
Marine Heterocyclic Compounds That Modulate Intracellular Calcium Signals: Chemistry and Synthesis Approaches

 , ,
, , 
Abstract
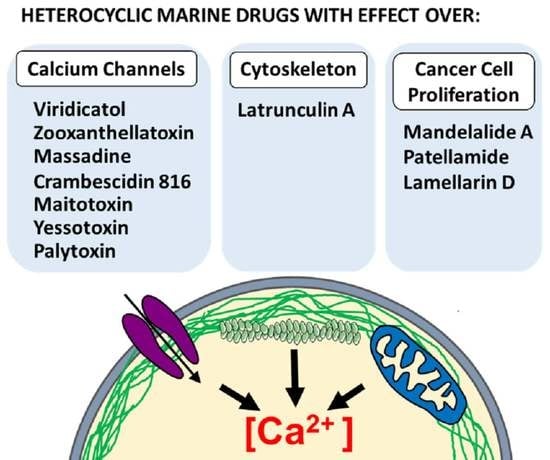
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Heterocyclic Marine Drugs with Effect over Calcium Channels
2.1. Viridicatol
2.2. Zooxanthellatoxin-A
2.3. Massadine
2.4. Crambescidin 816
2.5. Maitotoxin
2.6. Yessotoxin
2.7. Palytoxin
3. Heterocyclic Marine Drugs with a Direct Effect over Cytoskeleton
Latrunculin A
4. Heterocyclic Marine Drugs with Effect on Tumor Cell Proliferation
4.1. Mandelalides A–D
4.2. Patellamide
4.3. Lamellarins
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Humeau, J.; Bravo-San Pedro, J.M.; Vitale, I.; Nuñez, L.; Villalobos, C.; Kroemer, G.; Senovilla, L. Calcium signaling and cell cycle: Progression or death. Cell Calcium 2018, 70, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, C.; Faught, W.J.; Frawley, L.S. Dynamic changes in spontaneous intracellular free calcium oscillations and their relationship to prolactin gene expression in single, primary mammotropes. Mol. Endocrinol. 1998, 12, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J. Elementary and global aspects of calcium signalling. J. Physiol. 1997, 499, 291–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- González, D.R.; Treuer, A.V.; Castellanos, J.; Dulce, R.A.; Hare, J.M. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J. Biol. Chem. 2010, 285, 28938–28945. [Google Scholar] [CrossRef] [Green Version]
- Petersen, O.H.; Michalak, M.; Verkhratsky, A. Calcium signalling: Past, present and future. Cell Calcium 2005, 38, 161–169. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Rizzuto, R.; Bernardi, P.; Pozzan, T. Mitochondria as all-round players of the calcium game. J. Physiol. 2000, 529, 37–47. [Google Scholar] [CrossRef]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; de Strooper, B.; Bezprozvanny, I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Investig. 2007, 117, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- Joseph, N.; Reicher, B.; Mira Barda-Saad, M. The calcium feedback loop and T cell activation: How cytoskeleton networks control intracellular calcium flux. Biochim. Biophys. Acta 2014, 1838, 557–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, L.G.; Hernández-Morales, M.; Nuñez, L.; Villalobos, C. Inhibition of Polyamine Biosynthesis Reverses Ca2+ Channel Remodeling in Colon Cancer Cells. Cancers 2019, 11, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iamshanova, O.; Pla, A.F.; Prevarskaya, N. Molecular mechanisms of tumour invasion: Regulation by calcium signals. J. Physiol. 2017, 595, 3063–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Z.; Liu, Q.; Xing, C.; Zhang, Y.; Zhou, Y.; Zhang, J.; Liu, H.; Cao, M.; Yang, X.; Liu, G. Viridicatol Isolated from Deep-Sea Penicillium Griseofulvum Alleviates Anaphylaxis and Repairs the Intestinal Barrier in Mice by Suppressing Mast Cell Activation. Mar. Drugs 2020, 18, 517. [Google Scholar] [CrossRef] [PubMed]
- Luckner, M.; Mohammed, Y.S. About metabolic products of Penicillium viridicatum westling and Penicillium cyclopium westling; Synthesis of viridicatol, 3′-O-methylviridicatol and N-methyl-3′-O-methylviridicatol. Tetrahedron Lett. 1964, 5, 1987–1989. [Google Scholar] [CrossRef]
