
3-D Culture of Marine Sponge Cells for Production of Bioactive Compounds

 ,
,
Abstract
:1. Introduction
2. Results
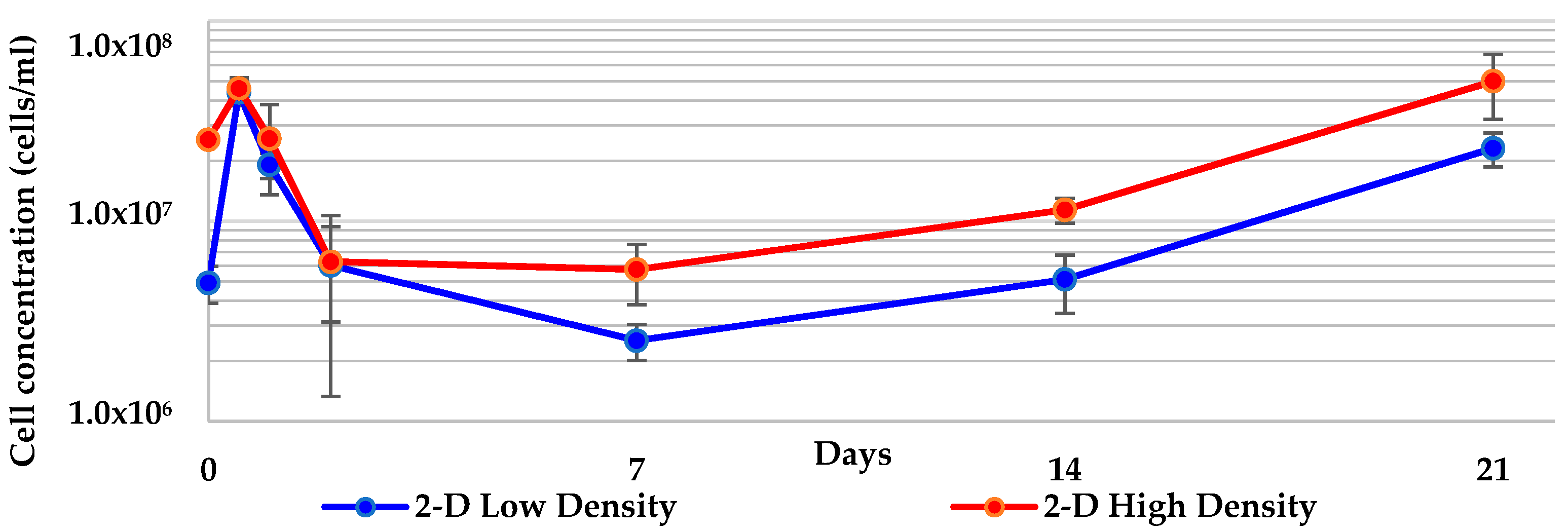
2.1. 2-D Cultures of Geodia neptuni Were Used as Controls
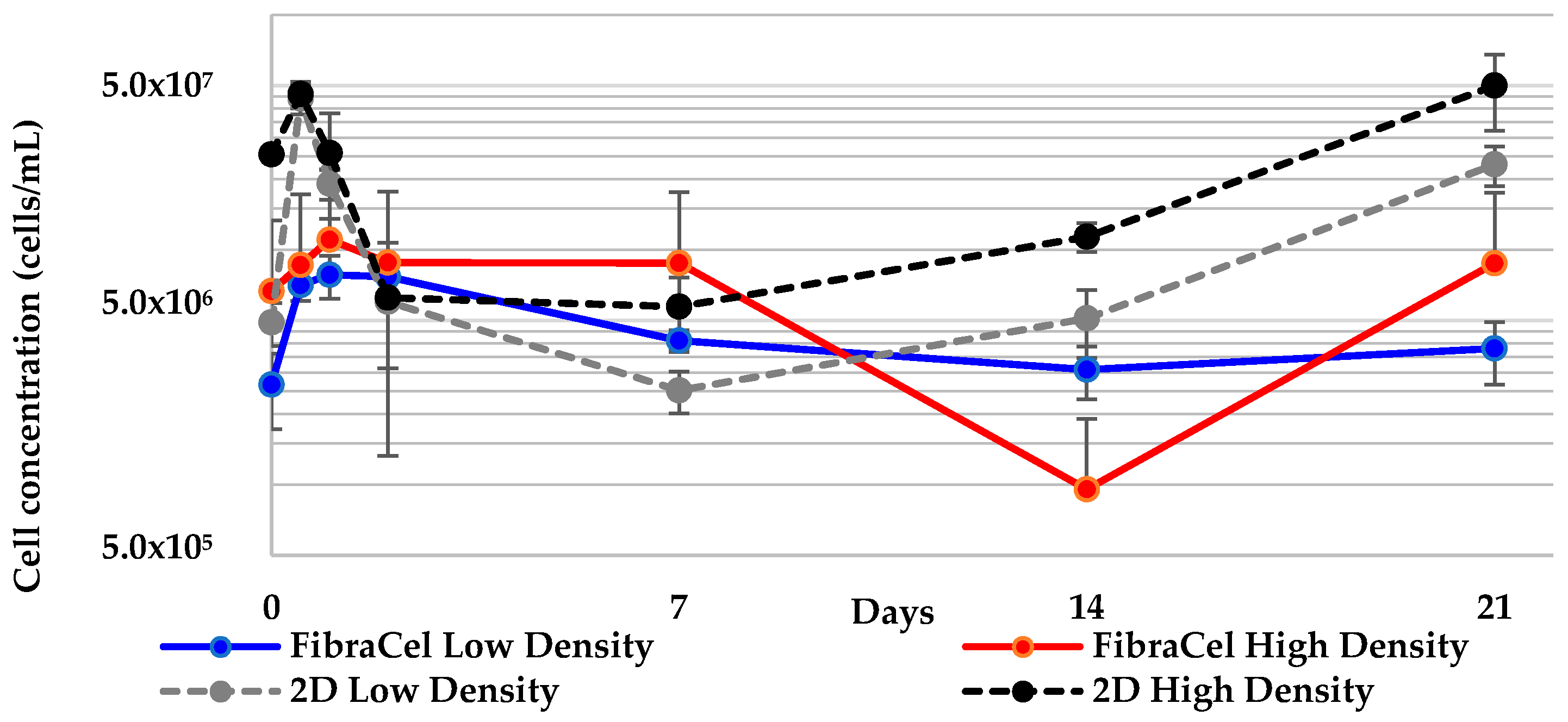
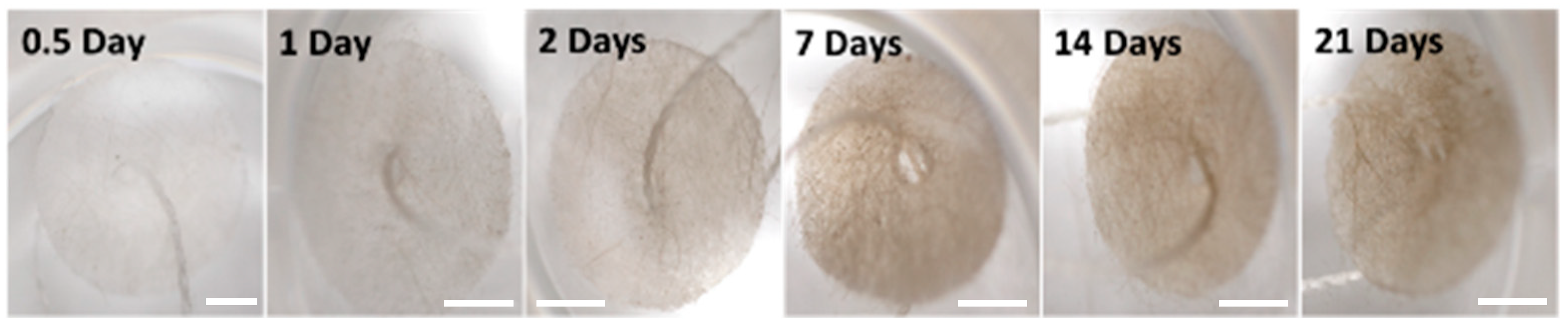
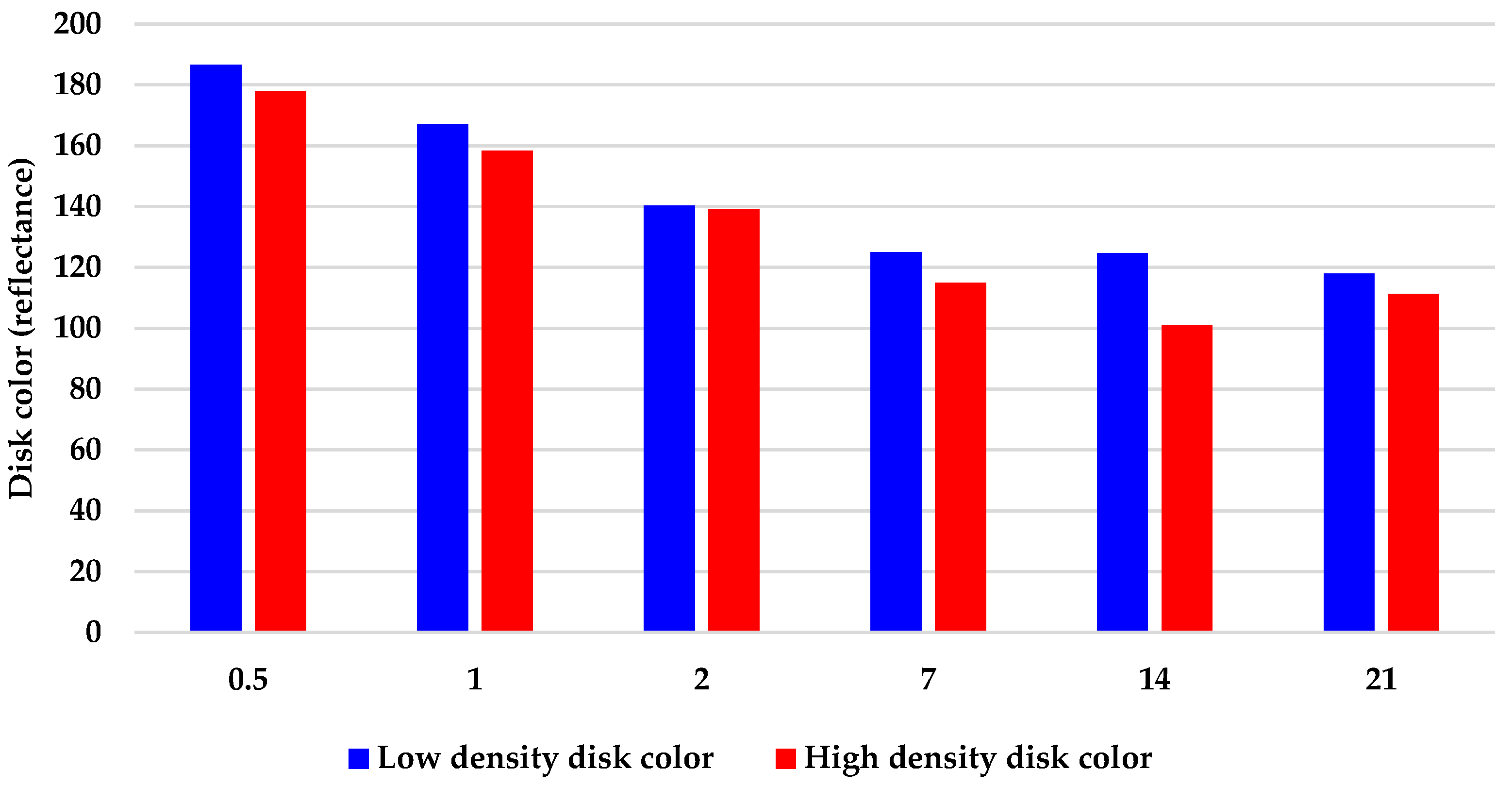
2.2. Geodia neptuni Cells Can Be Cultured in FibraCel® Disks
2.3. Porcine Gelatin Layers Dissolve in M1 Medium
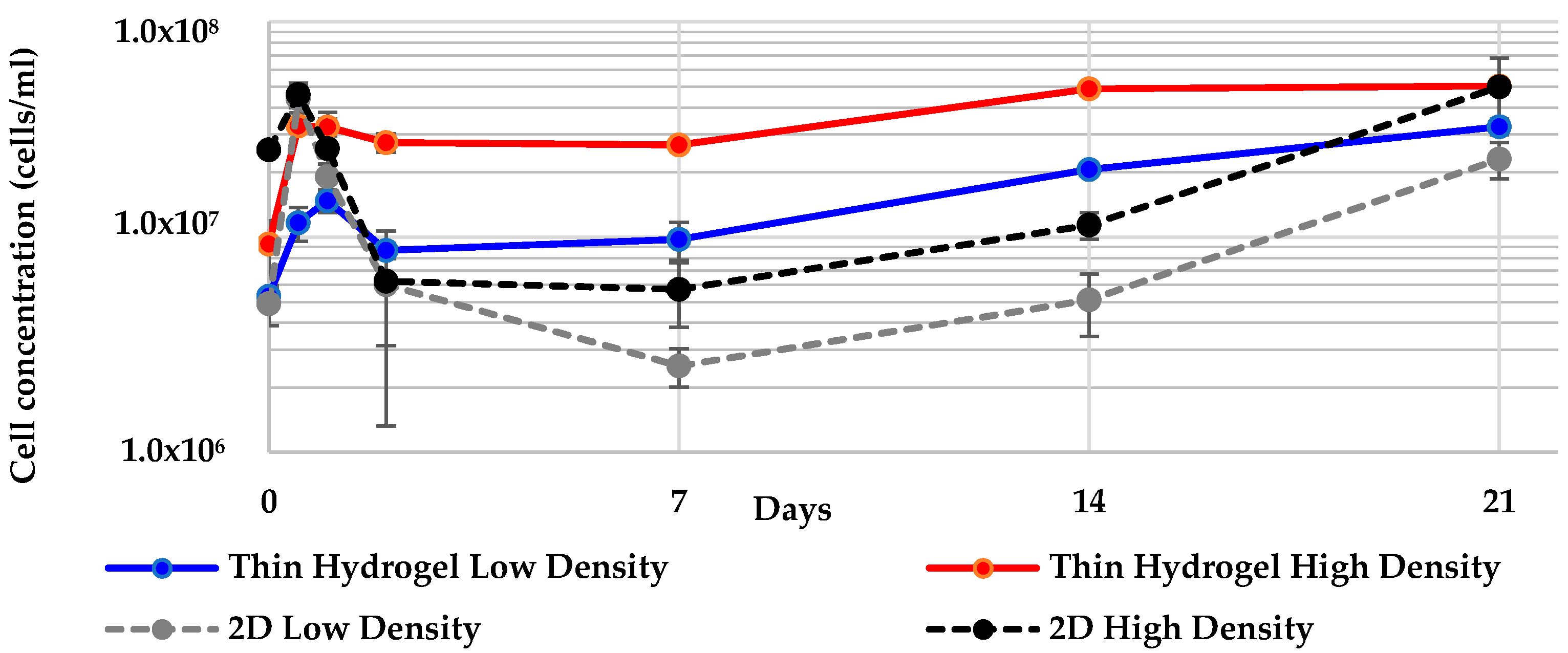
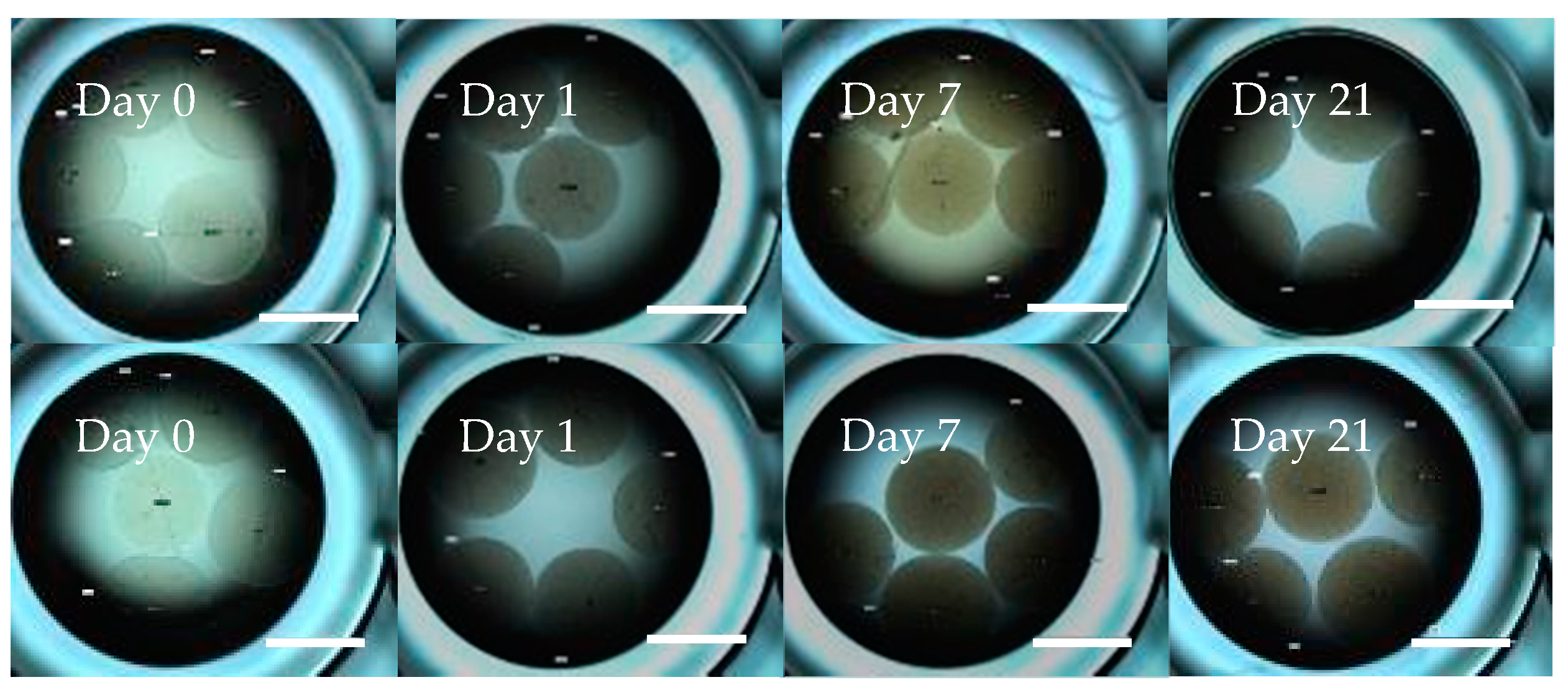
2.4. Geodia neptuni Cells Can Be Cultured in ULTA Thin Hydrogel Layers
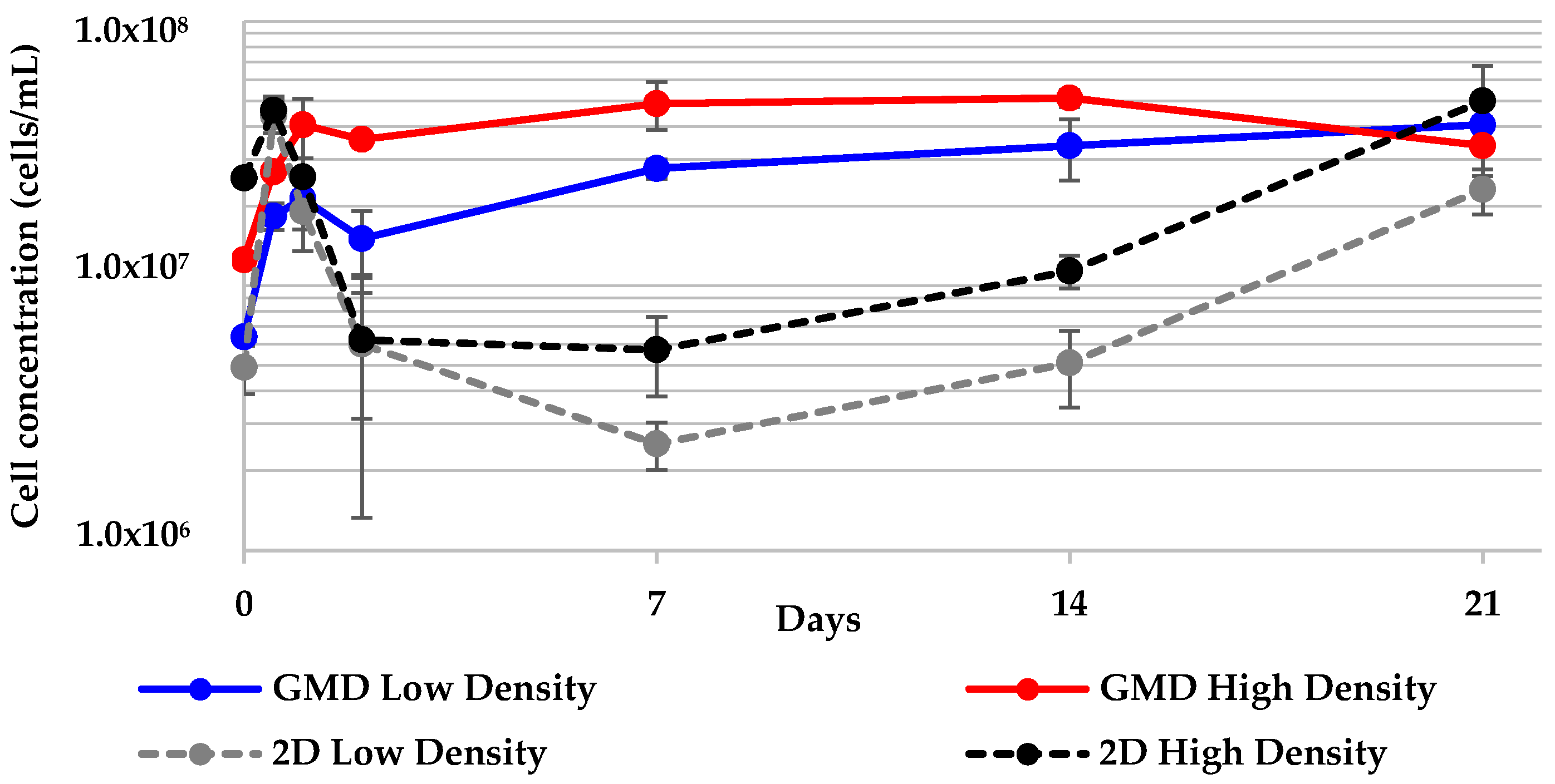
2.5. Geodia neptuni Cells Can Be Cultured in ULTA GMDs
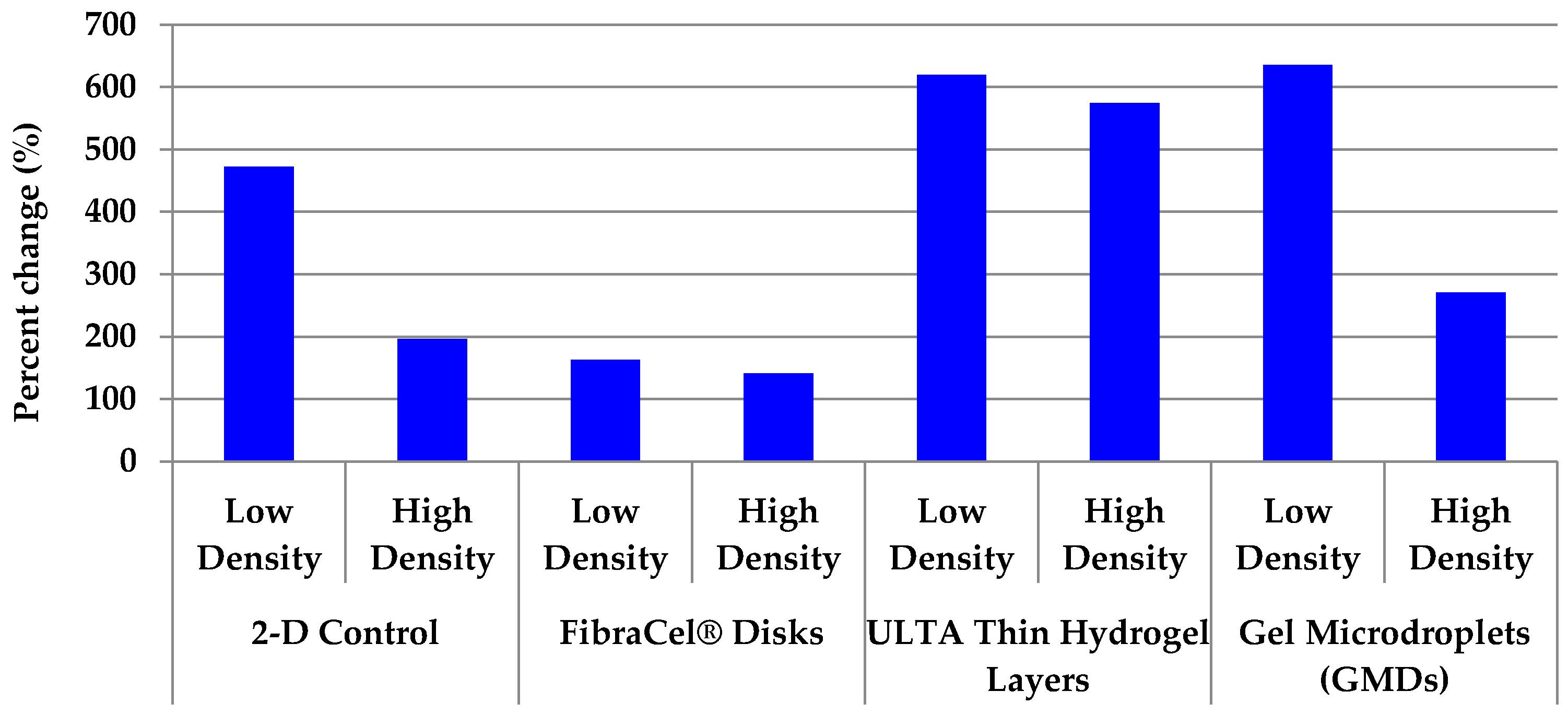
2.6. All 3-D Treatments Resulted in Substantial Increases in Cell Numbers
3. Discussion
4. Materials and Methods
4.1. Specimen Collection, Cell Dissociation and Cryopreservation
4.2. Cell Preparation, Counting, and Imaging
4.3. Solution Preparation
