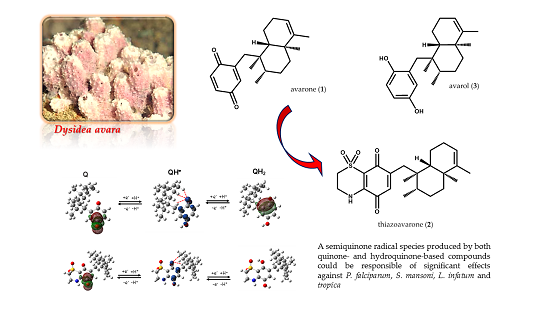
2.1. Chemistry
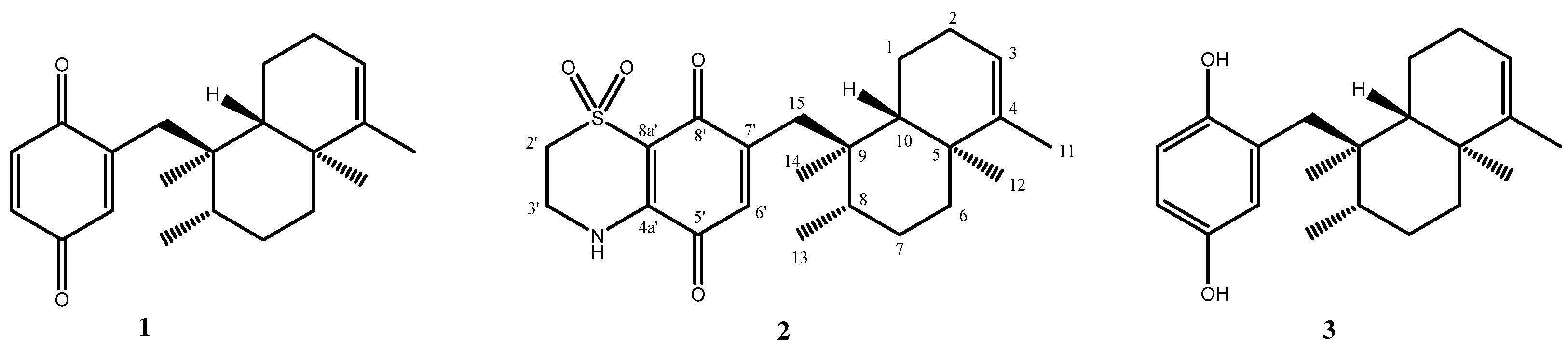
Avarone (
1) and avarol (
3) were isolated from the sponge
D. avara and purified according to the previously described procedures [
21,
22,
23]. They were easily identified by comparison of their spectroscopic properties (
1H and
13C NMR, HRESI-MS) with those reported in literature [
21,
22,
23].
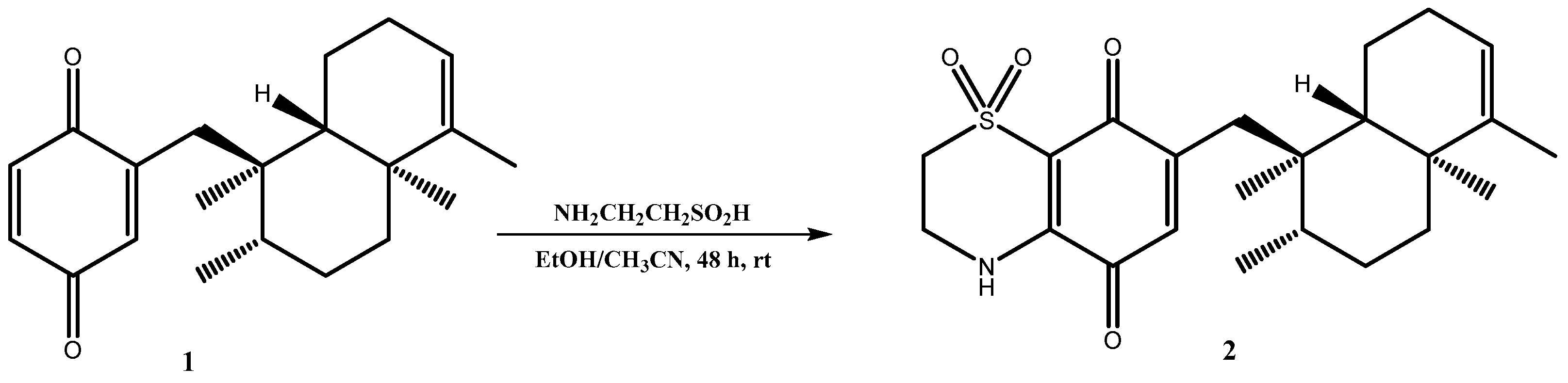
Thiazoavarone (
2) was prepared as reported in
Scheme 1. A portion of avarone was dissolved in a solution of CH
3CN/EtOH (1:1) and then hypotaurine and a catalytic amount of salcomine in portion were added. The mixture was stirred for 48 h at room temperature and then was extracted with diethyl ether. The crude material was purified by HPLC on a reverse phase column (Luna 3 µm, 150 × 3.00 mm) (MeOH/H
2O 75:25 v/v%) to afford the pure compound
2. The nucleophilic addition reaction is regioselective in unsymmetrical quinones but generally leads to the formation of both regioisomers one of which is obtained in large excess with respect to the other. In the case of avarone, a single isomeric product, thiazoavarone
2, has formed, as determined by MS and NMR spectroscopy; this could be reasonably due to the high steric hindrance of the heavy sesquiterpene moiety.
