
Diterpenoids from the Brown Alga Rugulopteryx okamurae and Their Anti-Inflammatory Activity

 , ,
, ,  and
and
Abstract
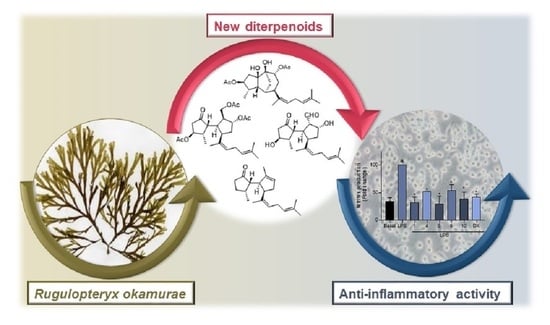
:
1. Introduction
2. Results and Discussion
2.1. Isolation and Structure Determination
2.2. Anti-Inflammatory Activity


3. Materials and Methods
3.1. General Experimental Procedures
3.2. Algae Collection
3.3. Extraction and Isolation
3.4. Characterization of Compounds
3.5. Cell Culture
3.6. Analysis of the Cellular Viability by Crystal Violeta Staining
3.7. Analysis of Nitrites (NO2−)
3.8. Quantitative Real-Time PCR (qPCR) Analysis
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gaysinski, M.; Ortalo-Magné, A.; Thomas, O.P.; Culioli, G. Extraction, purification, and NMR analysis of terpenes from brown algae. In Natural Products from Marine Algae: Methods and Protocols; Stengel, D.B., Connan, S., Eds.; Springer: New York, NY, USA, 2015; Volume 1308, pp. 207–223. [Google Scholar]
- De Paula, J.C.; Vallim, M.A.; Teixeira, V.L. What are and where are the bioactive terpenoids metabolites from Dictyotaceae (Phaeophyceae). Rev. Bras. Farmacogn. 2011, 21, 216–228. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, H.; Zhao, Z.; Xia, X.; Li, B.; Zhang, J.; Yan, X. Diterpenes from the marine algae of the genus Dictyota. Mar. Drugs 2018, 16, 159. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Li, B.; Hou, Y.; Zhang, J.; Yan, X. Diterpenes from the marine brown algae of the genus Dilophus. Nat. Prod. Commun. 2020, 15, 14. [Google Scholar] [CrossRef] [Green Version]
- Finer, J.; Clardy, J.; Fenical, N.; Minale, L.; Riccio, R.; Battaile, J.; Kirlap, M.; Moore, R.E. Structures of dictyodial and dictyolactone unusual marine diterpenoids. J. Org. Chem. 1979, 44, 2044–2047. [Google Scholar] [CrossRef]
- Amico, V.; Oriente, G.; Piatelli, M.; Tringali, C.; Fattorusso, E.; Magno, S.; Mayol, L. Diterpenes based on the dollabellane skeleton from Dictyota dichotoma. Tetrahedron 1980, 36, 1409–1414. [Google Scholar] [CrossRef]
- Tringali, C.; Oriente, G.; Piatelli, M.; Geraci, C.; Nicolosi, G.; Breitmaier, E. Crenuladial, an antimicrobial diterpenoid from the brown alga Dilophus ligulatus. Can. J. Chem. 1988, 66, 2799–2802. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Fenical, W. Spatane diterpenoids from the tropical marine algae Spatoglossum schmittii and Spatoglossum howleii (Dictyotaceae). J. Org. Chem. 1983, 48, 3325–3329. [Google Scholar] [CrossRef]
- Bouaicha, N.; Tringali, C.; Pesando, D.; Mallea, M.; Roussakis, C.; Verbist, J.F. Bioactive diterpenoids isolated from Dilophus ligulatus. Planta Med. 1993, 59, 256–258. [Google Scholar] [CrossRef]
