducted using an Agilent 1260 system equipped with a diode array and an ELSD detector (split flow) on a Kromasil 100 C18 5u (250 × 8 mm, 5 μm) HPLC column. The gradient elution using 55% MeCN for 13 min, increasing to 100% MeCN in 5 min and maintaining 100% MeCN for 20 min (solvent A: H2O, solvent B: MeCN), at a flow of 1.5 mL/min, afforded 1 (11.2 mg). Fr. C10 (118.0 mg) was subjected to RP-HPLC on the same system, using the same solvent gradient to afford pure 4 (1.0 mg, tR 25.9 min). Fr. C12 (66.0 mg) was separated by RP-HPLC under the same conditions to afford pure 8 (1.1 mg, tR 22.7 min), 3 (1.0 mg, tR 25.9 min), 5 (7.2 mg, tR 26.2 min) and 6 (1.7 mg, tR 26.8 min). Finally, the RP-HPLC purification of fr. C16 (49.0 mg) under the same conditions yielded 2 (0.9 mg).
3.4. VCD Spectroscopy
The IR and VCD spectra of 1 were recorded on a Bruker Vertex 70V spectrometer equipped with a PMA 50 module for polarisation-modulated measurements. Samples were held in a sealed BaF2 IR cell with 100 µm path length (Specac). Both IR and VCD spectra were recorded at 4 cm−1 spectral resolution by accumulating 32 and ~20,000 scans, respectively. Baseline correction of the spectra was done by subtraction of the solvent spectra measured under identical conditions.
Compound
1: colorless oil; [α]
D +9.4 (
c 0.32, MeOH), +4.6 (c 1.13, CHCl
3); UV (CHCl
3)
λmax (log
ε) 243 (2.19) nm; IR (thin film)
νmax 3369, 2917, 1690, 1642, 1437, 1247, 1157, 1022, 868 cm
−1;
1H NMR (500 MHz, CDCl
3) and
13C NMR (125 MHz, CDCl
3) see
Table 1 and
Table 2; HRESIMS
m/z 343.2247 [M + Na]
+ (calcd for C
20H
32O
3Na, 342.2244).
Compound
2: colorless oil; [α]
D −12.7 (
c 0.34, CHCl
3); UV (CHCl
3)
λmax (log
ε) 233 (2.02) nm; IR (thin film)
νmax 3422, 2936, 2872, 1656, 1459, 1385, 1193, 1060 cm
−1;
1H NMR (500 MHz, CDCl
3) and
13C NMR (125 MHz, CDCl
3) see
Table 1 and
Table 2; HRESIMS
m/z 345.2398 [M + Na]
+ (calcd for C
20H
34O
3Na, 345.2400).
Compound
3: colorless oil; [α]
D +6.0 (
c 0.10, MeOH); UV (CHCl
3)
λmax (log
ε) 245 (2.23) nm; IR (thin film)
νmax 3468, 2932, 1703, 1022 cm
−1;
1H NMR (600 MHz, C
6D
6) and
13C NMR (150 MHz, C
6D
6) see
Table 1 and
Table 2; HRESIMS
m/z 345.2402 [M + Na]
+ (calcd for C
20H
34O
3Na, 345.2400).
Compound
4: colorless oil; [α]
D −1.6 (
c 1.11, CHCl
3); UV (CHCl
3)
λmax (log
ε) 246 (2.26) nm; IR (thin film)
νmax 3492, 2957, 2874, 1718, 1456, 1369, 1240, 1030, 897, 753 cm
−1;
1H NMR (500 MHz, CDCl
3) and
13C NMR (125 MHz, CDCl
3) see
Table 1 and
Table 2; HRESIMS
m/z 343.2246 [M + Na]
+ (calcd for C
20H
32O
3Na, 343.2244).
Compound
5: colorless oil; [α]
D −12.7 (
c 0.34, CHCl
3); UV (CHCl
3)
λmax (log
ε) 236 (2.18) nm; IR (thin film)
νmax 3377, 2942, 2861, 1378, 1022 cm
−1;
1H NMR (500 MHz, CDCl
3) and
13C NMR (125 MHz, CDCl
3) see
Table 1 and
Table 2; HRESIMS
m/z 345.2405 [M + Na]
+ (calcd for C
20H
34O
3Na, 345.2400).
Compound
6: colorless oil; [α]
D −3.1 (
c 1.09, CHCl
3); UV (CHCl
3)
λmax (log
ε) 234 (1.98) nm; IR (thin film)
νmax 3448, 3225, 2952, 1683, 1454, 1378, 1019 cm
−1;
1H NMR (600 MHz, CDCl
3) and
13C NMR (150 MHz, CDCl
3) see
Table 1 and
Table 2; HRESIMS
m/z 345.2396 (calcd for C
20H
34O
3Na, 345.2400).
3.5. Anticancer Activity Assessments
The breast cancer cell line MDA-MB-231 (ATCC) was maintained in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Sigma-Aldrich) and incubated at 37 °C, 5% CO2. MDA-MB-231 cells were seeded in a 96-well plate (1 × 104 cells per well) and cultured for 24 h (37 °C, 5% CO2) before being treated with extracts at a final concentration of 0–100 μg/mL (vehicle control: 1% DMSO; positive control: 10 μM 5-Fluorouracil). After 72 h, cell viability was assessed using Alamar Blue assay. Briefly, 40 μL Alamar Blue (0.56 mM) was added to each well containing 200 μL of cell culture medium (93 μM final Alamar Blue concentration). After 6 h incubation, the fluorescence of each well was assessed (λex = 530 nm; λem = 595 nm) using a Victor 3V 1420 multilabel counter. Cell viability was calculated and expressed as a percentage of untreated control cells. The data are the mean ± SD of three experiments and GraphPad Prism software was used to plot the data and to determine the IC50 values.





{kind=link}
{kind=link}