Compound
2, obtained as yellow powder, [α]
D25 = +19.2 (c 0.004, MeOH), had a molecular formula of C
23H
31NO
4S as determined by the HRESI MS ion at m/z C
23H
31NO
4SNa [M + Na]
+ 440.1865 (calculated value: 440.1866). Molecular formula obtained from MS and a first survey of the 1D NMR spectra of
2 (CDCl
3) and the comparison with those of the known avarone
1 quickly allowed us to hypothesize that the condensation reaction with hypotaurine has occurred.
1H and
13C NMR data of
2 indicated the same decalin ring system of
1, and the only difference between the two compounds is in the quinonic portion. Indeed,
1H NMR spectrum of
2 lacked the signals at δ
H 6.51 and 6.71, whereas contained a quite deshielded methylene signals (δ
H3.30 and 4.05), resonating as two multiplets and each integrating for two protons. Likewise, the
13C spectrum of
2 contained two additional methilene carbon resonances at δ
C 39.8 and at δ
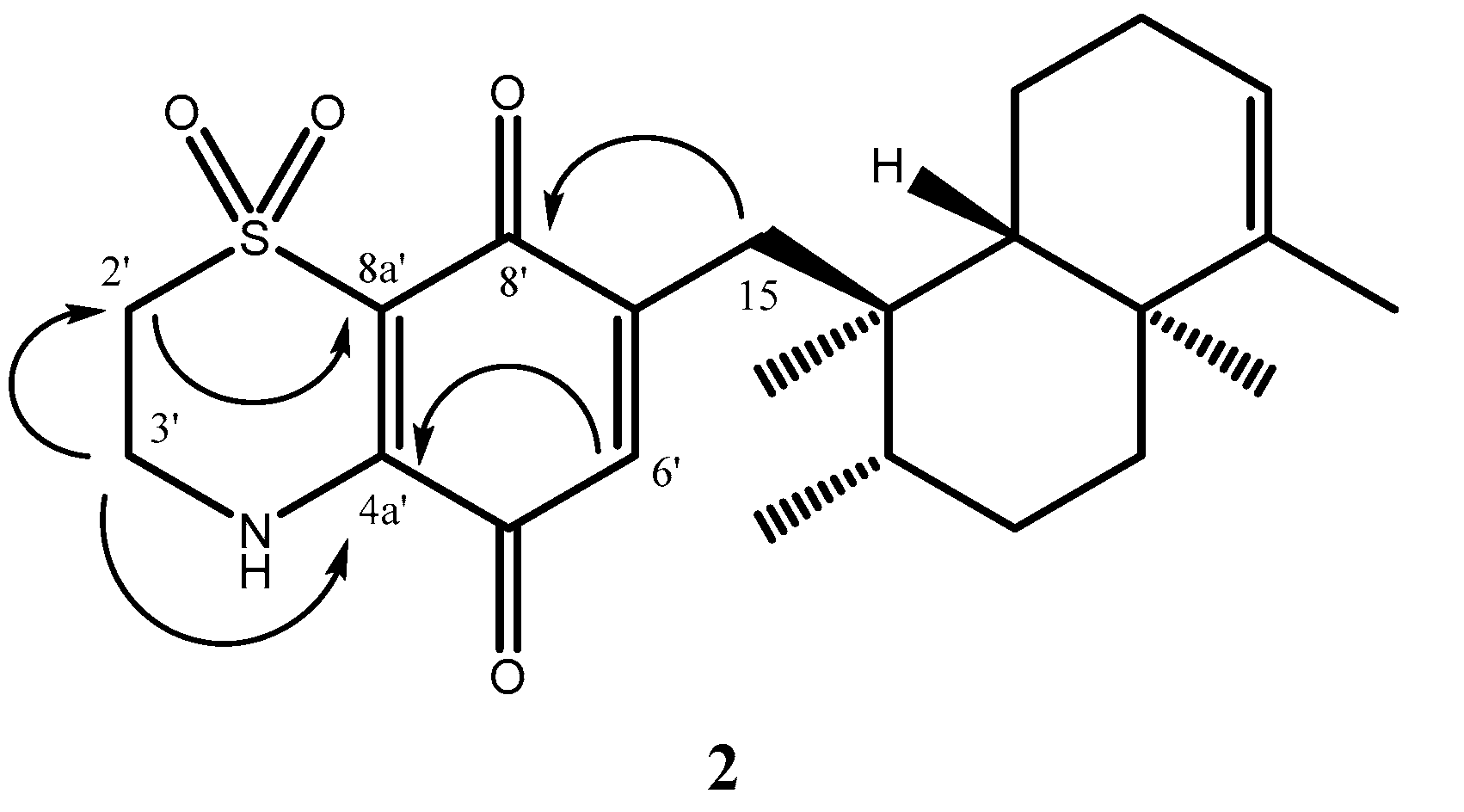
C 48.8 attributable to a nitrogen and sulfoxide-bearing carbons, respectively. The key HMBC cross-peaks (