4.3.1. Media
4.3.2. Hydrogels
Porcine Gelatin
ULTA
4.3.3. CMF
4.4. Incubation of Cell Cultures
4.5. Culture Methods
4.5.1. 2-D Culture Controls
4.5.2. 3-D Cultures: FibraCel® Disks
4.5.3. 3-D Cultures: Thin Hydrogel Layers
Porcine Gelatin Thin Hydrogel Layers
ULTA Thin Hydrogel Layers
4.5.4. 3-D Cultures: ULTA GMDs
4.6. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Analysis of Protein Concentration in Sponge Cells
References
- Calcabrini, C.; Catanzaro, E.; Bishayee, A.; Turrini, E.; Fimognari, C. Marine sponge natural products with anticancer potential: An updated review. Mar. Drugs 2017, 15, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.-E.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schippers, K.J.; Sipkema, D.; Osinga, R.; Smidt, H.; Pomponi, S.A.; Martens, D.E.; Wijffels, R.H. Cultivation of sponges, sponge cells and symbionts. Adv. Mar. Biol. 2012, 62, 273–337. [Google Scholar] [PubMed]
- Maslin, M.; Gaertner-Mazouni, N.; Debitus, C.; Joy, N.; Ho, R. Marine sponge aquaculture towards drug development: An ongoing history of technical, ecological, chemical considerations and challenges. Aquac. Res. 2021, 21, 100813. [Google Scholar] [CrossRef]
- Song, Y.; Qu, Y.; Cao, X.; Zhang, W.; Zhang, F.; Linhardt, R.J.; Yang, Q. Cultivation of fractionated cells from a bioactive-alkaloid-bearing marine sponge Axinella sp. Vitr. Cell. Dev. Biol.-Anim. 2021, 57, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, A.I.; Kosevich, I.A. Sponge cell reaggregation: Cellular structure and morphogenetic potencies of multicellular aggregates. J. Exp. Zool. 2016, 325A, 158–177. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Liu, J.R.; Patel, B.; Solomon, D.E. Microfluidics-based 3-D cell culture models: Utility in novel drug discovery and delivery research. Bioeng. Transl. Med. 2016, 1, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Markl, J.S.; Müller, W.E.G.; Sereno, D.; Elkhooly, T.A.; Kokkinopoulou, M.; Gardères, J.; Depoix, F.; Wiens, M. A