- Tangella, Y.; Manasa, K.L.; Krishna, N.H.; Sridhar, B.; Kamal, A.; Nagendra Babu, B. Regioselective Ring Expansion of Isatins with In Situ Generated α-Aryldiazomethanes: Direct Access to Viridicatin Alkaloids. Org. Lett. 2018, 20, 3639–3642. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Harayama, T. A Concise and Versatile Synthesis of Viridicatin Alkaloids from Cyanoacetanilides. Org. Lett. 2009, 11, 1603–1606. [Google Scholar] [CrossRef]
- Mamedov, V.A.; Mamedova, V.L.; Kadyrova, S.F.; Galimullina, V.R.; Khikmatova, G.Z.; Korshin, D.E.; Gubaidullin, A.T.; Krivolapov, D.B.; Rizvanov, I.K.; Bazanova, O.B.; et al. Synthesis of 3-Hydroxy-4-arylquinolin-2-ones Including Viridicatol via a Darzens Condensation/Friedel–Crafts Alkylation Strategy. J. Org. Chem. 2018, 83, 13132–13145. [Google Scholar] [CrossRef]
- Rho, M.C.; Nakahata, N.; Nakamura, H.; Murai, A.; Ohizumi, Y. Involvement of phospholipase C-gamma2 in activation of mitogen-activated protein kinase and phospholipase A2 by zooxanthellatoxin-A in rabbit platelets. J. Pharmacol. Exp. Ther. 1997, 282, 496–504. [Google Scholar]
- Moriya, T.; Furukawa, K.; Nakamura, H.; Murai, A.; Ohizumi, Y. The vaso-contractile action of zooxanthellatoxin-B from a marine dinoflagellate is mediated via Ca2+ influx in the rabbit aorta. Can. J. Physiol. Pharmacol. 2001, 79, 1030–1035. [Google Scholar] [CrossRef]
- Nakamura, H.; Asari, T.; Murai, A.; Kondo, T.; Yoshida, K.; Ohizumi, Y. Structure of periodate oxidation products with characteristic partial structures of zooxanthellatoxin-A, a potent vasoconstrictive polyol from a symbiotic dinoflagellate. J. Org. Chem. 1993, 58, 313–314. [Google Scholar] [CrossRef]
- Nakamura, H.; Sato, K.; Murai, A. Absolute configuration of the spiroacetal portion of zooxanthellatoxin-A. Tetrahedron Lett. 1996, 37, 7267–7270. [Google Scholar] [CrossRef]
- Nakamura, H.; Takahashi, M.; Murai, A. Synthesis and absolute configuration of an exomethylene portion of zooxanthellatoxin-A. Tetrahedron Asymmetry 1998, 9, 2571–2574. [Google Scholar] [CrossRef]
- Nakamura, H.; Fujimaki, K.; Murai, A. Synthetic studies on the common C25 long chain acid portion of zooxanthellatoxins from a symbiotic dinoflagellate Symbiodinium sp. Tetrahedron Lett. 1996, 37, 3153–3156. [Google Scholar] [CrossRef]
- Nakamura, H.; Maruyama, K.; Fujimaki, K.; Murai, A. Absolute configuration of the common terminal acid portion of zooxanthellatoxins from a symbiotic dinoflagellate Symbiodinium sp. established by the synthesis of its ozonolysis product. Tetrahedron Lett. 2000, 41, 1927–1930. [Google Scholar] [CrossRef]
- Onodera, K.-I.; Nakamura, H.; Oba, Y.; Ohizumi, Y.; Ojika, M. Zooxanthellamide Cs: Vasoconstrictive Polyhydroxylated Macrolides with the Largest Lactone Ring Size from a Marine Dinoflagellate of Symbiodinium sp. J. Am. Chem. Soc. 2005, 127, 10406–10411. [Google Scholar] [CrossRef]
- Nishimura, S.; Matsunaga, S.; Shibazaki, M.; Suzuki, K.; Furihata, K.; van Soest, R.W.; Fusetani, N. Massadine, a novel geranylgeranyltransferase type I inhibitor from the marine sponge Stylissa aff. massa. Org. Lett. 2003, 5, 2255–2257. [Google Scholar] [CrossRef]
- Bickmeyer, U.; Grube, A.; Klings, K.W.; Köck, M. Disturbance of voltage-induced cellular calcium entry by marine dimeric and tetrameric pyrrole-imidazole alkaloids. Toxicon 2007, 50, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Seiple, I.B.; Young, I.S.; Baran, P.S. Total Syntheses of (±)-Massadine and Massadine Chloride. J. Am. Chem. Soc. 2008, 130, 16490–16491. [Google Scholar] [CrossRef] [Green Version]