- Durán, R.; Zubía, E.; Ortega, M.J.; Salvá, J. New diterpenoids from the alga Dictyota dichotoma. Tetrahedron 1997, 53, 8675–8688. [Google Scholar] [CrossRef]
- Pereira, H.S.; Leao-Ferreira, L.R.; Moussatché, N.; Teixeira, V.L.; Cavalcanti, D.N.; Costa, L.J.; Diaz, R.; Frugulhetti, I.C.P.P. Antiviral activity of diterpenes isolated from the Brazilian marine alga Dictyota menstrualis against human immunodeficiency virus type 1 (HIV-1). Antivir. Res. 2004, 64, 69–76. [Google Scholar] [CrossRef]
- Barbosa, J.P.; Pereira, R.C.; Abrantes, J.L.; Dos Santos, C.C.C.; Rebello, M.A.; Frugulhetti, I.C.D.P.P.; Teixeira, V.L. In vitro antiviral diterpenes from the Brazilian brown alga Dictyota pfaffii. Planta Med. 2004, 70, 856–860. [Google Scholar] [CrossRef]
- Abrantes, J.L.; Barbosa, J.; Cavalcanti, D.; Pereira, R.C.; Fontes, C.F.L.; Teixeira, V.L.; Souza, T.M.; Paixao, I.C.P. The effects of the diterpenes isolated from the Brazilian brown algae Dictyota pfaffii and Dictyota menstrualis against the Herpes simplex Type-1 replicative cycle. Planta Med. 2010, 46, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Oliveira dos Santos, A.; Britta, E.A.; Bianco, E.M.; Ueda-Nakamura, T.; Filho, B.P.D.; Pereira, R.C.; Nakamura, C.V. 4-Acetoxydolastane diterpene from the Brazilian brown alga Canistrocarpus cervicornis as antileismanial agent. Mar. Drugs 2011, 9, 2369–2383. [Google Scholar] [CrossRef] [Green Version]
- Moura, L.A.; Marqui de Almeida, A.C.; Domingos, T.F.S.; Ortiz-Ramirez, F.; Cavalcanti, D.N.; Teixeira, V.L.; Fuly, A.L. Antiplatelet and anticoagulant effects of diterpenes isolated from the marine alga Dictyota menstrualis. Mar. Drugs 2014, 12, 2471–2484. [Google Scholar] [CrossRef]
- Chinnababu, B.; Reddy, P.; Rao, P.S.; Reddy, V.L.; Kumar, B.S.; Rao, J.V.; Prakasham, R.S.; Babu, H.S. Isolation, semi-synthesis and bio-evaluation of spatane derivatives from the brown algae Stoechospermum marginatum. Bioorg. Med. Chem. Lett. 2015, 25, 2479–2483. [Google Scholar] [CrossRef]
- Ayyad, S.-E.N.; Makki, M.S.; Al-kayal, N.S.; Basaif, S.A.; El-Foty, K.O.; Asiri, A.M.; Alarif, W.M.; Badria, F.A. Cytotoxic and protective DNA damage of three new diterpenoids from the brown alga Dictyota dichotoma. Eur. J. Med. Chem. 2011, 46, 175–182. [Google Scholar] [CrossRef]
- Niccolai, E.; Boem, F.; Emmi, G.; Amedei, A. The link “cancer and autoimmune diseases” in the light of microbiota: Evidence of a potential culprit. Immunol. Lett. 2020, 222, 12–28. [Google Scholar] [CrossRef]
- Scrivo, R.; Vaile, M.; Bartosiewicz, I.; Guido, V. Inflammation as “common soil” of the multifactorial diseases. Autoimmun. Rev. 2011, 10, 369–374. [Google Scholar] [CrossRef]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759. [Google Scholar] [CrossRef]
