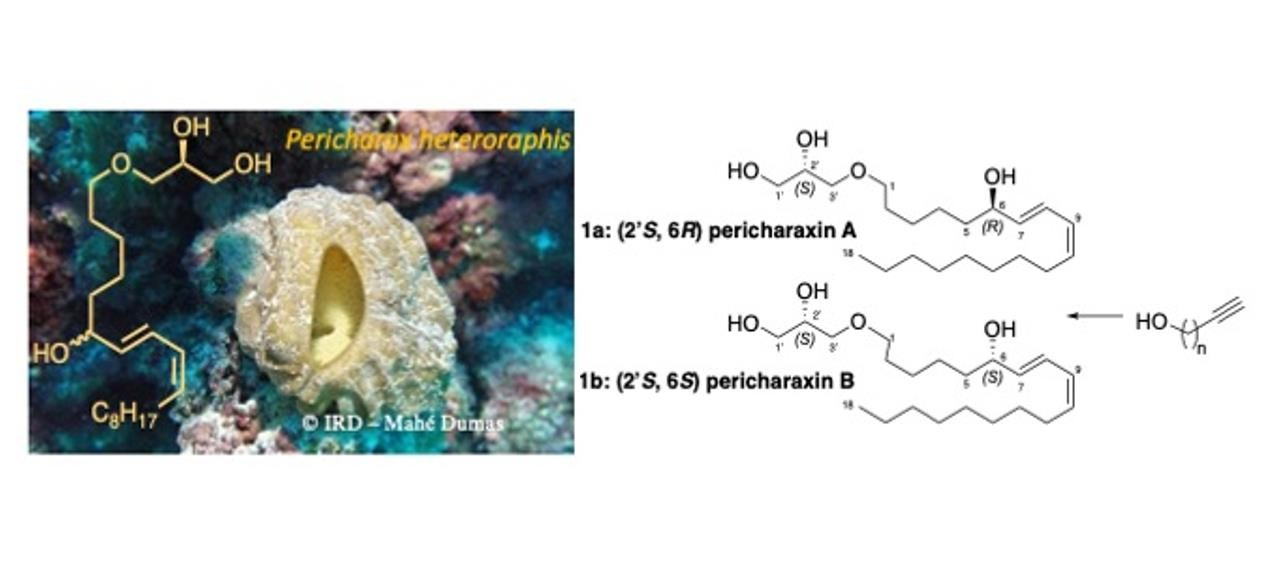
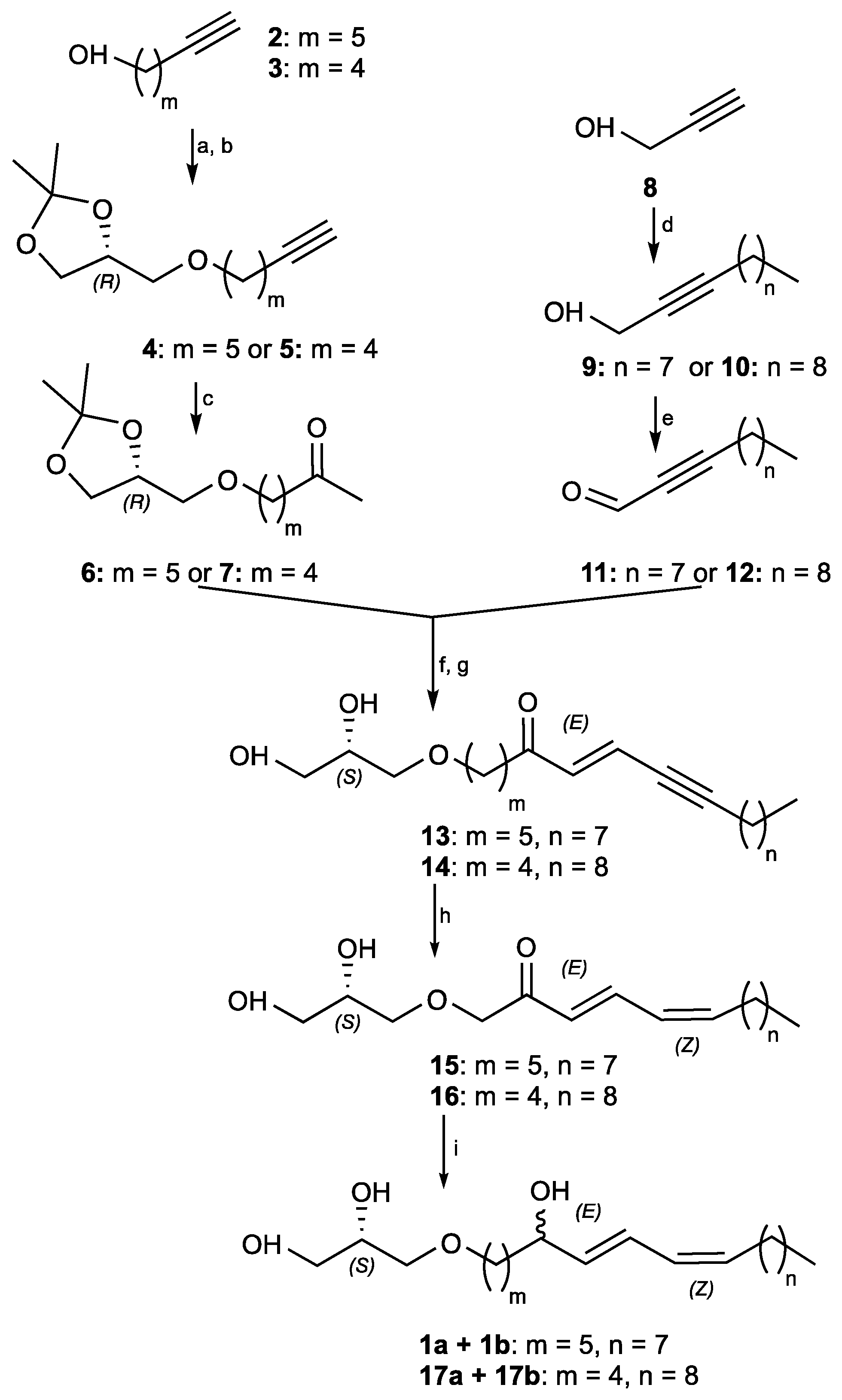
3.5. Synthesis of Pericharaxin (1)
(R)-4-((Hept-6-yn-1-yloxy)methyl)-2,2-dimethyl-1,3-dioxolane (
4): Prepared following the literature procedure described in the literature [
9]: (
R)-4-hydroxymethyl-2,2-dimethyl-1,3-dioxolane (4.3 g, 32.5 mmol) was dissolved in dimethylformamide (100 mL) at 0 °C under an inert atmosphere. Sodium hydride (60% wt, 1.56 g, 35 mmol) was added in several portions. The reaction mixture was allowed to stir 40 min at this temperature. 6-Heptyn-1-ol 2 was mesylated as described in the literature [
11] and the resulting mesylate (6.8 g, 35.7 mmol) in dimethylformamide (20 mL) was added. The reaction mixture was allowed to stir overnight at room temperature. The reaction was quenched by the addition of a saturated aqueous solution of ammonium chloride and extracted 3 times with diethyl ether. The combined organic layers were dried with MgSO
