
Cytotoxic N-Methylpretrichodermamide B Reveals Anticancer Activity and Inhibits P-Glycoprotein in Drug-Resistant Prostate Cancer Cells

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
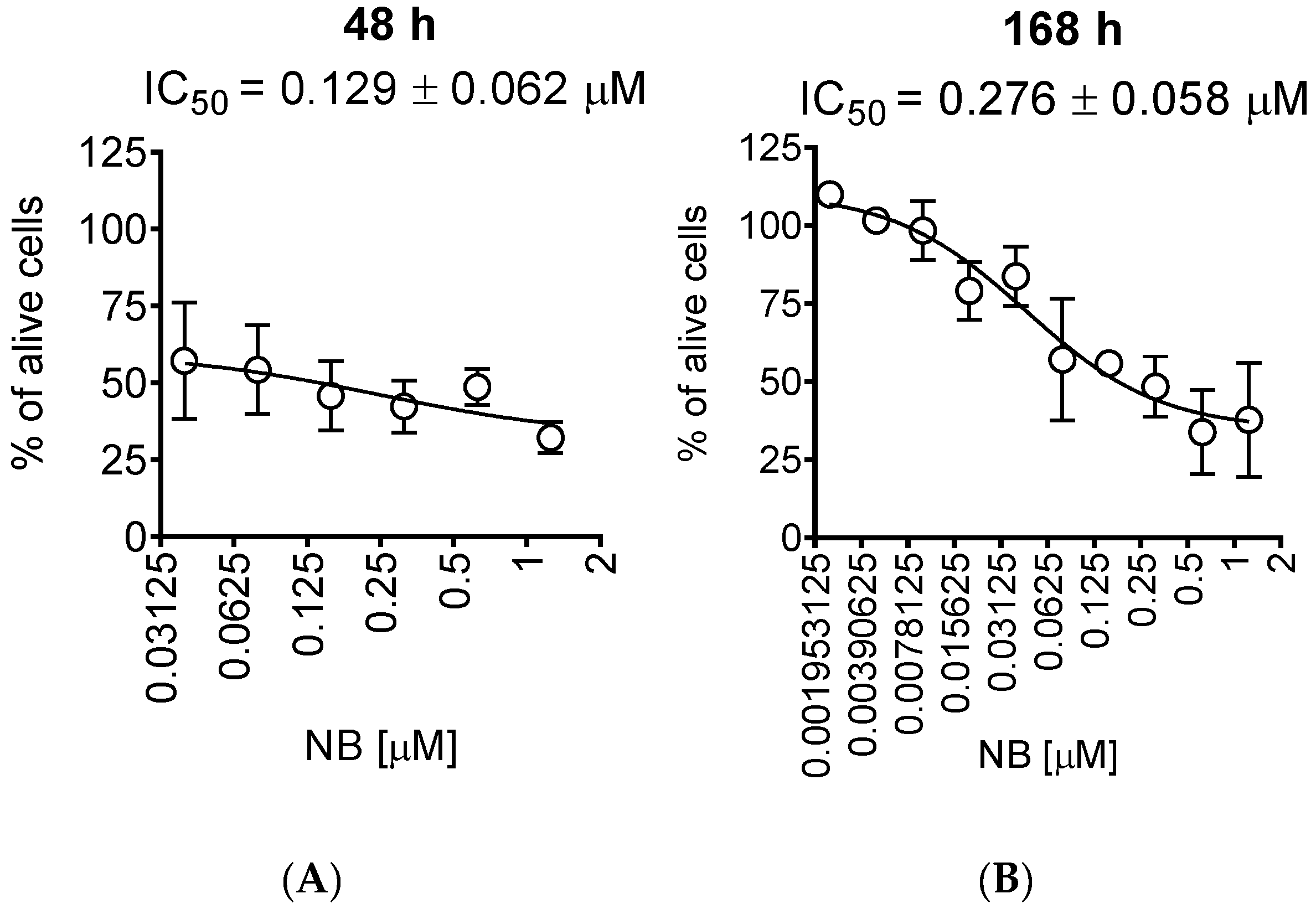
2.1. Cytotoxicity and Selectivity of NB in Human Prostate Cancer Cells
2.2. NB Affects Protein Tyrosine Kinases’ Activity
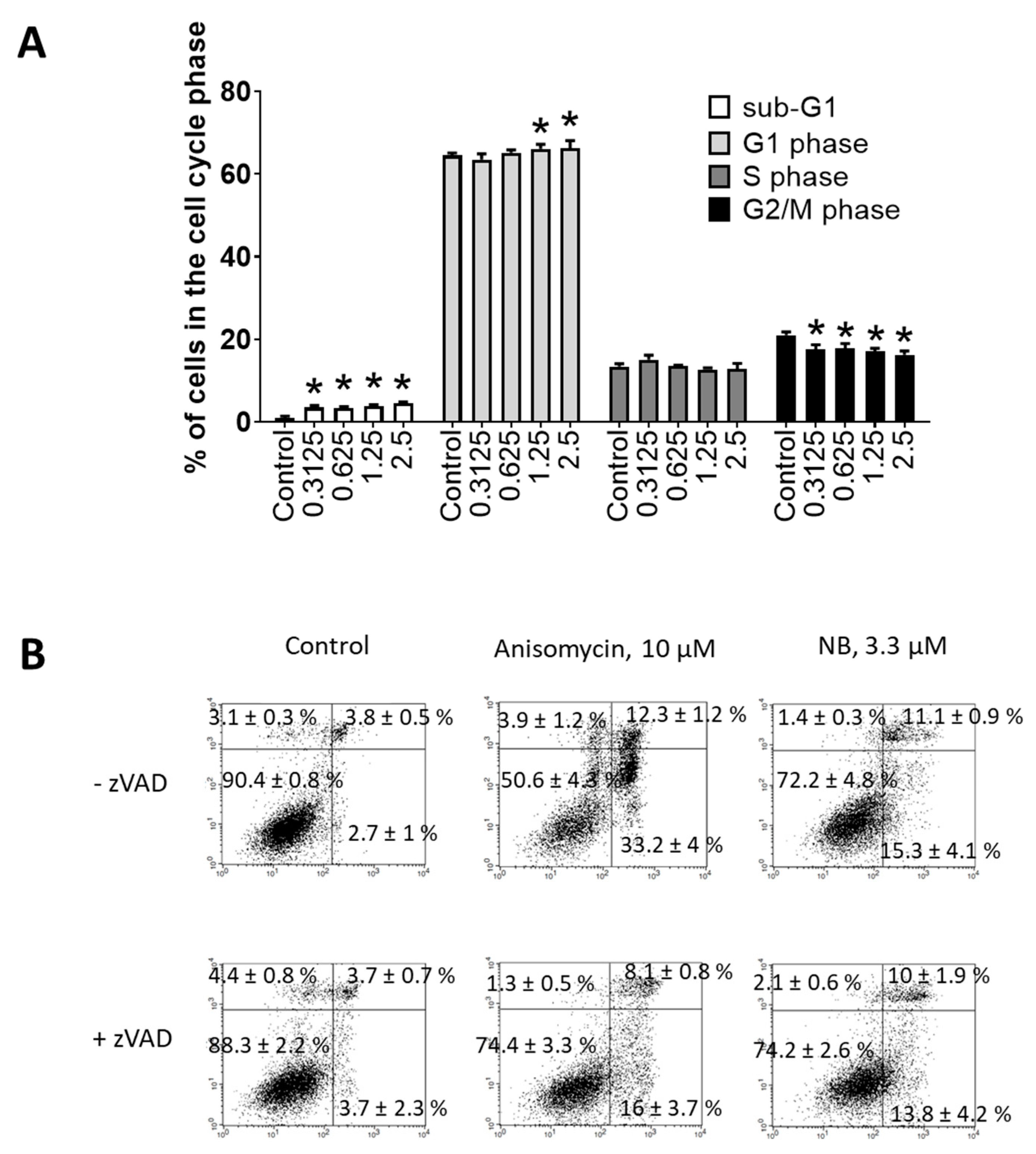
2.3. Effect of JNK1/2 and p38 MAPKs on the Cytotoxicity of NB
2.4. Inhibitory Effect of NB on P-Glycoprotein Activity and Synergism with Docetaxel
3. Materials and Methods
3.1. Isolation of NB
3.2. Reagents and Antibodies
3.3. Cell Lines and Culture Conditions
3.4. MTT Assay
3.5. In Vitro Trypan Blue Exclusion Assay
3.6. Profiling of Serine-/Threonine Kinase Activity
3.7. Western Blotting
3.8. Drug Combination Studies
3.9. Cell Cycle Progression and DNA Fragmentation Analysis
3.10. Annexin-V-FITC/PI Double Staining
3.11. P-Glycoprotein Activity Analysis
3.12. Data and Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, M.; Zhang, X.; Huang, X.; Wang, H.; Anjum, K.; Gu, Q.; Zhu, T.; Zhang, G.; Li, D. Irregularly Bridged Epipolythiodioxopiperazines and Related Analogues: Sources, Structures, and Biological Activities. J. Nat. Prod. 2020, 83, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- Stipanovic, R.D.; Howell, C.R. The structure of gliovirin, a new antibiotic from Gliocladium virens. J. Antibiot. 1982, 35, 1326–1330. [Google Scholar] [CrossRef]
- Miyamoto, C.; Yokose, K.; Furumai, T.; Maruyama, H.B. A new epidithiodiketopiperazine group antibiotic, FA-2097. J. Antibiot. 1982, 35, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Rotinsulu, H.; Narita, R.; Takahashi, R.; Namikoshi, M. Induced production of halogenated epidithiodiketopiperazines by a marine-derived Trichoderma cf. brevicompactum with sodium halides. J. Nat. Prod. 2015, 78, 2319–2321. [Google Scholar] [PubMed]
- Yamazaki, H.; Takahashi, O.; Murakami, K.; Namikoshi, M. Induced production of a new unprecedented epitrithiodiketopiperazine, chlorotrithiobrevamide, by a culture of the marine-derived Trichoderma cf. brevicompactum with dimethyl sulfoxide. Tetrahedron Lett. 2015, 56, 6262–6265. [Google Scholar] [CrossRef]