- Du, C.; Bhatia, M.; Tang, S.C.W.; Zhang, M.; Steiner, T. Mediators of inflammation; inflammation in cancer, chronic diseases, and wound healing. Mediat. Inflamm. 2015, 2015, 570653. [Google Scholar] [CrossRef]
- Cheng, S.; Zhao, M.; Sun, Z.; Yuan, W.; Zhang, S.; Xiang, Z.; Cai, Y.; Dong, J.; Huang, K.; Yan, P. Diterpenes from a chinese collection of the brown alga Dictyota plectens. J. Nat. Prod. 2014, 77, 2685–2693. [Google Scholar] [CrossRef]
- Zhao, M.; Cheng, S.; Yuan, W.; Dong, J.; Huang, K.; Sun, Z.; Yan, P. Further new xenicanes from a chinese collection of the brown alga Dictyota plectens. Chem. Pharm. Bull. 2015, 63, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.K.; Lee, W.J.; Kim, H.S.; De Clerck, O. Taxonomic reappraisal of Dilophus okamurae (Dictyotales, phaeophyta) from the western Pacific ocean. Phycologia 2009, 48, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Agatsuma, Y.; Kuwahara, Y.; Taniguchi, K. Life cycle of Dilophus okamurae (Phaeophyceae) and its associated invertebrate fauna in Onagawa Bay. Jpn. Fish. Sci. 2005, 71, 1107–1114. [Google Scholar] [CrossRef]
- García-Gómez, J.C.; Sempere-Valverdde, J.; Gonzalez, A.R.; Martínez-Chacón, M.; Olaya-Ponzone, L.; Sánchez-Moyano, E.; Ostalé-Valriberas, E.; Megina, C. From exotic to invasive in record time: The extreme impact of Rugulopteryx okamurae (Dictyotales, Ochrophyta) in the Strait of Gibraltar. Sci. Total Environ. 2020, 704, 135408. [Google Scholar] [CrossRef]
- García-Gómez, J.C.; Florido, M.; Olaya-Ponzone, L.; Díaz de Rada, J.R.; Donázar-Aramendia, I.; Chacón, M.; Quintero, J.J.; Magariño, S.; Megina, C. Monitoring extreme impacts of Rugulopteryx okamurae (Dictyotales, Ochrophyta) in El Estrecho Natural Park (biosphere reserve). Showing radical changes in the underwater seascape. Front. Ecol. Evol. 2021, 9, 639161. [Google Scholar] [CrossRef]
- Ochi, M.; Masui, N.; Kotsuki, H.; Miura, I.; Tokoroyama, T. The structures of fukurinolal and fukurinal, two new diterpenoids from the brown seaweed Dilophus okamurai Dawson. Chem. Lett. 1982, 11, 1927–1930. [Google Scholar] [CrossRef] [Green Version]
- Kurata, K.; Shiraishi, K.; Takato, T.; Taniguchi, K.; Suzuki, M. A new feeding-deterrent diterpenoid from the brown alga Dilophus okamurai Dawson. Chem. Lett. 1988, 17, 1629–1632. [Google Scholar] [CrossRef]
- Kurata, K.; Suzuki, M.; Shiraishi, K.; Taniguchi, K. Spatane-type diterpenes with biological activity from the brown alga Dilophus okamurai. Phytochemistry 1988, 27, 1321–1324. [Google Scholar] [CrossRef]