4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (elution by gradient heptane/EtOAc 100:0 to 60:40), to give
4 as a light-yellow oil (4.7 g, 58%). [α]
D20 −13.4 (
c 1.75, CH
2Cl
2); IR (neat) υ
max 3286, 2986, 2936, 2863, 2118, 1456 cm
−1;
1H NMR (500 MHz, CDCl
3)
δ ppm 4.23 (quint,
J = 6.1 Hz, 1H), 4.02 (dd,
J = 8.2, 6.6 Hz, 1H), 3.70 (dd,
J = 8.2, 6.6 Hz, 1H), 3.38–3.51 (m, 4H), 2.03 (dt,
J = 2.5, 6.8 Hz, 2H), 2.16 (td,
J = 7.0, 2.6 Hz, 2H), 1.91 (t,
J = 2.6 Hz, 1H), 1.48–1.60 (m, 4H), 1.41–1.46 (m, 2H), 1.39 (s, 3H), 1.33 (s, 3H);
13C NMR (500 MHz, CDCl
3)
δ ppm 109.4, 84.4, 74.8, 71.9, 71.6, 68.3, 67.0, 29.1, 28.3, 26.8, 25.5, 25.3, 18.4; HRESIMS
m/
z 227.1647 [M + H]
+ (calcd for C
12H
23O
3 227.1647).