- Capon, R.J.; Ratnayake, R.; Stewart, M.; Lacey, E.; Tennant, S.; Gill, J.H. Aspergillazines A-E: Novel heterocyclic dipeptides from an Australian strain of Aspergillus unilateralis. Org. Biomol. Chem. 2005, 3, 123–129. [Google Scholar] [CrossRef]
- Damour, H.; Okoye, F.B.C.; Proksch, P.; Hakiki, A.; Mosaddak, M.; Hegazy, M.F.; Debbab, A. Pretrichodermamide A and nafuredin from Trichoderma sp, an endophyte of Cola nitida. J. Mater. Environ. Sci. 2015, 6, 779–783. [Google Scholar]
- Orfali, R.S.; Aly, A.H.; Ebrahim, W.; Abdel-Aziz, M.S.; Müller, W.E.G.; Lin, W.; Daletos, G.; Proksch, P. Pretrichodermamide C and N-methylpretrichodermamide B, two new cytotoxic epidithiodiketopiperazines from hyper saline lake derived Penicillium sp. Phytochem. Lett. 2015, 11, 168–172. [Google Scholar] [CrossRef]
- Yurchenko, A.; Smetanina, O.; Ivanets, E.; Kalinovsky, A.; Khudyakova, Y.; Kirichuk, N.; Popov, R.; Bokemeyer, C.; von Amsberg, G.; Chingizova, E.; et al. Pretrichodermamides D–F from a Marine Algicolous Fungus Penicillium sp. KMM 4672. Mar. Drugs 2016, 14, 122. [Google Scholar] [CrossRef]
- Harwoko, H.; Daletos, G.; Stuhldreier, F.; Lee, J.; Wesselborg, S.; Feldbrügge, M.; Müller, W.E.G.; Kalscheuer, R.; Ancheeva, E.; Proksch, P. Dithiodiketopiperazine derivatives from endophytic fungi Trichoderma harzianum and Epicoccum nigrum. Nat. Prod. Res. 2021, 35, 257–265. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.M.; Meng, L.H.; Jiang, W.L.; Xu, G.M.; Huang, C.G.; Wang, B.G. Bisthiodiketopiperazines and acorane sesquiterpenes produced by the marine-derived fungus Penicillium adametzioides AS-53 on different culture media. J. Nat. Prod. 2015, 78, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Kajula, M.; Ward, J.M.; Turpeinen, A.; Tejesvi, M.V.; Hokkanen, J.; Tolonen, A.; Häkkänen, H.; Picart, P.; Ihalainen, J.; Sahl, H.-G.; et al. Bridged epipolythiodiketopiperazines from Penicillium raciborskii, an endophytic fungus of Rhododendron tomentosum Harmaja. J. Nat. Prod. 2016, 79, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Garo, E.; Starks, C.M.; Jensen, P.R.; Fenical, W.; Lobkovsky, E.; Clardy, J. Trichodermamides A and B, Cytotoxic Modified Dipeptides from the Marine-Derived Fungus Trichoderma virens. J. Nat. Prod. 2003, 66, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Jans, P.E.; Mfuh, A.M.; Arman, H.D.; Shaffer, C.V.; Larionov, O.V.; Mooberry, S.L. Cytotoxicity