synthetic biology approach for the fabrication of functional (fluorescent magnetic) bioorganic-inorganic hybrid materials in sponge primmorphs. Biotechnol. Bioeng. 2020, 117, 1789–1804. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Becken, U.; Sha, M. Vero perfusion, packed bed vessels intensify vaccine production: Eppendorf’s FibraCel and BioBLU 5p technologies show promise for high-titer vaccine production. Genet. Eng. Biotechnol. News 2018, 38, 16–18. [Google Scholar] [CrossRef]
- Clapp, K.P.; Castan, A.; Lindskog, E.K. Chapter 24—Upstream processing equipment. In Biopharmaceutical Processing; Jagschies, G., Lindskog, E., Lacki, K., Galliher, P., Eds.; Elsevier Academic Press: New York, NY, USA, 2018; pp. 47–476. [Google Scholar]
- Pomponi, S.A.; Willoughby, R.; Kaighn, M.E.; Wright, A.E. Development of techniques for in vitro production of bioactive natural products from marine sponges. In Invertebrate Cell Culture: Novel Directions and Biotechnology Applications; Maramorosch, K., Mitsuhashi, J., Eds.; Science Publishers: Enfield, NH, USA, 1997; pp. 231–237. [Google Scholar]
- Caliari, S.R.; Burdick, J.A. A practical guide to hydrogels for cell culture. Nat. Methods 2016, 13, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Xu, X.; Peng, K.; Zhang, Y.; Jiang, L.; Williams, T.; Paulsen, I.; Piper, J.; Li, M. Microdroplet enabled cultivation of single yeast cells correlates with bulk and reveals subpopulation phenomena. Biotechnol. Bioeng. 2020, 118, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Munroe, S.; Sandoval, K.; Martens, D.E.; Sipkema, D.; Pomponi, S.A. Genetic algorithm as an optimization tool for the development of sponge cell culture media. Vitr. Cell. Dev. Biol.-Anim. 2019, 55, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conkling, M.; Hesp, K.; Munroe, S.; Sandoval, K.; Martens, D.E.; Sipkema, D.; Wijffels, R.H.; Pomponi, S.A. Breakthrough in marine invertebrate cell culture: Sponge cells divide rapidly