- Seiple, I.B.; Su, S.; Young, I.S.; Nakamura, A.; Yamaguchi, J.; Jørgensen, L.; Rodriguez, R.A.; O’Malley, D.P.; Gaich, T.; Köck, M.; et al. Enantioselective Total Syntheses of (−)-Palau’amine, (−)-Axinellamines, and (−)-Massadines. J. Am. Chem. Soc. 2011, 133, 14710–14726. [Google Scholar] [CrossRef] [Green Version]
- Chinigo, G.M.; Breder, A.; Carreira, E.M. Ugi-4-Component Reaction Enabling Rapid Access to the Core Fragment of Massadine. Org. Lett. 2011, 13, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Breder, A.; Chinigo, G.M.; Waltman, A.W.; Carreira, E.M. Enantioselective Synthesis of the Carbocyclic D-Ring Subunit of Massadine. Angew. Chem. Int. Ed. Engl. 2008, 47, 8514–8517. [Google Scholar] [CrossRef] [PubMed]
- Breder, A.; Chinigo, G.M.; Waltman, A.W.; Carreira, E.M. Towards the Synthesis of Massadine: A Unified Strategy for the Stereoselective Synthesis of the Carbocyclic C,D-Ring Subunit. Chem. Eur. J. 2011, 17, 12405–12416. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Lee, H.; Lee, D. Synthesis of the Carbocyclic Core of Massadine. Org. Lett. 2015, 17, 5348–5351. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.S. A Nitrone Dipolar Cycloaddition Strategy toward an Enantioselective Synthesis of Massadine. Org. Lett. 2018, 20, 3883–3887. [Google Scholar] [CrossRef]
- Martín, V.; Vale, C.; Bondu, S.; Thomas, O.P.; Vieytes, M.R.; Botana, L.M. Differential Effects of Crambescins and Crambescidin 816 in Voltage-Gated Sodium, Potassium and Calcium Channels in Neurons. Chem. Res. Toxicol. 2013, 26, 169–178. [Google Scholar] [CrossRef]
- Berlinck, R.G.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Ferri, S.; Spampinato, S.; Speroni, E. Polycyclic guanidine alkaloids from the marine sponge Crambe crambe and Ca2+ channel blocker activity of crambescidin 816. J. Nat. Prod. 1993, 56, 1007–1015. [Google Scholar] [CrossRef]
- Méndez, A.G.; Juncal, A.B.; Silva, S.B.L.; Thomas, O.P.; Martín Vázquez, V.; Alfonso, A.; Vieytes, M.R.; Vale, C.; Botana, L.M. The Marine Guanidine Alkaloid Crambescidin 816 Induces Calcium Influx and Cytotoxicity in Primary Cultures of Cortical Neurons through Glutamate Receptors. ACS Chem. Neurosci. 2017, 8, 1609–1617. [Google Scholar] [CrossRef] [Green Version]
- Braekman, J.C.; Daloze, D.; Tavares, R.; Hajdu, E.; Van Soest, R.W.M. Novel Polycyclic Guanidine Alkaloids from Two Marine Sponges of the Genus Monanchora. J. Nat. Prod. 2000, 63, 193–196. [Google Scholar] [CrossRef]
- Moore, C.G.; Murphy, P.J.; Williams, H.L.; McGown, A.T.; Smith, N.K. Synthetic studies towards ptilomycalin A: Total synthesis of crambescidin 359. Tetrahedron 2007, 63, 11771–11780. [Google Scholar] [CrossRef]
- Nagasawa, K.; Hashimoto, Y. Synthesis of marine guanidine alkaloids and their application as chemical/biological tools. Chem. Rec. 2003, 3, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Aron, Z.D.; Overman, L.E. Total Synthesis and Properties of the Crambescidin Core Zwitterionic Acid and Crambescidin 359. J. Am. Chem. Soc. 2005, 127, 3380–3390. [Google Scholar] [CrossRef] [PubMed]