and Mechanism of Action of the Marine-Derived Fungal Metabolite Trichodermamide B and Synthetic Analogues. J. Nat. Prod. 2017, 80, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Reece, K.M.; Richardson, E.D.; Cook, K.M.; Campbell, T.J.; Pisle, S.T.; Holly, A.J.; Venzon, D.J.; Liewehr, D.J.; Chau, C.H.; Price, D.K.; et al. Epidithiodiketopiperazines (ETPs) exhibit in vitro antiangiogenic and in vivo antitumor activity by disrupting the HIF-1α/p300 complex in a preclinical model of prostate cancer. Mol. Cancer 2014, 13, 91. [Google Scholar] [CrossRef]
- Olsson, C.R.; Payette, J.N.; Cheah, J.H.; Movassaghi, M. Synthesis of Potent Cytotoxic Epidithiodiketopiperazines Designed for Derivatization. J. Org. Chem. 2020, 85, 4648–4662. [Google Scholar] [CrossRef]
- Sampson, N.; Neuwirt, H.; Puhr, M.; Klocker, H.; Eder, I.E. In vitro model systems to study androgen receptor signaling in prostate cancer. Endocr. Relat. Cancer 2013, 20, R49–R64. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Schäfer, G.; Erb, H.H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-mesenchymal transition leads to docetaxel resistance in prostate cancer and is mediated by reduced expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201. [Google Scholar] [CrossRef]
- Nelson, P.S. Targeting the androgen receptor in prostate cancer—A resilient foe. N. Engl. J. Med. 2014, 371, 1067–1069. [Google Scholar] [CrossRef]
- Kupcsik, L. Estimation of cell number based on metabolic activity: The MTT reduction assay. In Mammalian Cell Viability: Methods and Protocols; Stoddart, M.J., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 13–19. [Google Scholar]
- Louis, K.S.; Siegel, A.C. Cell viability analysis using trypan blue: Manual and automated methods. In Mammalian Cell Viability: Methods and Protocols; Stoddart, M.J., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 7–12. [Google Scholar]
- Ghavami, S.; Hashemi, M.; Ande, S.R.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.J.; Kadkhoda, K.; Wiechec, E.; Halayko, A.J.; et al. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [PubMed]