Figure 3) from H-3′ to C-2′ (δ
C 48.8), C-4a′ (δ
C 143.2), and from H-2′ to C-3′ (δ
C 39.8), C-8a′ (δ
C 111.6), from H-6′ to C-4a′ (δ
C 143.2), and from H-15a and H-15b′ to C-8′ (δ
C 177.1) indicated the regiochemistry of
2. It should be noted that only one of possible regioisomer has been obtained.
Chemical shifts and coupling patterns of the all signals of
2 were assigned by aid of COSY, HSQC, and HMBC experiments (
Table 1). Anyway, a purity higher than 99.8% has been determined by HPLC for compounds
1–
3.
2.2. In Vitro Activity on P. falciparum and Cytotoxicity
Avarone (
1) and the semisynthetic 1,1-dioxo-1,4-thiazine analogue (
2), as well as hydroquinone avarol (
3) were tested for their in vitro antiplasmodial activity against asexual and sexual (gametocytes stage V) stages of
P. falciparum (
Table 2). A chloroquine-sensitive (CQ-S) D10 and a chloroquine-resistant (CQ-R) W2 strains were used to determine the IC
50 against asexual stage of parasites. The most potent compound was thiazoavarone (
2) with an IC
50 value in the nanomolar range, higher than those exhibited by the previously identified synthetic lead [
13]. The high potency of the thiazinoquinone
2, specifically on the chloroquine-resistant strain W2 and compared with avarone (
1), lacking the heterocyclic moiety, confirmed once again the high potential of the thiazinoquinone scaffold for development of new antimalarial hits. Interestingly, both natural compounds
1 and
3 also exhibited a remarkable effect against sensitive and resistant
P. falciparum strains, showing no cross-resistance with chloroquine (see
Table 2). In particular, the reduced hydroquinone form (avarol,
3) resulted significantly more active than the oxidized quinone form (avarone,
1).
The evaluation of the effects of compounds
1–
3 on
Pf gametocytes stage V, the sexual stage circulating in the bloodstream, was performed in order to evaluate their transmission blocking potential. As evidenced in
Table 2, all the three compounds resulted less active against the gametocytes with respect to the parasite asexual stage; this finding is not unexpected since most of the current antimalarial drugs have no effect on the late stage of gametocytes. Avarol (
3) resulted the most potent in the series against
Pf gametocytes stage V with an IC
50 = 9.30 μM, comparable to that of OZ27, a drug in clinical development (IC
50 = 6.4 μM) [
42].
Finally, we tested compounds
1–
3 for their cytotoxic effects against two different human cell lines, microvascular endothelial (HMEC-1) and acute monocytic leukemia (THP-1) cells differentiated into macrophages; for each compound, we evaluated the selectivity index (SI,
Table 2), namely the ratio between the IC
50 on the human cells HMEC and that on the parasite strains (see
Table 3). Thiazoavarone (
2) exhibited a high toxicity against both mammalian cell lines, with IC
50 in the low micromolar concentration range and, consequently, a very low SI. Avarone and avarol (
1 and
3) were lowly and moderately cytotoxic, respectively (
Table 3) but the hydroquinone
3 exhibited a better SI.
2.4. In Vitro Activity on S. mansoni
Compounds
1–
3 were tested against larval stage (schistosomula), adult worm couples and eggs of the platyhelminth
S. mansoni. The most potent compound on schistosomula was thiazoavarone (
2) with a LC
50 value in the low micromolar range (
Table 5). The natural compounds (
1 and
3) showed comparable activity, with avarol (
3) slightly more active than avarone (
1), both showing a LC
50 in the high micromolar range (
Table 5). Therefore, the presence of a thiazine ring was proved to be very important for activity on schistosomula.