in improved nutrient medium. Sci. Rep. 2019, 9, 17321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Deinsen-Hesp, K. Dawn of a New Era in Sponge Biotechnology. Ph.D. Thesis, Wageningen University & Research, Wageningen, The Netherlands, February 2021. [Google Scholar]
- Munroe, S.; Martens, D.E.; Sipkema, D.; Pomponi, S.A. Comparison of cryopreservation techniques for cells of the marine sponge Dysidea etheria. Cryoletters 2018, 39, 269–278. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p-Values | ||
|---|---|---|
| Final Density | Maximum Density | |
| 2-D low density to 2-D high density | 0.113 | 0.624 |
| p-Values | ||
|---|---|---|
| Final Concentration | Maximum Concentration | |
| FibraCel® low density to 2-D low density | 0.011 * | 0.005 * |
| FibraCel® high density to 2-D high density | 0.055 | 0.059 |
| FibraCel® high density to FibraCel® low density | 0.039 | 0.235 |
| p-Values | ||
|---|---|---|
| Final Concentration | Maximum Concentration | |
| ULTA thin layer low density to 2-D low density | 0.047 | 0.066 |
| ULTA thin layer high density to 2-D high density | 0.977 | 0.977 |
| ULTA thin layer high density to ULTA thin layer low density | 0.004 * | 0.004 * |
| p-Values | ||
|---|---|---|
| Final Concentration | Maximum Concentration | |
| GMD low density to 2-D low density | 0.036 | 0.582 |
| GMD high density to 2-D high density | 0.254 | 0.919 |
| GMD low density to GMD high density | 0.344 | 0.118 |
| Culture Method | Advantages | Disadvantages | Effort | Expense | Recommended Applications |
|---|---|---|---|---|---|
| FibraCel® disks | Hardy matrix retains cells | Unable to count cells | Low | $ | Cell culture scale-up, aquaculture scale-up |
| ULTA thin hydrogel layers | Easy to visualize cells/aggregates | Delicate matrix | Mid | $$ | Cell characterization and differentiation, aquaculture scale-up |