- Kurata, K.; Taniguchi, K.; Shiraishi, K.; Suzuki, M. Structures of secospatane-type diterpenes with feeding-deterrent activity from the brown alga Dilophus okamurai. Tetrahedron Lett. 1989, 30, 1567–1570. [Google Scholar] [CrossRef]
- Kurata, K.; Taniguchi, K.; Shiraishi, K.; Suzuki, M. Feeding-deterrent diterpenes from the brown alga Dilophus okamurai. Phytochemistry 1990, 29, 3453–3455. [Google Scholar] [CrossRef]
- Ninomiya, M.; Hirohara, H.; Onishi, J.I.; Kusumi, T. Chemical study and absolute configuration of a new marine secospatane from the brown alga Dilophus okamurae. J. Org. Chem. 1999, 64, 5436–5440. [Google Scholar] [CrossRef] [PubMed]
- Yamase, H.; Umemoto, K.; Ooi, T.; Kusumi, T. Structures and absolute stereochemistry of five new secospatanes and a spatane isolated from the brown alga Dilophus okamurai Dawson. Chem. Pharm. Bull. 1999, 47, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Yamada, H.; Kurata, K. Dictyterpenoids A and B, two novel diterpenoids with feeding-deterrent activity from the brown alga Dilophus okamurae. J. Nat. Prod. 2002, 65, 121–125. [Google Scholar] [CrossRef]
- Casal-Porras, I.; Zubía, E.; Brun, F.G. Dilkamural: A novel chemical weapon involved in the invasive capacity of the alga Ruguloptexyx okamurae in the Strait of Gibraltar. Estuar. Coast. Shelf Sci. 2021, 257, 107398. [Google Scholar] [CrossRef]
- Ravi, B.N.; Wells, R.J. A series of new diterpenes from the brown alga Dilophus marginatus. Aust. J. Chem. 1982, 35, 129–144. [Google Scholar] [CrossRef]
- Zhang, X.; Mosser, D.M. Macrophage activation by endogenous danger signals. J. Pathol. 2008, 214, 161–178. [Google Scholar] [CrossRef]
- Ohashi, W.; Hattori, K.; Hattori, Y. Control of macrophage dynamics as a potential therapeutic approach for clinical disorders involving chronic inflammation. J. Pharmacol. Exp. Ther. 2015, 354, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef]
- Sharma, J.N.; Al-Omram, A.; Paryathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- González, Y.; Torres-Mendoza, D.; Jones, G.E.; Fernandez, P.L. Marine diterpenoids as potential anti-inflammatory agents. Mediat. Inflamm. 2015, 2015, 263543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signalling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, e17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar-Lorenzo, A.; Ardiles, A.E.; Arroba, A.I.; Hernández-Jiménez, E.; Pardo, V.; López-Collazo, E.; Jiménez, I.A.; Bazzochi, I.L.; González-Rodíguez, A.; Valverde, A.M. Fridelenae-type triterpenoids as selective anti-inflammatory agents by regulation of differential signaling pathways in LPS-stimulated macrophages. Toxicol. Appl. Pharmacol. 