- Li, J.; Zhang, Y.; Da Silva Sil Dos Santos, B.; Wang, F.; Ma, Y.; Perez, C.; Yang, Y.; Peng, J.; Cohen, S.M.; Chou, T.F.; et al. Epidithiodiketopiperazines Inhibit Protein Degradation by Targeting Proteasome Deubiquitinase Rpn11. Cell Chem. Biol. 2018, 25, 1350–1358.e9. [Google Scholar] [CrossRef] [PubMed]
- Waring, P.; Sjaarda, A.; Lin, Q.H. Gliotoxin inactivates alcohol dehydrogenase by either covalent modification or free radical damage mediated by redox cycling. Biochem. Pharmacol. 1995, 49, 1195–1201. [Google Scholar] [CrossRef]
- Amatov, T.; Jahn, U. Gliotoxin: Nature’s way of making the epidithio bridge. Angew. Chem. Int. Ed. Engl. 2014, 53, 3312–3314. [Google Scholar] [CrossRef]
- Kroll, M.; Arenzana-Seisdedos, F.; Bachelerie, F.; Thomas, D.; Friguet, B.; Conconi, M. The secondary fungal metabolite gliotoxin targets proteolytic activities of the proteasome. Chem. Biol. 1999, 6, 689–698. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Struve, N.; Binder, Z.A.; Stead, L.F.; Brend, T.; Bagley, S.J.; Faulkner, C.; Ott, L.; Müller-Goebel, J.; Weik, A.-S.; Hoffer, K.; et al. EGFRvIII upregulates DNA mismatch repair resulting in increased temozolomide sensitivity of MGMT promoter methylated glioblastoma. Oncogene 2020, 39, 3041–3055. [Google Scholar] [CrossRef]
- Xu, R.; Hu, J. The role of JNK in prostate cancer progression and therapeutic strategies. Biomed. Pharmacother. 2020, 121, 109679. [Google Scholar] [CrossRef]
- Rodríguez-Berriguete, G.; Fraile, B.; Martínez-Onsurbe, P.; Olmedilla, G.; Paniagua, R.; Royuela, M. MAP Kinases and Prostate Cancer. J. Signal Transduct. 2012, 2012, 169170. [Google Scholar] [CrossRef]
- Yang, Y.-M.; Bost, F.; Charbono, W.; Dean, N.; McKay, R.; Rhim, J.S.; Depatie, C.; Mercola, D. C-Jun NH2-terminal Kinase Mediates Proliferation and Tumor Growth of Human Prostate Carcinoma. Clin. Cancer Res. 2003, 9, 391. [Google Scholar] [PubMed]
- Hu, J.; Wang, G.; Sun, T. Dissecting the roles of the androgen receptor in prostate cancer from molecular perspectives. Tumour Biol. 2017, 39, 1010428317692259. [Google Scholar] [CrossRef]