®-4-((Hex-5-yn-1-yloxy)methyl)-2,2-dimethyl-1,3-dioxolane (
5): Prepared following the literature procedure described in the literature [
9]: (
R)-4-hydroxymethyl-2,2-dimethyl-1,3-dioxolane (1.7 g, 12.9 mmol) was dissolved in dimethylformamide (75 mL) at 0 °C under inert atmosphere. Sodium hydride (60% wt, 620 mg, 15.5 mmol) was added in several portions. The reaction mixture was allowed to stir 40 min at this temperature. 6-Hexyn-1-ol 2 was mesylated as described in the literature [
11] and the resulting mesylate (2.5 g, 14.2 mmol) in dimethylformamide (20 mL) was added. Reaction mixture was allowed to stir overnight at room temperature. The reaction was quenched by the addition of a saturated aqueous solution of ammonium chloride and extracted 3 times with diethyl ether. The combined organic layers were dried with MgSO
4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (elution by gradient heptane/EtOAc 100:0 to 60:40), to give 5 as a clear oil (1.62 g, 54%).
1H NMR (500 MHz, CDCl
3)
δ ppm 4.24 (quint,
J = 6.1 Hz, 1H), 4.04 (dd,
J = 8.2, 6.5 Hz, 1H), 3.72 (dd,
J = 8.3, 6.5 Hz, 1H), 3.39–3.54 (m, 4H), 2.03 (dt,
J = 2.5, 6.8 Hz, 2H), 1.93 (t,
J = 2.5 Hz, 1H), 1.69 (2t,
J = 6.6, 6.3 Hz,2H), 1.60 (t,
J = 6.8 Hz, 1H), 1.57 (td,
J = 2.5, 7.3 Hz, 2H), 1.41 (s, 3H), 1.35 (s, 3H).
(R)-7-((2,2-Dimethyl-1,3-dioxolan-4-yl)methoxy)heptan-2-one (6): To a stirred solution of (R)-4-((hept-6-yn-1-yloxy)methyl)-2,2-dimethyl-1,3-dioxolane (4) (4.7 g, 20.8 mmol) in tetrahydrofurane (100 mL, pH 7 buffer 9:1) at 0 °C were added pyridinium p-toluenesulfonate (8.7 g, 35 mmol) and mercuric acetate (734 mg, 2.3 mmol). The reaction mixture was warmed to 50 °C and stirred 5 h. The reaction was quenched by the addition of a saturated solution of sodium bicarbonate and extracted 3 times with ethyl acetate. The combined organic extracts were dried with MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (elution by gradient heptane/EtOAc 100:0 to 50:50), to give 6 as a light-yellow oil (2.37 g, 60%). −8.5 (c 3.37, CH2Cl2); IR (neat) υmax 2987, 2935, 2864, 1714, 1455 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 4.21 (quint, J = 6.0 Hz, 1H), 4.01 (dd, J = 8.2, 6.6 Hz, 1H), 3.67 (dd, J = 8.2, 6.6 Hz, 1H), 3.35–3.48 (m, 4H), 2.39 (t, J = 7.4 Hz, 2H), 2.08 (s, 3H), 1.54 (m,2H), 1.37 (s, 3H), 1.32 (s, 3H); 13C NMR (500 MHz, CDCl3) δ ppm 208.9, 109.4, 74.8, 71.9, 71.5, 66.9, 43.6, 29.9, 29.4, 26.8, 25.7, 25.4, 23.6; HRESIMS m/z 245.1748 [M + H]+ (calcd for C13H25O4 245.1753).
(R)-6-((2,2-Dimethyl-1,3-dioxolan-4-yl)methoxy)hexan-2-one (7): To a stirred solution of (R)-4-((hex-5-yn-1-yloxy)methyl)-2,2-dimethyl-1,3-dioxolane (5) (4.9 g, 23 mmol) in tetrahydrofurane (100 mL, pH 7 buffer 9:1) at 0 °C were added pyridinium p-toluenesulfonate (8.7 g, 35 mmol) and mercuric acetate (734 mg, 2.3 mmol). The reaction mixture was warmed to 50 °C and stirred 5 h. The reaction was quenched by the addition of a saturated solution of sodium bicarbonate, and extracted 3 times with ethyl acetate. The combined organic extracts were dried with MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (elution by gradient heptane/EtOAc 100:0 to 50:50), to give 7 as a yellow oil (2.3 g, 43%). [α]D20 −17.9 (c 0.8, CH2Cl2); IR (neat) υmax 2986, 2926, 28,577, 1714, 1456 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 4.25 (quint, J = 6.0 Hz, 1H), 4.04 (dd, J = 8.2, 6.6 Hz, 1H), 3.72 (dd, J = 8.2, 6.6 Hz, 1H), 3.38–3.54 (m, 4H), 2.45 (t, J = 7.0 Hz, 2H), 2.13 (s, 3H), 1.54–1.70 (m, 4H), 1.41 (s, 3H), 1.35 (s, 3H); 13C NMR (500 MHz, CDCl3) δ ppm 208.1, 108.9, 74.5, 71.6, 71.0, 66.5, 43.0, 29.5, 28.7, 26.5, 25.1, 20.2; HRESIMS m/z 231.1528 [M + H]+ (calcd for C12H23O4 231.1596).