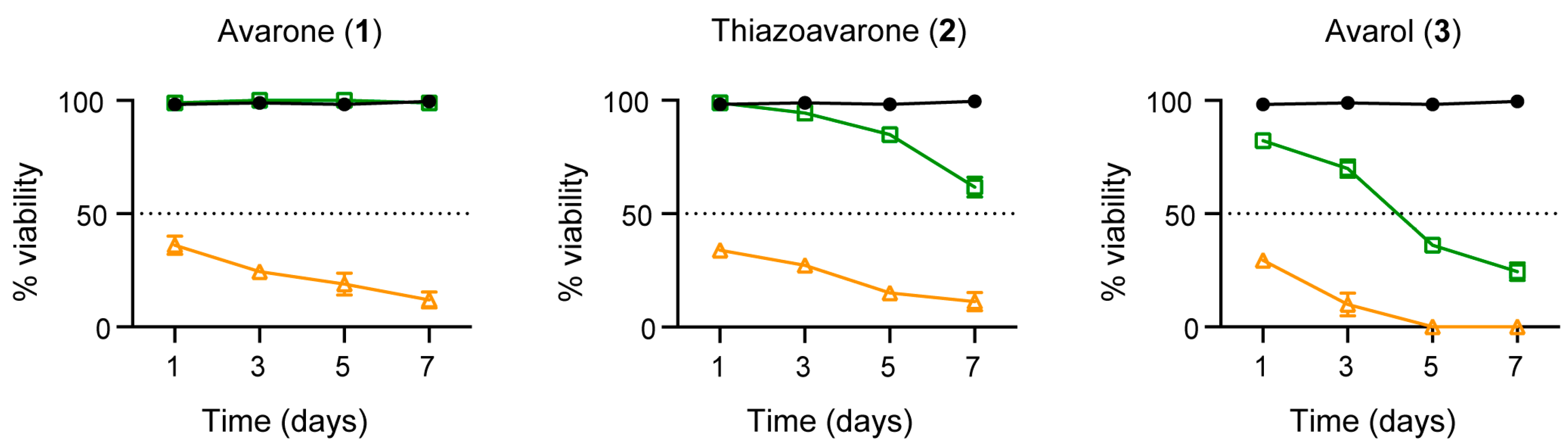
All three compounds
1–
3 were also very active on adult worm pairs at 50 μM leading to parasites death 7 days after treatment (
Figure 4). However, when used at lower concentration (20 μM), only avarol (
3) strongly impaired parasites viability (only 20% survival), while thiazoavarone (
2) was poorly effective against the adult stage (
Figure 4), despite its strong lethal effect on the larval stage (
Table 5). These results suggest the possibility that the double lipid bilayer coating the adult worms, namely the tegument [
45], can interfere with compound
2 uptake by adult parasites.
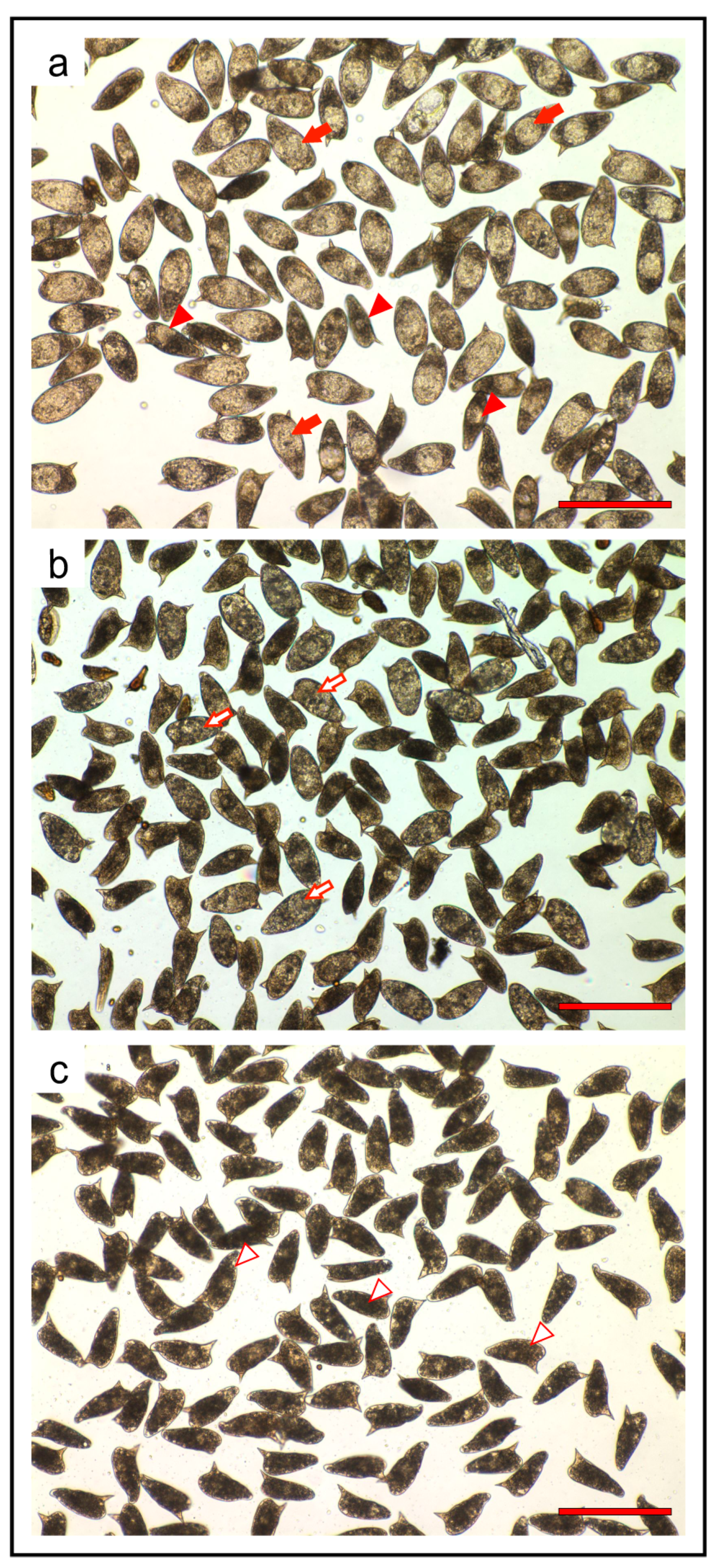
In the process of drug discovery for schistosomiasis, strategy involving any impairment in egg production and/or development must also be taken into account. In fact, upon mating with males, mature
S. mansoni adult females, residing in the mesenteric veins of the definitive host, can lay hundreds of eggs each day. The eggs secreted in stool or trapped in the liver respectively cause disease transmission and, as a result of inflammatory granulomas reactions, intestinal and hepato-splenic diseases [
46]. Therefore, compounds
1–
3 were also assayed against the in vitro laid eggs (IVLEs). The IVLEs produced in the first 48 h by
S. mansoni pairs and treated for 3 days with vehicle or compounds
1–
3 were classified by microscopic observation according to the Vogel and Prata staging system of egg maturation [
47]. The thiazoavarone (
2) resulted the most effective compound, impairing eggs maturation already at 5 μM and resulting in undeveloped and severely damaged eggs at 20 μM (
Figure 5). Similar results were obtained with compounds
1 and
3 at 50 μM.