- Liu, P.-Y.; Lin, S.-Z.; Sheu, J.J.-C.; Lin, C.-T.; Lin, P.-C.; Chou, Y.-W.; Huang, M.-H.; Chiou, T.-W.; Harn, H.-J. Regulation of androgen receptor expression by Z-isochaihulactone mediated by the JNK signaling pathway and might be related to cytotoxicity in prostate cancer. Prostate 2013, 73, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Kokontis, J.; Lin, Y.; Liao, S.; Lin, A.; Xiang, J. Androgen via p21 inhibits tumor necrosis factor alpha-induced JNK activation and apoptosis. J. Biol. Chem. 2009, 284, 32353–32358. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lin, A. Role of JNK activation in apoptosis: A double-edged sword. Cell Res. 2005, 15, 36–42. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose–Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Kaune, M.; Hauschild, J.; Kriegs, M.; Hoffer, K.; Busenbender, T.; Smirnova, P.A.; Zhidkov, M.E.; Poverennaya, E.V.; Oh-Hohenhorst, S.J.; et al. Efficacy and mechanism of action of marine alkaloid 3,10-dibromofascaplysin in drug-resistant prostate cancer cells. Mar. Drugs 2020, 18, 609. [Google Scholar] [CrossRef]
- O’Neill, A.J.; Prencipe, M.; Dowling, C.; Fan, Y.; Mulrane, L.; Gallagher, W.M.; O’Connor, D.; O’Connor, R.; Devery, A.; Corcoran, C.; et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol. Cancer 2011, 10, 126. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Shubina, L.K.; Makarieva, T.N.; Hauschild, J.; Strewinsky, N.; Guzii, A.G.; Menshov, A.S.; Popov, R.S.; Grebnev, B.B.; Busenbender, T.; et al. New diterpenes from the marine sponge Spongionella sp. overcome drug resistance in prostate cancer by inhibition of P-glycoprotein. Sci. Rep. 2022, 12, 13570. [Google Scholar] [CrossRef]
- Weidner, L.D.; Fung, K.L.; Kannan, P.; Moen, J.K.; Kumar, J.S.; Mulder, J.; Innis, R.B.; Gottesman, M.M.; Hall, M.D. Tariquidar is an inhibitor and not a substrate of human and mouse p-glycoprotein. Drug Metab. Dispos. 2016, 44, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, R.J.; Draper, D.; Chen, C.C.; Robey, R.W.; Figg, W.D.; Piekarz, R.L.; Chen, X.; Gardner, E.R.; Balis, F.M.; Venkatesan, A.M.; et al. A pharmacodynamic study of docetaxel in combination with the p-glycoprotein antagonist tariquidar (XR9576) in patients with lung, ovarian, and cervical cancer. Clin. Cancer Res. 2011, 17, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Staud, F.; Ceckova, M.; Micuda, S.; Pavek, P. Expression and function of p-glycoprotein in normal tissues: Effect on pharmacokinetics. Methods Mol. Biol. 2010, 596, 199–222. [Google Scholar] [PubMed]