Undec-2-yn-1-ol (
9): Prepared according to the literature from propargyl alcohol (5.5 g, 82%) [
12].
Dodec-2-yn-1-ol (
10): Prepared according to the literature from propargyl alcohol (4.1 g, 56%) [
12].
Undec-2-ynal (11): To a cooled solution of undec-2-yn-1-ol (9) (4.9 g, 29.2 mmol) in dry dichloromethane (100 mL) was added Dess–Martin periodinane (11.8 g, 28 mmol) at 0 °C under inert atmosphere. The mixture was stirred for 30 min at the same temperature and filtered through a pad of celite using ethyl acetate as eluent. The filtered solution was concentrated and purified by flash chromatography (elution by a gradient heptane/EtOAc 100:0 to 75:25) to give 11 (4.9 g, quant.) as a light-yellow oil. IR (neat) υmax 2925, 2855, 2200, 1685 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 9.17 (s, 1H), 2.40 (t, J = 7.0, Hz, 2H), 1.59 (tt, J = 7.2, 7.0 Hz, 2H), 1.40 (m, 2H), 1.28 (m, 8H), 0.88 (t, J = 6.6 Hz, 3H); 13C NMR (500 MHz, CDCl3) δ ppm 177.5, 99.7, 81.8, 31.9, 29.2, 29.1, 28.9, 27.7, 22.7, 19.2, 14.2; HRESIMS m/z 167.1427 [M + H]+ (calcd for C11H19O 167.1436).
Dodec-2-ynal (12): To a cooled solution of dodec-2-yn-1-ol (10) (3.5 g, 19 mmol) in dry dichloromethane (100 mL) was added Dess–Martin periodinane (7.7 g, 18 mmol) at 0 °C under an inert atmosphere. The mixture was stirred for 30 min at the same temperature and filtered through a pad of celite using ethyl acetate as eluent. The filtered solution was concentrated and purified by flash chromatography (elution by a gradient heptane/EtOAc 100:0 to 75:25) to give 12 (2.95 g, 86%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ ppm 9.18 (s, 1H), 2.41 (t, J = 7.2, Hz, 2H), 1.60 (tt, J = 7.3, 7.2 Hz, 2H), 1.41 (m, 2H), 1.28 (m, 12H), 0.88 (t, J = 6.6 Hz, 3H); 13C NMR (500 MHz, CDCl3) δ ppm 177.3, 99.5, 81.9, 32.0, 29.5, 29.4, 29.1, 28.9, 27.7, 22.7, 19.2, 14.2.
(S,E)-1-(2,3-dihydroxypropoxy)octadec-7-en-9-yn-6-one (13): LDA was prepared in situ: diisopropylamine (855 μL, 6.1 mmol) was dried on MgSO4 under an inert atmosphere and was dissolved in tetrahydrofuran (3 mL) at −78 °C. n-Butyllithium (5.1 mL, 6.4 mmol) was added turning the reaction mixture light yellow. Freshly prepared lithium diisopropylamide was dissolved in tetrahydrofuran (10 mL) at −78 °C and (R)-7-((2,2-dimethyl-1,3-dioxolan-4-yl)methoxy)heptan-2-one (6) (1.5 g, 6.1 mmol) was added dropwise. The reaction mixture was allowed to stir 5 min. Undec-2-ynal (11) (1.5 g, 9.2 mmol) was then added dropwise and the reaction mixture was allowed to warm to room temperature and to stir 25 min. The reaction was quenched by the addition of a saturated aqueous solution of ammonium chloride and extracted 3 times with diethyl ether. The combined organic layers were dried with MgSO4, filtered, and concentrated under reduced pressure to provide 3.8 g of crude product. The obtained acetonide was not purified and was used directly in the next step.