2.5. Computational Studies and DFT Calculations
To rationalize the observed SARs, the steric and electronic features of compounds 1–3 were investigated by means of computational studies, including conformational analysis and DFT calculations.
A systematic conformational search considering all rotatable bonds was applied to generate all possible conformations of the compounds, which were, then, subjected to molecular mechanic (MM) geometry optimization using the CFF force field and a distance dependent dielectric constant value of 80 (Discovery Studio 2017, BIOVIA, San Diego USA; see the experimental Section for details) [
48]. The global minimum energy conformer (GM) was identified for each compound. All the generated conformers presented an energy difference from the GM (Δ
EGM) ≤ 3 kcal/mol. MM conformers were, then, subjected to density functional theory (DFT) calculations. In order to mimic an aqueous environment, all DFT calculations were performed using the conductor-like polarizable continuum model (C-PCM) as solvent model [
49]. Moreover, to characterize every structure as minimum, a vibrational analysis was carried out (see the experimental Section for details). Fully optimized DFT conformers were classified into families according to the values of their torsion angles (
Table 6 and
Table 7 and
Table S1).
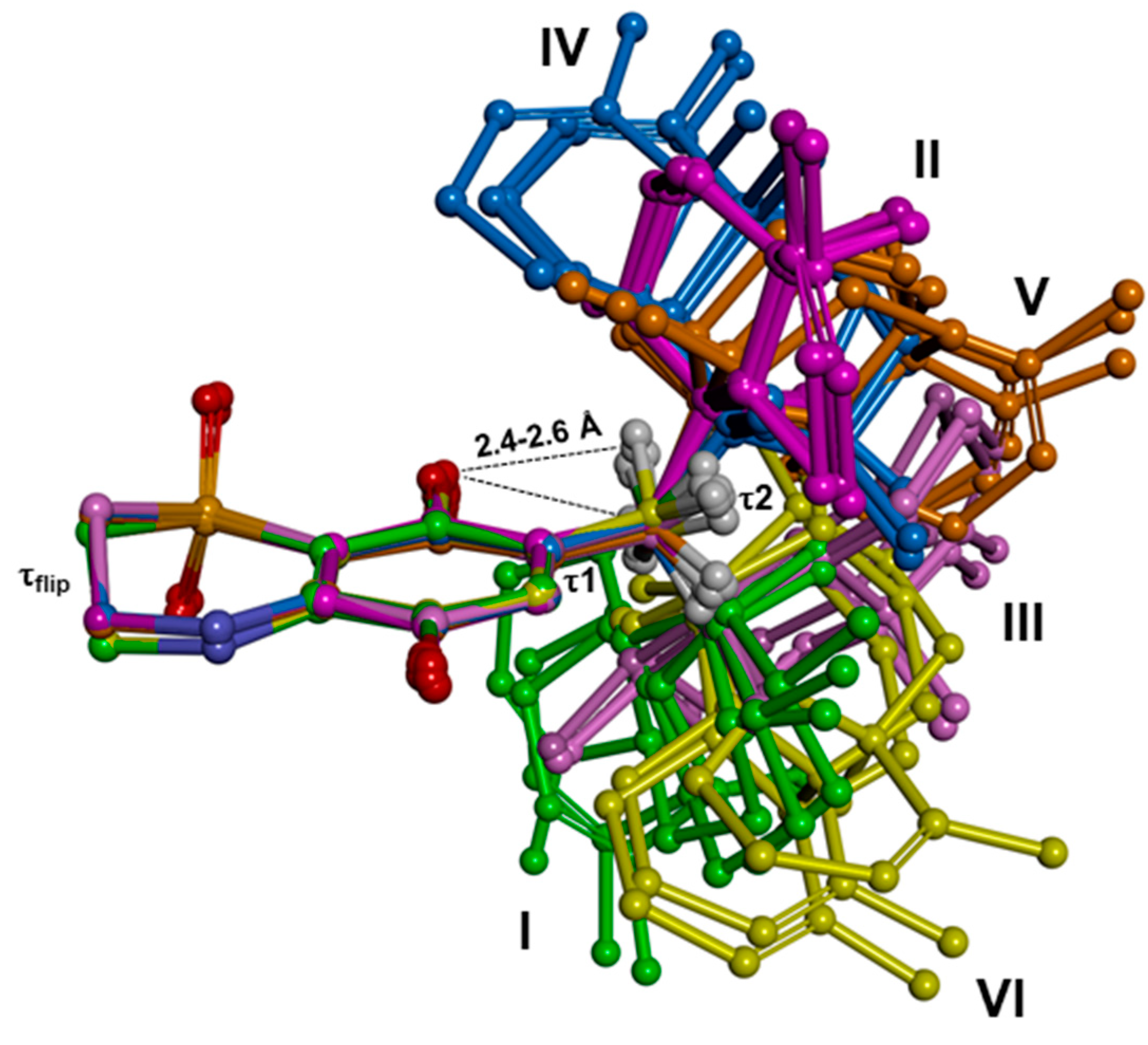
Results evidenced that compounds
1–
3 present common conformational features, characterized by the electronic attraction between the hydrogen atoms of the first methylene group of the alkyl substituent and the nearby quinone oxygen, which limits the conformational freedom of R′ (
Figure 6). Accordingly, the torsional angle τ1 showed just two sets of possible values (~±100°;
Table 6 and
Table 7 and
Table S1), and for each of them the rigid sesquiterpene ring could assume three orientations with respect to the thiazinoquinone/quinone/quinol ring (τ2 = ~60°, ~−60° and ~180°;
Table 6 and
Table 7 and
Table S1). This determined a total number of six conformers (named I–VI;
Figure 6). In the case of the thiazinoquinone derivative
2, due to the presence of the two opposite flips of the thiazinoquinone ring (τ
flip ~ ±60°), we obtained two specular sets of conformers with the same conformational energy (i.e., conformational enantiomers;
Figure S10), as previously reported for other thiazinoquinone derivatives [
13,
14].