- Flores, P.L.; Rodríguez, E.; Zapata, E.; Carbó, R.; Farías, J.M.; Martínez, M. Maitotoxin Is a Potential Selective Activator of the Endogenous Transient Receptor Potential Canonical Type 1 Channel in Xenopus laevis Oocytes. Mar. Drugs 2017, 15, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaou, K.C.; Gelin, C.F.; Seo, J.H.; Huang, Z.; Umezawa, T. Synthesis of the QRSTU Domain of Maitotoxin and Its 85-epi- and 86-epi-Diastereoisomers. J. Am. Chem. Soc. 2010, 132, 9900–9907. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, K.C.; Baker, T.M.; Nakamura, T. Synthesis of the WXYZA’ Domain of Maitotoxin. J. Am. Chem. Soc. 2011, 133, 220–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaou, K.C.; Seo, J.H.; Nakamura, T.; Aversa, R.J. Synthesis of the C’D’E’F’ Domain of Maitotoxin. J. Am. Chem. Soc. 2011, 133, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, K.C.; Aversa, R.J. Maitotoxin: An Inspiration for Synthesis. Isr. J. Chem. 2011, 51, 359–377. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, K.C.; Heretsch, P.; Nakamura, T.; Rudo, A.; Murata, M.; Konoki, K. Synthesis and Biological Evaluation of QRSTUVWXYZA’ Domains of Maitotoxin. J. Am. Chem. Soc. 2014, 136, 16444–16451. [Google Scholar] [CrossRef] [Green Version]
- Oishi, T.; Hasegawa, F.; Torikai, K.; Konoki, K.; Matsumori, N.; Murata, M. Convergent Synthesis and Biological Activity of the WXYZA’B’C’ Ring System of Maitotoxin. Org. Lett. 2008, 10, 3599–3602. [Google Scholar] [CrossRef]
- Kunitake, M.; Oshima, T.; Konoki, K.; Ebine, M.; Torikai, K.; Murata, M.; Oishi, T. Synthesis and Biological Activity of the C’D’E’F’ Ring System of Maitotoxin. J. Org. Chem. 2014, 79, 4948–4962. [Google Scholar] [CrossRef]
- Onoue, H.; Baba, T.; Konoki, K.; Torikai, K.; Ebine, M.; Oishi, T. Synthesis and Biological Activity of the QRS Ring System of Maitotoxin. Chem. Lett. 2014, 43, 1904–1906. [Google Scholar] [CrossRef]
- Onoue, H.; Marubayashi, R.; Ishikawa, E.; Konoki, K.; Torikai, K.; Ebine, M.; Murata, M.; Oishi, T. Syntheses and Biological Activities of the LMNO, ent-LMNO, and NOPQR(S) Ring Systems of Maitotoxin. J. Org. Chem. 2017, 82, 9595–9618. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Ishiyama, S.; Koshino, H.; Nakata, T. Synthetic Studies on Maitotoxin. 1. Stereoselective Synthesis of the C’D’E’F’-Ring System Having a Side Chain. Org. Lett. 2008, 10, 1675–1678. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Haketa, T.; Koshino, H.; Nakata, T. Synthetic Studies on Maitotoxin. 2. Stereoselective Synthesis of the WXYZA’-Ring System. Org. Lett. 2008, 10, 1679–1682. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Koshino, H.; Nakata, T. Synthetic Studies on Maitotoxin. 3. Stereoselective Synthesis of the BCDE-Ring System. Org. Lett. 2008, 10, 1683–1685. [Google Scholar] [CrossRef]
- Saito, T.; Morita, M.; Koshino, H.; Sodeoka, M.; Nakata, T. Convergent Synthesis of the ent-ZA’B’C’D’-Ring System of Maitotoxin. Org. Lett. 2017, 19, 3203–3206. [Google Scholar] [CrossRef]
- Pérez-Gómez, A.; Ferrero-Gutierrez, A.; Novelli, A.; Franco, J.M.; Paz, B.; Fernández-Sánchez, M.T. Potent Neurotoxic Action of the Shellfish Biotoxin Yessotoxin on Cultured Cerebellar Neurons. Toxicol. Sci. 2005, 90, 168–177. [Google Scholar] [CrossRef] [Green Version]
- de la Rosa, L.A.; Alfonso, A.; Vilariño, N.; Vieytes, M.R.; Botana, L.M. Modulation of cytosolic calcium levels of human lymphocytes by yessotoxin, a novel marine phycotoxin. Biochem. Pharmacol. 2001, 61, 827–833. [Google Scholar] [CrossRef]