- Dyshlovoy, S.A.; Tabakmakher, K.M.; Hauschild, J.; Shchekaleva, R.K.; Otte, K.; Guzii, A.G.; Makarieva, T.N.; Kudryashova, E.K.; Fedorov, S.N.; Shubina, L.K.; et al. Guanidine alkaloids from the marine sponge Monanchora pulchra show cytotoxic properties and prevent EGF-induced neoplastic transformation in vitro. Mar. Drugs 2016, 14, 133. [Google Scholar] [CrossRef]
- Arni, S.; Le, T.H.N.; de Wijn, R.; Garcia-Villegas, R.; Dankers, M.; Weder, W.; Hillinger, S. Ex vivo multiplex profiling of protein tyrosine kinase activities in early stages of human lung adenocarcinoma. Oncotarget 2017, 8, 68599–68613. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Kaune, M.; Kriegs, M.; Hauschild, J.; Busenbender, T.; Shubina, L.K.; Makarieva, T.N.; Hoffer, K.; Bokemeyer, C.; Graefen, M.; et al. Marine alkaloid monanchoxymycalin C: A new specific activator of JNK1/2 kinase with anticancer properties. Sci. Rep. 2020, 10, 13178. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Pelageev, D.N.; Hauschild, J.; Sabutskii, Y.E.; Khmelevskaya, E.A.; Krisp, C.; Kaune, M.; Venz, S.; Borisova, K.L.; Busenbender, T.; et al. Inspired by sea urchins: Warburg effect mediated selectivity of novel synthetic non-glycoside 1,4-naphthoquinone-6S-glucose conjugates in prostate cancer. Mar. Drugs 2020, 18, 251. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 [µM] | Mean IC50, Cancer Cells | Mean IC50, Noncancer Cells | Selectivity Index (SI) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prostate Cancer Cells | Noncancer Cells | ||||||||||||
| PC3 | PC3-DR | DU145 | 22Rv1 | VCaP | LNCaP | PNT2 | RWPE-1 | HEK293 | MRC-9 | ||||
| NB [µM] | 6.03 ± 1.18 | 3.63 ± 1.03 | 12.97 ± 2.69 | 3.02 ± 2.5 | 0.143 ± 0.145 | 12.15 ± 2.97 | 25.13 ± 5.92 | 16.4 ± 1.87 | 9.3 ± 1.57 | 11.12 ± 4.3 | 6.46 | 15.49 | 2.4 |
| Doce-taxel [nM] | 7.49 ± 7.09 | 324.9 ± 21.7 | 2.2 ± 0.6 | 0.6 ± 0.12 | 0.23 ± 0.22 | 5.06 ± 0.58 | >500 | 0.42 ± 0.23 | 11.49 ± 3.22 | >500 | 56.75 | 252.98 | 4.5 |
| Antibodies | Clonality | Source | Cat.-No. | Dilution | Manufacturer |
|---|---|---|---|---|---|
| anti-ERK1/2 | mAb | mouse | #9107 | 1:2000 | Cell Signaling |
| anti-JNK1/2 | mAb | rabbit | #9258 | 1:1000 | Cell Signaling |
| anti-p38 | mAb | rabbit | #9212 | 1:1000 | Cell Signaling |
| anti-phospho-ERK1/2 | mAb | rabbit | #4377 | 1:1000 | Cell Signaling |