The fixed position of the methylene group combined with the presence of the rigid sesquiterpene moiety, place in the putative semiquinone radical produced upon one electron reduction/oxidation several hydrogen atoms at a distance (≤3 Å) suitable for an intramolecular radical shift from the oxygen atom to a carbon atom of R′ (see below).
Then, starting from the DFT minima, the redox properties of
1–
3 were calculated. At this aim, we considered the two electrons/two protons quinone reduction pathway in a protic solvent (
Scheme S1) and all the species involved in the pathway (Q
•−, QH
•, QH
−, QH
2) were generated and DFT optimized using as starting structures the energetically favored DFT minima I and II.
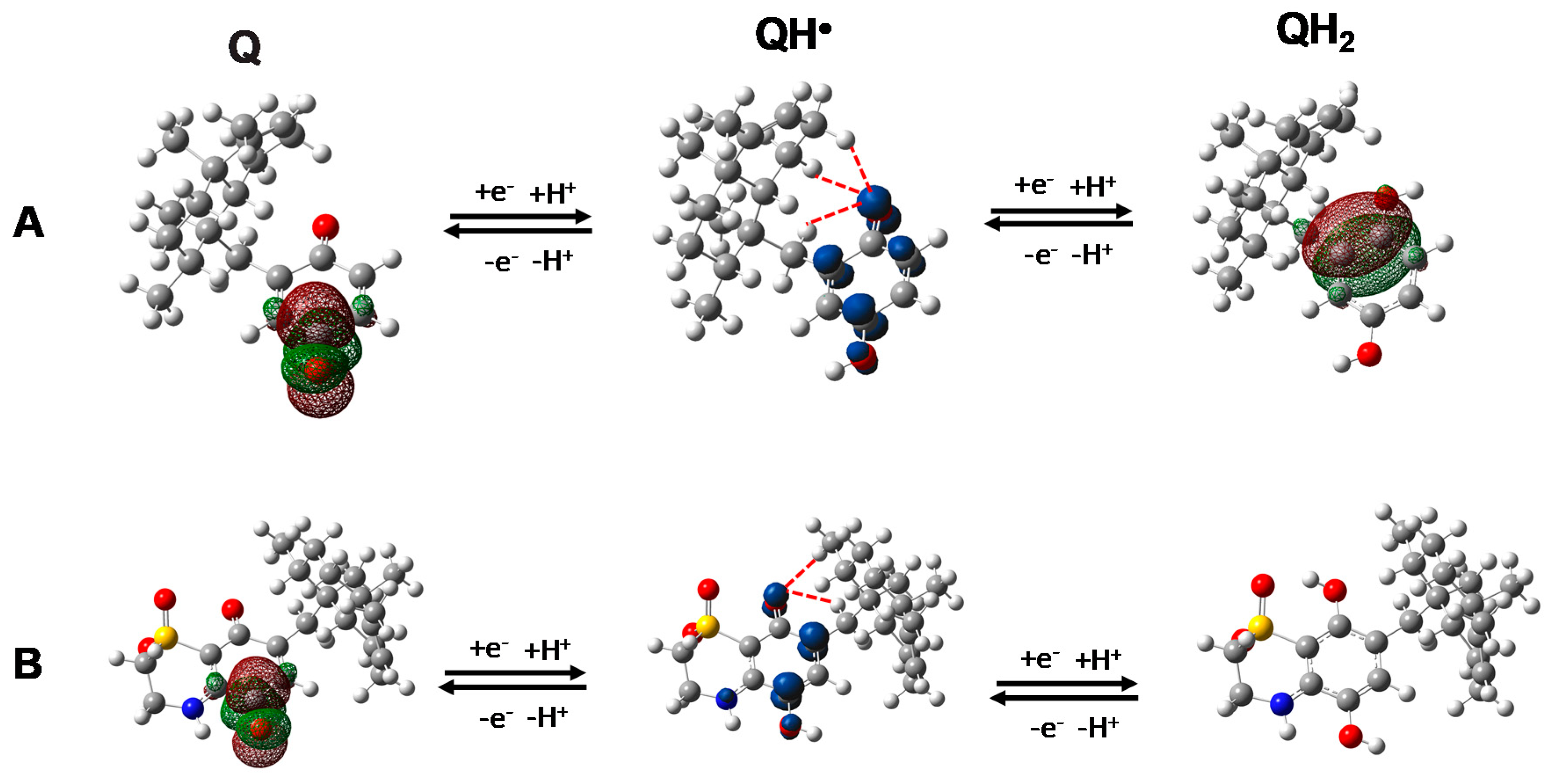
Two possible protonated semiquinone species may be formed, depending on which of the two quinone oxygen atoms is reduced/oxidized at first. The location of the lowest unoccupied molecular orbital (LUMO) in
1 and
2 and of the highest occupied molecular orbital (HOMO) in
3 indicated the oxygen opposite to the alkyl chain as the most probable site to be reduced and the one close to the alkyl chain as the most probable site to be oxidized, respectively (
Figure 7). It is worthy to be mentioned that, regarding the most probable oxidation pathway of
3 to its quinone form, since the deprotonation step is supposed to be the first event in protic solvents (
Scheme S1), we calculated the pKa values of the two hydroxyl groups, too. Results indicated the hydroxyl group nearby the alkyl substituent as the first to lose the proton (
Figure S11), further supporting the formation of the hydroquinone radical reported in
Figure 7.
The standard redox potential (E°) and the standard Gibbs free energy (ΔG
0red, aq) of each electron-transfer reaction (see
Scheme S1) alongside with the standard Gibbs free energy required for the protonation of the resulting reduced species (ΔG
0H+) of the redox couple
1,
3 and
2 were calculated (for details see the experimental Section). To further evaluate the propensity of
1 and
2 to undergo a one-electron reduction, the energy of the lowest unoccupied molecular orbital (E
LUMO) was also taken into account. Similarly, we calculated the ionization potential (IP; i.e., −E
HOMO) of the radical anion QH
− as indicative of the tendency of the deprotonated species of
3 to undergo a one-electron oxidation. Finally, we considered the energy of the single occupied molecular orbital (E