- Pang, M.; Qu, P.; Gao, C.-L.; Tang, X.; Wang, Z.-L. Effect of yessotoxin on cytosolic calcium levels in human hepatocellular carcinoma cells in vitro. Biomed. Rep. 2014, 2, 93–96. [Google Scholar] [CrossRef]
- Mori, Y.; Hayashi, H. Synthetic studies of yessotoxin, a polycyclic ether implicated in diarrhetic shellfish poisoning: Convergent synthesis of the BCDE ring system via an alkyne intermediate. Tetrahedron 2002, 58, 1789–1797. [Google Scholar] [CrossRef]
- Sakai, T.; Sugimoto, A.; Tatematsu, H.; Mori, Y. Divergent synthesis of trans-fused polycyclic ethers by a convergent oxiranyl anion strategy. J. Org. Chem. 2012, 77, 11177–11191. [Google Scholar] [CrossRef] [PubMed]
- Kadota, I.; Ueno, H.; Sato, Y.; Yamamoto, Y. Convergent synthesis of the FGHI ring segment of yessotoxin. Tetrahedron Lett. 2006, 47, 89–92. [Google Scholar] [CrossRef]
- Kadota, I.; Abe, T.; Sato, Y.; Kabuto, C.; Yamamoto, Y. Stereocontrolled synthesis of the IJK ring segment of yessotoxin. Tetrahedron Lett. 2006, 47, 6545–6548. [Google Scholar] [CrossRef]
- Oishi, T.; Suzuki, M.; Watanabe, K.; Murata, M. Synthesis of the ABC and IJ ring fragments of yessotoxin. Tetrahedron Lett. 2006, 47, 3975–3978. [Google Scholar] [CrossRef]
- Watanabe, K.; Suzuki, M.; Murata, M.; Oishi, T. Convergent synthesis of the FGHI ring system of yessotoxin: Stereoselective construction of the G ring. Tetrahedron Lett. 2005, 46, 3991–3995. [Google Scholar] [CrossRef]
- Torikai, K.; Watanabe, K.; Minato, H.; Imaizumi, T.; Murata, M.; Oishi, T. Convergent Synthesis of the A-J Ring System of Yessotoxin. Synlett 2008, 2008, 2368–2372. [Google Scholar] [CrossRef]
- Oishi, T.; Imaizumi, T.; Murata, M. Reductive Etherification under Microfluidic Conditions: Application to Practical Synthesis of the FGHIJ-Ring System of Yessotoxin. Chem. Lett. 2010, 39, 108–109. [Google Scholar] [CrossRef]
- Zhang, Y.; Rohanna, J.; Zhou, J.; Iyer, K.; Rainier, J.D. Total Synthesis of Brevenal. J. Am. Chem. Soc. 2011, 133, 3208–3216. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Rainier, J.D. Synthesis of the ABCDEF and FGHI ring system of yessotoxin and adriatoxin. J. Antibiot. 2016, 69, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Hirai, Y.; Yokoyama, H.; Kusumoto, Y.; Sumiyoshi, K.; Miyazawa, M. Synthetic Studies of Yessotoxin: Iterative Synthesis of the AB Ring System via Pd(II)-Catalyzed Cyclization of Alcohol. Heterocycles 2014, 89, 353–358. [Google Scholar] [CrossRef]
- Czabaniuk, L.C.; Jamison, T.F. Hydroxyl-Substituted Ladder Polyethers via Selective Tandem Epoxidation/Cyclization Sequence. Org. Lett. 2015, 17, 774–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, H.; Nishida, K.; Togawa, T.; Yamagami, M.; Miyazawa, M.; Hirai, Y. Stereoselective synthesis of the KJ ring system of yessotoxin by Pd(II)-catalyzed cyclization. Tetrahedron Lett. 2016, 57, 4379–4381. [Google Scholar] [CrossRef]
- Trost, B.M.; Rhee, Y.H. A Flexible Approach toward trans-Fused Polycyclic Tetrahydropyrans. A Synthesis of Prymnesin and Yessotoxin Units. Org. Lett. 2004, 6, 4311–4313. [Google Scholar] [CrossRef] [PubMed]
- Habermann, E. Palytoxin acts through Na+, K+-ATPase. Toxicon 1989, 27, 1171–1187. [Google Scholar] [CrossRef]
- Shimahara, T.; Molgó, J. Palytoxin enhances quantal acetylcholine release from motor nerve terminals and increases cytoplasmic calcium levels in a neuronal hybrid cell line. Life Sci. Adv. Pharmacol. 1990, 9, 785–792. [Google Scholar]