To the crude acetonide (2.5 g, 6.1 mmol) in THF/H2O 5:1 (60 mL) was added hydrochloric acid (2.9 mL, 30.5 mmol, 37% wt). The reaction mixture was warmed to 65 °C and allowed to stir for 12 h. The reaction was quenched by the addition of a saturated aqueous solution of sodium bicarbonate and extracted 3 times with dichloromethane. The combined organic extracts were dried with MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (elution by gradient heptane/EtOAc 80:20 to 20:80), to give 13 as a white amorphous solid (1 g, 47% over two steps). + 2.1 (c 0.75, CH2Cl2); IR (neat) υmax 3353, 2925, 2855, 2211, 1685, 1460 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 6.65 (dt, J = 16.1, 2.3 Hz, 1H), 6.44 (d, J = 16.1 Hz, 1H), 3.86 (m, 1H), 3.72 (dd, J = 11.3, 3.9 Hz, 1H), 3.65 (dd, J = 11.1, 5.2 Hz, 1H), 3.46–3.56 (m, 4H), 2.54 (t, J = 7.2 Hz, 2H), 2.38 (td, J = 7.0, 2.2 Hz, 2H), 1.60 (m, 6H), 1.34–1.43 (m, 4H), 1.28 (s, 8H), 0.89 (t, J = 7.0 Hz, 3H); 13C NMR (500 MHz, CDCl3) δ ppm 199.5, 136.6, 124.3, 102.2, 78.6, 72.6, 71.5, 70.6, 64.3, 40.8, 31.9, 29.4, 29.3, 29.19, 29.0, 28.5, 25.9, 23.8, 22.8, 20.0, 14.2; HRESIMS m/z 353.2633 [M + H]+ (calcd for C21H37O4 353.2692).
1-(((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)methoxy)-7-hydroxyoctadec-8-yn-5-one (14′): LDA was prepared in situ: diisopropylamine (175 μL, 1.25 mmol) was dried on MgSO4 under an inert atmosphere and was dissolved in tetrahydrofurane (3 mL) at −78 °C. n-Butyllithium (1.2 mL, 1.31 mmol) was added turning the reaction mixture turned light yellow. Freshly prepared Lithium diisopropylamide was dissolved in tetrahydrofurane (5 mL) at −78 °C and (R)-6-((2,2-dimethyl-1,3-dioxolan-4-yl)methoxy)hexan-2-one (7) (230 mg, 1 mmol) was added dropwise. The reaction mixture was allowed to stir for 5 min. Dodec-2-ynal (12) (227 mg, 1.25 mmol) was then added dropwise and the reaction mixture was allowed to warm to room temperature and to stir for 25 min. The reaction was quenched by the addition of a saturated aqueous solution of ammonium chloride and extracted 3 times with diethyl ether. The combined organic layers were dried with MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (elution by gradient heptane/EtOAc 100:0 to 50:50), to give 14’ as a white amorphous solid (360 mg, 70%). +1.2 (c 0.75, CH2Cl2); IR (neat) υmax 3380, 2924, 2855, 2204, 1706, 1657, 1592, 1458 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 4.77 (m, 1H, H7), 4.25 (quint, J = 6.0 Hz, 1H, H2’), 4.04 (dd, J = 8.2, 6.5 Hz, 1H, H3’), 3.72 (dd, J = 8.2, 6.5 Hz, 1H, H3’), 3.39–3.52 (m, 4H, H1’ & H1), 2.85 (dd, J = 17.0, 7.9 Hz, 1H, H6), 2.75 (dd, J = 17.0, 3.7 Hz, 1H, H6), 2.45 (t, J = 7.0 Hz, 2H), 2.18 (m, 2H), 1.62–1.68 (m, 2H), 1.56–1.61 (m, 2H), 1.42 (s, 3H), 1.36 (s, 3H), 1.26 (s, 14H), 0.88 (t, J = 6.6 Hz, 3H); 13C NMR (500 MHz, CDCl3) δ ppm 210.1, 109.5, 86.0, 79.7, 74.9, 72.0, 71.4, 67.0, 58.8, 49.8, 43.3, 32.0, 29.6, 29.4, 29.2, 29.0, 28.9, 28.7, 26.9, 25.5, 22.8, 20.3, 18.8, 14.2; HRESIMS m/z 411.3133 [M + H]+ (calcd for C24H42O5 411.3110).