2016, 313, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano-Cano, F.; Alcalde-Estévez, E.; Gómez-Jaramillo, L.; Iturregui, M.; Sánchez-Fernández, E.M.; García-Fernández, J.M.; Ortiz-Mellet, C.; Campos-Caro, A.; López-Tinoco, C.; Aguilar-Diosdado, M.; et al. Anti-inflammatory (M2) response is induced by a sp2-iminosugar glycolipid sulfoxide in diabetic retinopathy. Front. Immunol. 2021, 12, 632132. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Guha, M.; Mackman, N. LPS induction of gene expression in human monocytes. Cell. Signal. 2021, 13, 85–94. [Google Scholar] [CrossRef]
- Monagail, M.M.; Cronish, L.; Morrison, L.; Araújo, R.; Critchley, A.T. Sustainable harvesting of wild seaweed resources. Eur. J. Phycol. 2017, 52, 371–390. [Google Scholar] [CrossRef]
- Hafting, J.T.; Craigie, J.S.; Stengel, D.B.; Loureiro, R.R.; Buschmann, A.H.; Yarish, C.; Edwards, M.D.; Critchley, A.T. Prospects and challenges for industrial production of seaweed bioactives. J. Phycol. 2015, 51, 821–837. [Google Scholar] [CrossRef]
- Milledge, J.J.; Nielsen, B.V.; Baile, D. High-value products from macroalgae: The potential uses of the invasive brown seaweed, Sargassum muticum. Rev. Environ. Sci. Biotechnol. 2015, 15, 67–88. [Google Scholar] [CrossRef]
- Stabili, L.; Fraschetti, S.; Acquaviva, M.I.; Cavallo, R.A.; De Pascali, S.A.; Fanizzi, F.P.; Gerardi, C.; Narraci, M.; Rizzo, L. The potential exploitation of the mediterranean invasive alga Caulerpa cylindracea: Can the invasión be transformed into a gain? Mar. Drugs 2016, 14, 210. [Google Scholar] [CrossRef]
- Máximo, P.; Ferreira, L.M.; Branco, P.; Lima, P.; Lourenço, A. Secondary metabolites and biological activity of invasive macroalgae of southern Europe. Mar. Drugs 2018, 16, 265. [Google Scholar] [CrossRef] [Green Version]
- Pereira, A.G.; Fraga-Corral, M.; Garcia-Oliveira, P.; Lourenço-Lopes, C.; Carpena, M.; Prieto, M.A.; Simal-Gandara, J. The uses of invasive algae species as a source of secondary metabolites and biological activities: Spain as case-study. Mar. Drugs 2021, 19, 178. [Google Scholar] [CrossRef]
- Griess, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite and [15N] in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC, Type | δH, m (J in Hz) | Position | δC, Type | δH, m (J in Hz) |
|---|---|---|---|---|---|
| 1 | 42.7, CH | 2.53, m | 11 | 13.4, CH3 | 0.92, d (7.0) |
| 2 | 82.2, CH | 4.66, ddd (8.6, 8.0, 7.5) | 12 | 39.8, CH2 | 1.93, dd (12.1, 2.0) 1.49, ddd (12.1, 5.8, 1.9) |
| 3 | 43.6, CH2 | 2.91, dd (14.5, 7.5) 1.57, dd (14.5, 8.0) | 13 | 138.3, C | |
| 4 | 82.1, C | 14 | 22.4, CH3 | 1.70, d (1.2) | |
| 5 | 76.3, CH | 5.23, ddd (4.0, 1.9, 1.9) | 15 | 127.2, CH | 5.18, br t (7.4) |