- Rakowski, R.F.; Artigas, P.; Palma, F.; Holmgren, M.; De Weer, P.; Gadsby, D.C. Sodium Flux Ratio in Na/K Pump-Channels Opened by Palytoxin. J. Gen. Physiol. 2007, 130, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiki, H.; Suganuma, M.; Nakayasu, M.; Hakii, H.; Horiuchi, T.; Takayama, S.; Sugimura, T. Palytoxin is a non-12-O-tetradecanoylphorbol-13-acetate type tumor promoter in two-stage mouse skin carcinogenesis. Carcinogenesis 1986, 7, 707–710. [Google Scholar] [CrossRef]
- Ko, S.S.; Finan, J.M.; Yonaga, M.; Kishi, Y.; Uemura, D.; Hirata, Y. Stereochemistry of palytoxin. Part 2. C1-C6, C47-C74, and C77-C83 segments. J. Am. Chem. Soc. 1982, 104, 7364–7367. [Google Scholar] [CrossRef]
- Kishi, Y. Natural products synthesis: Palytoxin. Pure Appl. Chem. 1989, 61, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Suh, E.M.; Kishi, Y. Synthesis of Palytoxin from Palytoxin Carboxylic Acid. J. Am. Chem. Soc. 1994, 116, 11205–11206. [Google Scholar] [CrossRef]
- Still, W.C.; Galynker, I. Stereospecific synthesis of the C30-C43 segment of palytoxin by macrocyclically controlled remote asymmetric induction. J. Am. Chem. Soc. 1982, 104, 1774–1776. [Google Scholar] [CrossRef]
- Hodgson, R.; Nelson, A. A two-directional synthesis of the C58–C71 fragment of palytoxin. Org. Biomol. Chem. 2004, 2, 373–386. [Google Scholar] [CrossRef]
- Morton, W.M.; Ayscough, K.R.; McLaughlin, P.J. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat. Cell Biol. 2000, 2, 376–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F. Latrunculin A depolarizes starfish oocytes. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2007, 148, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Gokina, N.I.; Osol, G. Actin cytoskeletal modulation of pressure-induced depolarization and Ca2+ influx in cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1410–H1420. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.T.; Limatola, N.; Vasilev, F.; Santella, L. Early events of fertilization in sea urchin eggs are sensitive to actin-binding organic molecules. Biochem. Biophys. Res. Commun. 2014, 450, 1166–1174. [Google Scholar] [CrossRef]
- Spector, I.; Shochet, N.R.; Blasberger, D.; Kashman, Y. Latrunculins-novel marine macrolides that disrupt microfilament organization and affect cell growth: Comparison with cytochalasin D. Cell Motil. Cytoskelet. 1989, 13, 127–144. [Google Scholar] [CrossRef]
- Fürstner, A.; Turet, L. Concise and Practical Synthesis of Latrunculin A by Ring-Closing Enyne–Yne Metathesis. Angew. Chem. Int. Ed. Engl. 2005, 44, 3462–3466. [Google Scholar] [CrossRef]
- Fürstner, A.; De Souza, D.; Parra-Rapado, L.; Jensen, J.T. Catalysis-Based Total Synthesis of Latrunculin B. Angew. Chem. Int. Ed. Engl. 2003, 42, 5358–5360. [Google Scholar] [CrossRef]
- Fürstner, A.; Souza, D.; Turet, L.; Fenster, M.; Parra-Rapado, L.; Wirtz, C.; Mynott, R.; Lehmann, C. Total Syntheses of the Actin-Binding Macrolides Latrunculin A, B, C, M, S and 16-epi-Latrunculin B. Chem. Eur. J. 2007, 13, 115–134. [Google Scholar] [CrossRef]
- White, J.D.; Kawasaki, M. Total synthesis of (+)-latrunculin A, an ichthyotoxic metabolite of the sponge Latrunculia magnifica and its C-15 epimer. J. Org. Chem. 1992, 57, 5292–5300. [Google Scholar] [CrossRef]