(S,E)-1-(2,3-Dihydroxypropoxy)11ctadic-6-en-8-yn-5-one (14): To acetonide 14’ (360 mg, 0.9 mmol) in THF/H2O 5:1 (15 mL) was added hydrochloric acid (430 μL, 4.4 mmol, 37% wt). The reaction mixture was heated to 65 °C and allowed to stir for 12 h. The reaction was quenched by the addition of a saturated aqueous solution of sodium bicarbonate and extracted 3 times with dichloromethane. The combined organic extracts were dried with MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (elution by gradient heptane/EtOAc 80:20 to 0:100), to give 14 as a white amorphous solid (125 mg, 40%). + 0.71 (c 0.70, CH2Cl2); IR (neat) υmax 3348, 2922, 2849, 2235, 1740, 1642, 1598 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 6.65 (dt, J = 16.1, 2.3 Hz, 1H), 6.44 (d, J = 16.1 Hz, 1H), 3.86 (m, 1H), 3.72 (dd, J = 11.1, 3.9 Hz, 1H), 3.65 (dd, J = 11.1, 5.2 Hz, 1H), 3.49 (m, 4H), 2.56 (t, J = 7.0 Hz, 2H), 2.38 (td, J = 7.1, 2.3 Hz, 2H), 1.70 (quint, J = 7.0 Hz, 2H), 1.53–1.63 (m, 4H), 1.34–1.43 (m,2H), 1.26 (s, 10H), 0.88 (t, J = 7.0 Hz, 3H); 13C NMR (500 MHz, CDCl3) δ ppm 199.3, 136.5, 124.4, 102.2, 78.6, 72.6, 71.4, 70.6, 64.3, 40.5, 32.0, 29.6, 29.4, 29.2, 29.1, 29.0, 28.5, 22.8, 20.8, 20.0, 14.2; HRESIMS m/z 353.2661 [M + H]+ (calcd for C21H37O4 353.2692).
(2S)-3-(((6E,8Z)-5-Hydroxyoctadeca-6,8-dien-1-yl)oxy)propane-1,2-diol (17): To (S,E)-1-(2,3-dihydroxypropoxy)octadec-6-en-8-yn-5-one (14) (70 mg, 0.2 mmol) dissolved in heptane/EtOAc 80:20 (5 mL) at 0 °C was added quinoline (129 mg, 1 mmol) and Lindlar catalyst (42 mg, 0.02 mmol, 5% wt). The reaction mixture was allowed to stir under an H2 atmosphere for 1.5 h. The reaction mixture was filtered on celite, eluted with ethyl acetate, and concentrated. The crude product was dissolved in methanol (5 mL) at 0 °C and sodium borohydride (11 mg, 0.3 mmol) was added. The reaction mixture was allowed to stir until complete consumption of the starting material (30 min). The reaction was quenched by the addition of water, the excess methanol was evaporated, and the aqueous layer was extracted 3 times with ethyl acetate. The combined organic extracts were dried with MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by HPLC (preparative C18 Sunfire column, elution by gradient H2O/CH3CN 80:20 to 0:100), to give 17 as a colorless oil (6 mg, 8% over two steps). [α]D20 + 4.0 (c 0.65, CH2Cl2); IR (neat) υmax 3351, 2925, 2853, 1653, 1459 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 6.50 (dd, J = 15.2, 11.2 Hz, 1H), 5.97 (t, J = 10.9 Hz, 1H), 5.66 (dd, J = 15.2, 6.7 Hz, 1H), 5.46 (dt, J = 10.9, 7.6 Hz, 1H), 4.18 (dt, J = 6.7, 6.7 Hz, 1H), 3.87 (m, 1H), 3.72 (dd, J = 11.4, 3.9 Hz, 1H), 3.65 (dd, J = 11.4, 5.1 Hz, 1H), 3.45–3.56 (m, 4H), 2.18 (q, J = 7.2 Hz, 2H), 1.54–1.66 (m, 2H), 1.44–1.53 (m, 2H), 1.35–1.42 (m, 2H), 1.27 (s, 14H), 0.89 (t, J = 6.6 Hz, 3H); 13C NMR (500 MHz, CDCl3) δ ppm 135.6, 133.4, 127.7, 126.2, 72.8, 72.6, 71.6, 70.6, 64.3, 37.0, 32.0, 29.8, 29.7, 29.6, 29.5, 29.4, 29.4, 27.9, 22.8, 22.1, 14.2.