SOMO) of the radical species as indicative of the ability to delocalize the unpaired electron. The resulting data are reported in
Table 8 and
Table 9.
A first consideration can be derived comparing the redox properties of the quinone-based compounds
1 and
2. With respect to
1,
2 showed either a higher tendency to acquire one electron (lower E
LUMO and ΔG
0(red, aq); higher E°;
Table 8) and a higher stability of the QH
• radical (lower E
SOMO;
Table 8). While the
ESOMO values of the anion radical species showed little difference (E
SOMO Q
•−;
Table 8), on the contrary, the differences became evident after the protonation step (E
SOMO QH
•). These results, on one hand confirm the key role played by the 1,1-dioxo-1,4-thiazine ring on the electron affinity [
11,
13,
14]; on the other hand, support the hypothesis that the compound activity is related to the formation of the semiquinone radical species. Indeed, the thiazinoquinone derivative
2 resulted overall more potent than the quinone derivative
1.
A second consideration is that, as evidenced in
Figure 7 and
Figure S12, all the calculated semiquinone radicals showed a hydrogen atom of the first methylene group together with, at least, another hydrogen atom of the rigid sesquiterpene ring, at a distance suitable for an intra-molecular hydrogen radical shift to the semi- reduced/oxidized oxygen atom (≤3 Å) (
Table S2). By consequence, as above mentioned, the presence of the sesquiterpene moiety as alkyl substituent is expected to promote the putative “through space” intramolecular hydrogen radical shift leading to the formation of the toxic radical species. In line with this hypothesis,
2 resulted the most potent thiazinoquinone developed by us against
P. falciparum D10 and W2 strains as well as against stage V gametocytes. In addition,
1, although lacking the 1,1-dioxo-1,4-thiazine ring of
2, resulted still active on
P. falciparum D10 and W2 strains as well as on schistosomula, contrarily to what previously observed by us for other 1,4-benzoquinone derivatives when compared to the corresponding thiazinoquinone analogues [
13,
14].
Finally, the hydroquinone
3 showed a higher propensity to be oxidized to the semiquinone radical (i.e., lower E° and IP of the QH
− anion) with respect to the corresponding reduced form of
2 (
Table 9). Thus, the absence of the 1,1-dioxo-1,4-thiazine ring favors the one-electron oxidation reaction of
3.