| 6 | 30.6, CH2 | 2.37, ddd (14.8, 13.4, 4.0) 1.58, m | 16 | 27.7, CH2 | 2.68, m |
| 7 | 40.1, CH | 2.88, dd (13.4, 4.8) | 17 | 124.2, CH | 5.06, br t (7.2) |
| 8 | 38.3, CH | 1.97, br d (5.5) | 18 | 132.0, C | |
| 9 | 53.1, CH | 2.41, br d (8.5) | 19 | 25.9, CH3 | 1.67, d (1.1) |
| 10 | 89.3, C | 20 | 17.9, CH3 | 1.64, br s | |
| CH3COO (2) | 172.9, C | ||||
| CH3COO (2) | 21.0, CH3 | 2.03, s | |||
| CH3COO (5) | 172.8, C | ||||
| CH3COO (5) | 21.5, CH3 | 2.09, s |
| Position | 2 | 3 | 4 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | |
| 1 | 41.5, CH | 3.07, m | 41.3, CH | 3.06, m | 43.6, CH | 2.51, m |
| 2 | 170.7, CH | 7.73, dd (5.8, 3.0) | 170.7, CH | 7.68, ddd (5.8, 3.1, 0.6) | 73.6, CH | 4.11, d (5.8) |
| 3 | 132.3, CH | 5.98, dd (5.8, 1.6) | 132.2, CH | 5.96, dd (5.8, 1.5) | 45.2, CH2 | 2.42, dd (19.0, 5.7) 2.06, br d (19.0) |
| 4 | 58.6, CH | 3.76, ddd (10.8, 6.8, 1.8) | 58.3, CH | 3.78, ddd (11.2, 7.3, 1.8) | 61.6, CH | 3.18, ddd (9.8, 6.6, 2.6) |
| 5 | 77.8, CH | 5.67, ddd (6.8, 6.8, 3.5) | 77.8, CH | 5.69, ddd (7.3, 6.7, 4.1) | 75.9, CH | 4.76, ddd (6.2, 6.2, 3.5) |
| 6 | 37.8, CH2 | 2.25, ddd (14.6, 6.3, 5.6) 1.93, m | 37.2, CH | 2.27, ddd (14.7, 6.7, 4.7) 1.95, m | 41.0, CH2 | 2.02, m 1.79, m |
| 7 | 42.0, CH | 3.63, ddd (8.8, 8.6, 5.6) | 42.4, CH | 3.84, ddd (8.7, 8.7, 4.7) | 41.6, CH | 3.64, m |
| 8 | 39.5, CH | 3.01, ddd (10.8, 9.1, 8.8) | 40.0, CH | 3.06, m | 38.6, CH | 2.97, ddd (9.8, 9.8, 9.8) |
| 9 | 49.6, CH | 2.28, dd (9.1, 6.1) | 50.1, CH | 2.21, dd (10.0, 6.0) | 51.6, CH | 2.63, dd (9.8, 8.4) |
| 10 | 212.8, C | 212.7, C | 220.7, C | |||
| 11 | 17.5, CH3 | 1.19, d (7.2) | 17.6, CH3 | 1.20, d (7.2) | 14.5, CH3 | 0.96, d (7.4) |
| 12 | 201.0, C | 9.60, d (1.8) | 200.8, C | 9.62, d (1.8) | 204.9, CH | 9.64, d (2.6) |
| 13 | 139.0, C | 138.8, C | 136.4, C | |||
| 14 | 22.2, CH3 | 1.68, br s | 22.5, CH3 | 1.75, d (0.7) | 22.4, CH3 | 1.66, br s |
| 15 | 125.9, CH | 5.29, br t (7.7) | 130.9, CH | 6.01, br d (11.0) | 129.3, CH | 5.22, br t (7.0) |
| 16 | 32.6, CH2 | 2.59, m 2.35, m | 126.1, CH | 6.59, dd (15.5, 11.0) | 28.1, CH2 | 2.84, m 2.73, m |
| 17 | 78.3, CH | 5.12, dd (7.6, 5.2) | 138.7, CH | 5.76, d (15.5) | 124.0, CH | 5.09, br t (7.1) |
| 18 | 144.5, C | 82.6, C | 132.5, C | |||
| 19 | 113.2, CH2 | 4.94, br s 4.91, dq (1.6, 1.6) | 24.6, CH3 | 1.36, s | 25.9, CH3 | 1.69, br s |
| 20 | 18.7, CH3 | 1.76, br s | 25.3, CH3 | 1.31, s | 17.9, CH3 | 1.64, br s |
| CH3COO (5) | 172.2 | 172.2 | ||||
| CH3COO (5) | 20.9 | 1.96, s | 20.9 | 1.96, s | ||
| CH3COO (17) | 172.1 | |||||
| CH3COO (17) | 21.0 | 2.03, s | ||||
| Position | 5 | 6 | 7 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | |
| 1 | 40.2, CH | 2.77, m | 42.7, CH | 2.67, m | 40.3, CH | 2.67, m |
| 2 | 76.8, CH | 5.01, d (6.3) | 73.4, CH | 4.10, d (6.3) | 76.7, CH | 5.01, d (6.4) |
| 3 | 41.6, CH2 | 2.56, dd (19.9, 6.3) 2.31, br d (19.9) | 44.2, CH2 | 2.43, dd (19.4, 5.8) 2.11, br d (19.4) | 41.8, CH2 | 2.57, dd (19.7, 6.4) 2.37, br d (19.7) |
| 4 | 47.7, CH | 3.06, m | 47.8, CH | 3.06, m | 153.8, C | |
| 5 | 79.8, CH | 5.23, br d (4.3) | 79.8, CH | 5.22, br d (4.7) | 78.2, CH | 5.63, br dd (7.5, 4.8) |
| 6 | 35.0, CH2 | 1.96, m 1.76, m | 35.2, CH2 | 1.95 ddd (15.0, 9.2, 4.7) 1.76 m | 38.0, CH2 | 2.09, ddd (14.5, 7.5, 3.7) 1.98, m |
| 7 | 39.54c, CH | 3.65, ddd (11.1, 9.2, 9.2) | 39.6, CH2 | 3.65, ddd (11.0, 9.2, 9.2) | 41.4, CH | 3.51, m |
| 8 | 39.52c, CH | 2.65, ddd (12.6, 11.1, 7.7) | 39.7, CH | 2.64, ddd (12.8, 11.0, 7.7) | 43.4, CH | 2.87, m |
| 9 | 50.5, CH | 2.86, ddd (12.6, 6.9, 0.9) | 49.5, CH | 2.99, dd (12.8, 7.0, 0.8) | 53.1, CH | 2.87, m |
| 10 | 217.2, C | 219.7, C | 216.0, C | |||
| 11 | 13.7, CH3 | 0.92, d (7.3) | 14.3, CH3 | 0.86, d (7.4) | 14.2, CH3 | 0.99, d (7.3) |
| 12 | 64.0, CH2 | 4.09, dd (10.8, 5.0) 4.03, dd (10.8, 10.8) | 64.1, CH2 | 4.09, m 4.04, dd (10.8, 10.8) | 113.4, CH2 | 5.27, br s 5.03, br s |
| 13 | 135.9, C | 136.3, C | 137.9, C | |||
| 14 | 22.0, CH3 | 1.73, d (1.2) | 22.2, CH3 | 1.76, d (1.2) | 22.4, CH3 | 1.57, br s |
| 15 | 130.0, CH | 5.23, br t (7.4) | 129.6, CH | 5.24, br t (7.3) | 128.4, CH | 5.12, br t (7.2) |
| 16 | 28.0, CH2 | 2.81, m 2.70, m | 28.0, CH2 | 2.83, ddd (16.0, 7.3, 7.3) 2.72, m | 27.7, CH2 | 2.76, m 2.67, m |
| 17 | 123.8, CH | 5.05 br t, (7.2) | 124.0, CH | 5.07, br t (7.1) | 123.7, CH | 5.05, br t (6.9) |
| 18 | 132.6, C | 132.5, C | 132.8, C | |||
| 19 | 25.8, CH3 | 1.68, d (1.1) | 25.9, CH3 | 1.68, d (1.1) | 25.8, CH3 | 1.68, br s |
| 20 | 17.8, CH3 | 1.63, br s | 17.8, CH3 | 1.63, d (0.6) | 17.9, CH3 | 1.63, br s |
| CH3COO (2) | 172.2, C | 172.1, C | ||||
| CH3COO (2) | 20.8 d | 2.04 c, s | 21.0 c, CH3 | 2.03 c, s | ||
| CH3COO (5) | 172.3, C | 172.4, C | 172.7, C | |||
| CH3COO (5) | 21.0 d | 2.048 c, s | 20.8 c, CH3 | 2.04 c, (s) | 21.3 c, CH3 | 2.04 c, s |
| CH3COO (12) | 172.9, C | 172.9, C | ||||
| CH3COO (12) | 21.3 d | 2.045 c, s | 21.3 c, CH3 | 2.05 c, (s) | ||
| Position | 8 | 9 | 10 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | |
| 1 | 34.6 c, CH | 2.68, m | 34.8, CH | 2.64, m | 42.3, CH | 3.05, m |
| 2 | 28.6, CH2 | 2.05, m 1.66, m | 28.8, CH2 | 2.05, m 1.65, m | 170.9, CH | 7.72, dd (5.8, 2.8) |
| 3 | 34.5 c, CH2 | 2.25, m 2.09, m | 35.0, CH2 | 2.22, m 2.11, dd (9.0, 9.0) | 133.2, CH | 6.06, dd (5.8, 1.9) |