| ULTA GMDs | Rapid diffusion of nutrients, gases and products | Delicate matrix | High | $$ | Cell metabolism studies, cell culture scale-up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urban-Gedamke, E.; Conkling, M.; McCarthy, P.J.; Wills, P.S.; Pomponi, S.A. 3-D Culture of Marine Sponge Cells for Production of Bioactive Compounds. Mar. Drugs 2021, 19, 569. https://doi.org/10.3390/md19100569
Urban-Gedamke E, Conkling M, McCarthy PJ, Wills PS, Pomponi SA. 3-D Culture of Marine Sponge Cells for Production of Bioactive Compounds. Marine Drugs. 2021; 19(10):569. https://doi.org/10.3390/md19100569
Chicago/Turabian StyleUrban-Gedamke, Elizabeth, Megan Conkling, Peter J. McCarthy, Paul S. Wills, and Shirley A. Pomponi. 2021. "3-D Culture of Marine Sponge Cells for Production of Bioactive Compounds" Marine Drugs 19, no. 10: 569. https://doi.org/10.3390/md19100569
APA StyleUrban-Gedamke, E., Conkling, M., McCarthy, P. J., Wills, P. S., & Pomponi, S. A. (2021). 3-D Culture of Marine Sponge Cells for Production of Bioactive Compounds. Marine Drugs, 19(10), 569. https://doi.org/10.3390/md19100569