The hydroquinone 3 resulted more active than the corresponding quinone 1 against all considered parasites, while presenting the highest selectivity index with respect to mammalian cells. Both the oxidized and the reduced forms could produce the same putative toxic radical species upon a one-electron transfer reaction. In this view, our results suggest the presence in the parasite cell of a bioactivation reaction partner preferentially binding the hydroquinone (reduced) form (3) rather than the quinone (oxidized) form (1). This putative bioactivation partner seems not to be present in human cells.
To investigate the role played by molecular pharmacokinetics on the observed SARs, we calculated the distribution coefficient values of
1–
3 (clogD,
Table S3) (ACD/Percepta 2017). According to Lipinski’s rules for drug absorption [
50] the thiazinoquinone
2 showed a better cLogD value for cell membrane passive diffusion (cLogD ~ 4) with respect to
1 and
3 (cLogD > 5). However, in specific developmental stages (i.e.,
Pf late gametocytes, promastigote of
L. infantum and
L. tropica, amastigote of
L. infantum, and adult worms of
S. mansoni)
3 resulted more active than
2 (
Table 2,
Table 4 and
Table 5;
Figure 4 and
Figure 5). According to what previously reported by us [
14], this peculiar activity profile could be due to the significant morphological changes in the parasite during the above-mentioned developmental stages [
51,
52,
53], which are likely to impair compound ability to penetrate into the parasite by passive diffusion, while it could still penetrate by exploiting the large number of transport proteins expressed on the parasite membrane.
Taken together, the results of our computational investigation indicate that a toxic semiquinone radical species [
11,
13,
14] which can be produced starting both from quinone- and hydroquinone-based compounds could mediate the anti-parasitic effects of the tested compounds
1–
3.

 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
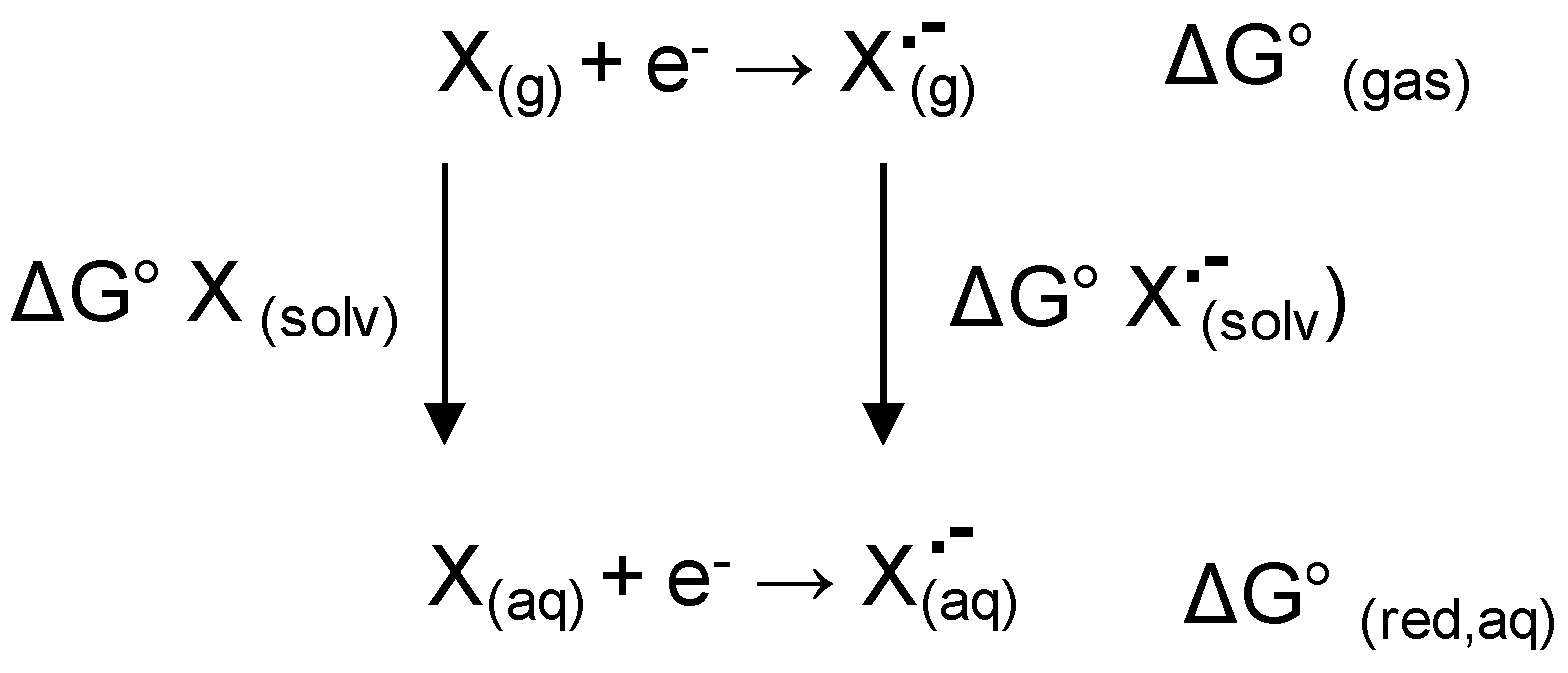{kind=link}