| 4 | 141.8, C | 142.9, C | 47.6, CH | 2.76, m | ||
| 5 | 130.5, CH | 5.76, br s | 126.6, CH | 5.39, br s | 77.6, CH | 5.44, ddd (5.8, 5.8, 2.5) |
| 6 | 37.9, CH2 | 2.57, m 2.05, m | 37.9, CH2 | 2.48, m 2.01, m | 36.9, CH2 | 2.24, ddd (14.0, 9.2, 5.8) 1.81, ddd (14.0, 7.8, 2.5) |
| 7 | 43.7, CH | 3.68, br t (7.3) | 44.2, CH | 3.57, ddd (7.5, 7.5, 3.0) | 42.6, CH | 3.48, m |
| 8 | 45.9, CH | 2.97, m | 47.4, CH | 2.88, m | 40.9, CH | 2.32, ddd (9.4, 9.4, 4.4) |
| 9 | 55.5, CH | 2.68, m | 55.1, CH | 2.57, ddd (8.4, 7.0, 1.3) | 49.0, CH | 2.54, dd (6.1, 4.4) |
| 10 | 220.9, C | 221.0, C | 213.2, C | |||
| 11 | 15.5, CH3 | 0.91, d (6.6) | 15.8, CH3 | 0.93, d (7.1) | 17.9, CH3 | 1.13, d (7.4) |
| 12 | 65.0, CH2 | 4.74, d (13.7) 4.67, d (13.7) | 16.6, CH3 | 1.68, br s | 62.0, CH2 | 3.46, m 3.42, dd (10.7, 4.7) |
| 13 | 139.2, C | 139.7, C | 136.5, C | |||
| 14 | 23.1, CH3 | 1.58, br s | 23.0, CH3 | 1.58, br s | 22.0, CH3 | 1.71, d (1.2) |
| 15 | 127.4, CH | 5.07, br t (7.2) | 126.9, CH | 5.05, br t (7.6) | 129.4, CH | 5.35, br t (7.3) |
| 16 | 27.9, CH2 | 2.80, m 2.68, m | 27.9, CH2 | 2.78, m 2.68, m | 28.0, CH2 | 2.73, m |
| 17 | 124.0, CH | 5.07, br t (7.2) | 124.2, CH | 5.07, br t (7.1) | 124.0, CH | 5.05, br t (7.0) |
| 18 | 132.5, C | 132.3, C | 132.5, C | |||
| 19 | 25.9, CH3 | 1.68, br s | 25.9, CH3 | 1.68, br s | 25.9, CH3 | 1.68, d (1.2) |
| 20 | 17.8, CH3 | 1.63, br s | 17.8, CH3 | 1.62, br s | 17.5, CH3 | 1.64, br s |
| CH3COO | 172.6, C | 172.8, C | ||||
| CH3COO | 20.9, CH3 | 2.01, s | 21.2, CH3 | 2.05, s | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuevas, B.; Arroba, A.I.; de los Reyes, C.; Gómez-Jaramillo, L.; González-Montelongo, M.C.; Zubía, E. Diterpenoids from the Brown Alga Rugulopteryx okamurae and Their Anti-Inflammatory Activity. Mar. Drugs 2021, 19, 677. https://doi.org/10.3390/md19120677
Cuevas B, Arroba AI, de los Reyes C, Gómez-Jaramillo L, González-Montelongo MC, Zubía E. Diterpenoids from the Brown Alga Rugulopteryx okamurae and Their Anti-Inflammatory Activity. Marine Drugs. 2021; 19(12):677. https://doi.org/10.3390/md19120677
Chicago/Turabian StyleCuevas, Belén, Ana I. Arroba, Carolina de los Reyes, Laura Gómez-Jaramillo, M. Carmen González-Montelongo, and Eva Zubía. 2021. "Diterpenoids from the Brown Alga Rugulopteryx okamurae and Their Anti-Inflammatory Activity" Marine Drugs 19, no. 12: 677. https://doi.org/10.3390/md19120677
APA StyleCuevas, B., Arroba, A. I., de los Reyes, C., Gómez-Jaramillo, L., González-Montelongo, M. C., & Zubía, E. (2021). Diterpenoids from the Brown Alga Rugulopteryx okamurae and Their Anti-Inflammatory Activity. Marine Drugs, 19(12), 677. https://doi.org/10.3390/md19120677