- Smith, A.B.; Noda, I.; Remiszewski, S.W.; Liverton, N.J.; Zibuck, R. Total synthesis of (+)-latrunculin A. J. Org. Chem. 1990, 55, 3977–3979. [Google Scholar] [CrossRef]
- Smith, A.B.; Leahy, J.W.; Noda, I.; Remiszewski, S.W.; Liverton, N.J.; Zibuck, R. Total synthesis of the latrunculins. J. Am. Chem. Soc. 1992, 114, 2995–3007. [Google Scholar] [CrossRef]
- White, J.D.; Kawasaki, M. Total synthesis of (+)-latrunculin A. J. Am. Chem. Soc. 1990, 112, 4991–4993. [Google Scholar] [CrossRef]
- Sikorska, J.; Hau, A.M.; Anklin, C.; Parker-Nance, S.; Davies-Coleman, M.T.; Ishmael, J.E.; McPhail, K.L. Mandelalides A-D, cytotoxic macrolides from a new Lissoclinum species of South African tunicate. J. Org. Chem. 2012, 77, 6066–6075. [Google Scholar] [CrossRef] [Green Version]
- Nazari, M.; Serrill, J.D.; Wan, X.; Nguyen, M.H.; Anklin, C.; Gallegos, D.A.; Smith, A.B.; Ishmael, J.E.; McPhail, K.L. New Mandelalides Expand a Macrolide Series of Mitochondrial Inhibitors. J. Med. Chem. 2017, 60, 7850–7862. [Google Scholar] [CrossRef]
- Villalobos, C.; Gutiérrez, L.G.; Hernández-Morales, M.; del Bosque, D.; Nuñez, L. Mitochondrial control of store-operated Ca2+ channels in cancer: Pharmacological implications. Pharmacol. Res. 2018, 135, 136–143. [Google Scholar] [CrossRef]
- Hamada, Y.; Shibata, M.; Shioiri, T. New methods and reagents in organic synthesis. 55. Total syntheses of patellamides B and C, cytotoxic cyclic peptides from a tunicate 1. Their proposed structures should be corrected. Tetrahedron Lett. 1985, 26, 5155–5158. [Google Scholar] [CrossRef]
- Williams, A.B.; Jacobs, R.S. A marine natural product, patellamide D, reverses multidrug resistance in a human leukemic cell line. Cancer Lett. 1993, 71, 97–102. [Google Scholar] [CrossRef]
- Hamada, Y.; Shibata, M.; Shioiri, T. New methods and reagents in organic synthesis. 56. Total syntheses of patellamides B and C, cytotoxic cyclic peptides from a tunicate 2. Their real structures have been determined by their syntheses. Tetrahedron Lett. 1985, 26, 5159–5162. [Google Scholar] [CrossRef]
- Hamada, Y.; Shibata, M.; Shioiri, T. New methods and reagents in organic synthesis. 58: A synthesis of patellamide a, a cytotoxic cyclic peptide from a tunicate. Revision of its proposed structure. Tetrahedron Lett. 1985, 26, 6501–6504. [Google Scholar] [CrossRef]
- Schmidt, U.; Griesser, H. Total synthesis and structure determination of patellamide B. Tetrahedron Lett. 1986, 27, 163–166. [Google Scholar] [CrossRef]
- García-Reynaga, P.; VanNieuwenhze, M.S. A New Total Synthesis of Patellamide A. Org. Lett. 2008, 10, 4621–4623. [Google Scholar] [CrossRef] [Green Version]
- Bailly, C. Anticancer Properties of Lamellarins. Mar. Drugs 2015, 13, 1105–1123. [Google Scholar] [CrossRef] [Green Version]
- Ballot, C.; Kluza, J.; Lancel, S.; Martoriati, A.; Hassoun, S.M.; Mortier, L.; Vienne, J.-C.; Briand, G.; Formstecher, P.; Bailly, C.; et al. Inhibition of mitochondrial respiration mediates apoptosis induced by the anti-tumoral alkaloid lamellarin D. Apoptosis 2010, 15, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J.; Faulkner, D.J.; He, C.H.; Van Duyne, G.D.; Clardy, J. Metabolites of the marine prosobranch mollusk Lamellaria sp. J. Am. Chem. Soc. 1985, 107, 5492–5495. [Google Scholar] [CrossRef]
- Pla, D.; Marchal, A.; Olsen, C.A.; Albericio, F.; Alvarez, M. Modular total synthesis of lamellarin D. J. Org. Chem. 2005, 70, 8231–8234. [Google Scholar] [CrossRef]