| anti-phospho-JNK1/2 | mAb | rabbit | #4668 | 1:1000 | Cell Signaling |
| anti-phospho-p38 | mAb | rabbit | #4511 | 1:1000 | Cell Signaling |
| anti-β-Actin-HRP | pAb | goat | sc-1616 | 1:10,000 | Santa Cruz |
| anti-MDR1 (p-gp) | mAb | rabbit | #13342 | 1:1000 | Cell Signaling |
| anti-α-Tubulin | mAb | mouse | T5168 | 1:5000 | Sigma-Aldrich |
| anti-LC3B-I/II | pAb | rabbit | #2775 | 1:1000 | Cell Signaling |
| anti-cleaved Caspase-3 | mAb | rabbit | #9664 | 1:1000 | Cell Signaling |
| anti-PARP | pAb | rabbit | #9542 | 1:1000 | Cell Signaling |
| anti-Survivin | pAb | rabbit | NB500-201 | 1:1000 | Novus |
| anti-mouse IgG-HRP | sheep | NXA931 | 1:10,000 | GE Healthcare | |
| anti-rabbit IgG-HRP | goat | #7074 | 1:5000 | Cell Signaling |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyshlovoy, S.A.; Busenbender, T.; Hauschild, J.; Girich, E.V.; Kriegs, M.; Hoffer, K.; Graefen, M.; Yurchenko, A.N.; Bokemeyer, C.; von Amsberg, G. Cytotoxic N-Methylpretrichodermamide B Reveals Anticancer Activity and Inhibits P-Glycoprotein in Drug-Resistant Prostate Cancer Cells. Mar. Drugs 2022, 20, 597. https://doi.org/10.3390/md20100597
Dyshlovoy SA, Busenbender T, Hauschild J, Girich EV, Kriegs M, Hoffer K, Graefen M, Yurchenko AN, Bokemeyer C, von Amsberg G. Cytotoxic N-Methylpretrichodermamide B Reveals Anticancer Activity and Inhibits P-Glycoprotein in Drug-Resistant Prostate Cancer Cells. Marine Drugs. 2022; 20(10):597. https://doi.org/10.3390/md20100597
Chicago/Turabian StyleDyshlovoy, Sergey A., Tobias Busenbender, Jessica Hauschild, Elena V. Girich, Malte Kriegs, Konstantin Hoffer, Markus Graefen, Anton N. Yurchenko, Carsten Bokemeyer, and Gunhild von Amsberg. 2022. "Cytotoxic N-Methylpretrichodermamide B Reveals Anticancer Activity and Inhibits P-Glycoprotein in Drug-Resistant Prostate Cancer Cells" Marine Drugs 20, no. 10: 597. https://doi.org/10.3390/md20100597
APA StyleDyshlovoy, S. A., Busenbender, T., Hauschild, J., Girich, E. V., Kriegs, M., Hoffer, K., Graefen, M., Yurchenko, A. N., Bokemeyer, C., & von Amsberg, G. (2022). Cytotoxic N-Methylpretrichodermamide B Reveals Anticancer Activity and Inhibits P-Glycoprotein in Drug-Resistant Prostate Cancer Cells. Marine Drugs, 20(10), 597. https://doi.org/10.3390/md20100597