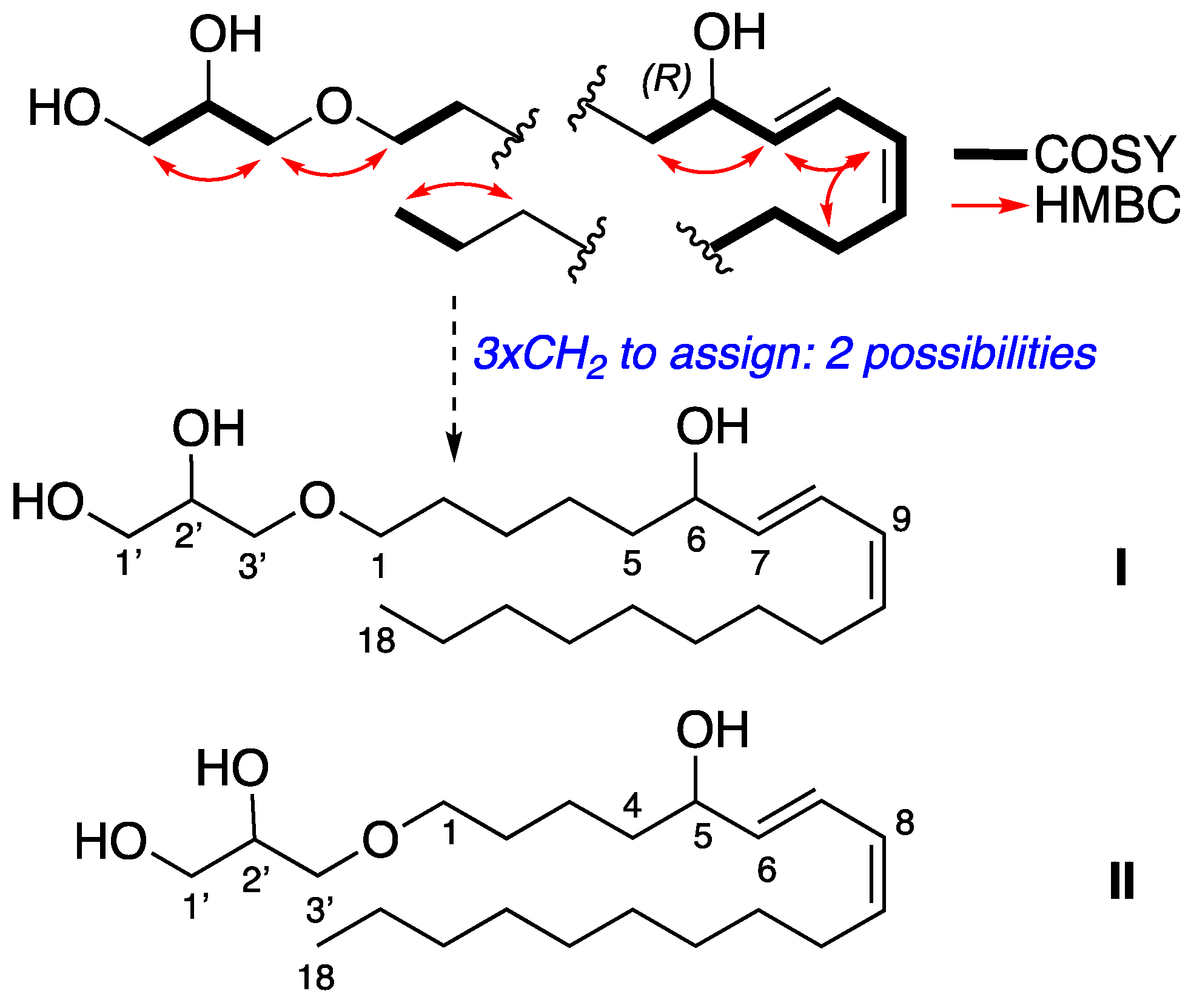
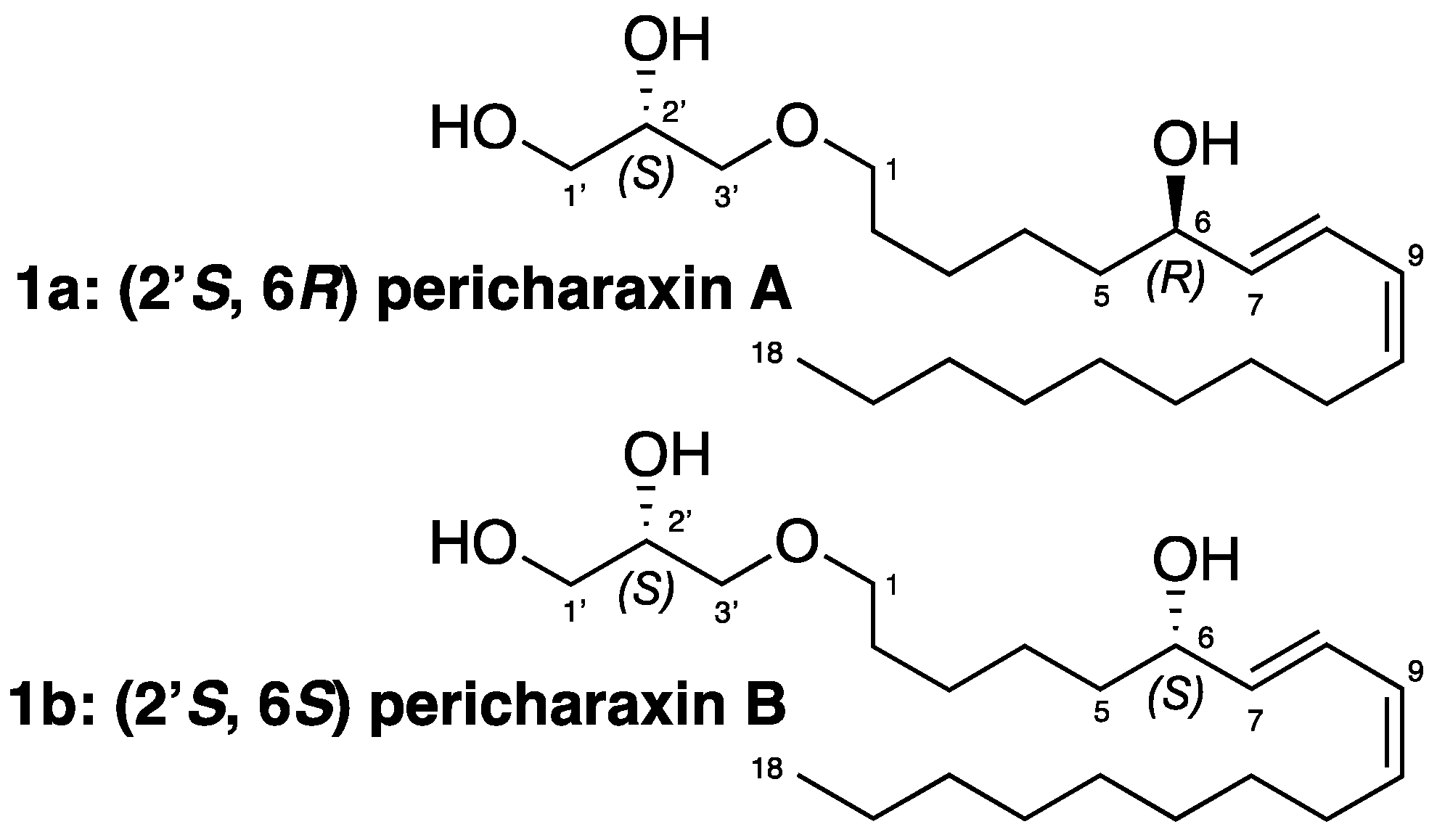
Synthetic epimers Pericharaxins A (1a) and B (1b): To (S,E)-1-(2,3-dihydroxypropoxy)octadec-7-en-9-yn-6-one (13) (300 mg, 0.85 mmol) dissolved in heptane/EtOAc 8:2 (15 mL) at 0 °C was added quinoline (504 μL, 4.3 mmol) and Lindlar catalyst (179 mg, 0.09 mmol, 5% wt). The reaction mixture was allowed to stir under an H2 atmosphere for 1.5 h. The reaction mixture was filtered on celite, eluted with ethyl acetate, and concentrated. The crude product was dissolved in methanol (15 mL) at 0 °C and sodium borohydride (48.5 mg, 1.28 mmol) was added. Reaction mixture was allowed to stir until complete consumption of the starting material (30 min). The reaction was quenched by the addition of water, the excess methanol was evaporated, and the aqueous layer was extracted 