- Manjappa, K.B.; Lin, J.-M.; Yang, D.-Y. Construction of Pentacyclic Lamellarin Skeleton via Grob Reaction: Application to Total Synthesis of Lamellarins H and D. J. Org. Chem. 2017, 82, 7648–7656. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Total Synthesis of Lamellarins D, H, and R and Ningalin B. Org. Lett. 2011, 13, 312–315. [Google Scholar] [CrossRef]
- Ploypradith, P.; Petchmanee, T.; Sahakitpichan, P.; Litvinas, N.D.; Ruchirawat, S. Total Synthesis of Natural and Unnatural Lamellarins with Saturated and Unsaturated D-Rings. J. Org. Chem. 2006, 71, 9440–9448. [Google Scholar] [CrossRef]
- Liermann, J.C.; Opatz, T. Synthesis of Lamellarin U and Lamellarin G Trimethyl Ether by Alkylation of a Deprotonated α-Aminonitrile. J. Org. Chem. 2008, 73, 4526–4531. [Google Scholar] [CrossRef] [PubMed]
- Colligs, V.C.; Dialer, C.; Opatz, T. Synthesis of Lamellarin G Trimethyl Ether by von Miller–Plöchl-Type Cyclocondensation. Eur. J. Org. Chem. 2018, 2018, 4064–4070. [Google Scholar] [CrossRef]
- Imbri, D.; Tauber, J.; Opatz, T. A High-Yielding Modular Access to the Lamellarins: Synthesis of Lamellarin G Trimethyl Ether, Lamellarin η and Dihydrolamellarin η. Chem. Eur. J. 2013, 19, 15080–15083. [Google Scholar] [CrossRef] [PubMed]
- Dialer, C.; Imbri, D.; Hansen, S.P.; Opatz, T. Synthesis of Lamellarin D Trimethyl Ether and Lamellarin H via 6π-Electrocyclization. J. Org. Chem. 2015, 80, 11605–11610. [Google Scholar] [CrossRef]
- Lade, D.M.; Pawar, A.B.; Mainkar, P.S.; Chandrasekhar, S. Total Synthesis of Lamellarin D Trimethyl Ether, Lamellarin D, and Lamellarin H. J. Org. Chem. 2017, 82, 4998–5004. [Google Scholar] [CrossRef]
- Kumar, V.; Awasthi, A.; Salam, A.; Khan, T. Scalable Total Syntheses of Some Natural and Unnatural Lamellarins: Application of a One-Pot Domino Process for Regioselective Access to the Central 1,2,4-Trisubstituted Pyrrole Core. J. Org. Chem. 2019, 84, 11596–11603. [Google Scholar] [CrossRef]
- Klintworth, R.; de Koning, C.B.; Michael, J.P. Demethylative Lactonization Provides a Shortcut to High-Yielding Syntheses of Lamellarins. J. Org. Chem. 2020, 85, 1054–1061. [Google Scholar] [CrossRef]
























































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Andrés, P.; Fernández-Peña, L.; Díez-Poza, C.; Villalobos, C.; Nuñez, L.; Barbero, A. Marine Heterocyclic Compounds That Modulate Intracellular Calcium Signals: Chemistry and Synthesis Approaches. Mar. Drugs 2021, 19, 78. https://doi.org/10.3390/md19020078
González-Andrés P, Fernández-Peña L, Díez-Poza C, Villalobos C, Nuñez L, Barbero A. Marine Heterocyclic Compounds That Modulate Intracellular Calcium Signals: Chemistry and Synthesis Approaches. Marine Drugs. 2021; 19(2):78. https://doi.org/10.3390/md19020078
Chicago/Turabian StyleGonzález-Andrés, Paula, Laura Fernández-Peña, Carlos Díez-Poza, Carlos Villalobos, Lucía Nuñez, and Asunción Barbero. 2021. "Marine Heterocyclic Compounds That Modulate Intracellular Calcium Signals: Chemistry and Synthesis Approaches" Marine Drugs 19, no. 2: 78. https://doi.org/10.3390/md19020078
APA StyleGonzález-Andrés, P., Fernández-Peña, L., Díez-Poza, C., Villalobos, C., Nuñez, L., & Barbero, A. (2021). Marine Heterocyclic Compounds That Modulate Intracellular Calcium Signals: Chemistry and Synthesis Approaches. Marine Drugs, 19(2), 78. https://doi.org/10.3390/md19020078