3 times with ethyl acetate. The combined organic extracts were dried with MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by HPLC (preparative C18 Sunfire column, elution by gradient H2O/CH3CN 80:20 to 0:100), to give 1 as a yellow oil (32 mg, 10% over two steps). +1.8 (c 0.62, CH2Cl2); IR (neat) υmax 3360, 2950, 2855, 1460 cm−1; 1H NMR (500 MHz, CDCl3) δ ppm 6.49 (dd, J = 15.4, 11.0 Hz, 1H, H-8), 5.97 (t, J = 11.0 Hz, 1H, H-9), 5.66 (dd, J = 15.4, 6.7 Hz, 1H, H-7), 5.46 (dt, J = 11.0, 7.6 Hz, 1H, H-10), 4.16 (dt, J = 6.7, 6.4 Hz, 1H, H-6), 3.86 (m, 1H, H-2’), 3.72 (dd, J = 11.3, 3.8 Hz, 1H, H-3’a), 3.64 (dd, J = 11.3, 5.1 Hz, 1H, H-3’b), 3.44–3.55 (m, 4H, H-1’ and H-1), 2.18 (q, J = 7.2 Hz, 2H, H-11), 1.60 (m, 2H, H-2), 1.48–1.57 (m, 2H, H-5), 1.38–1.47 (m, 2H, H-12), 1.34–1.41 (m, 4H, H-3 and H-4), 1.24–1.32 (m, 10H, H-13 to H-17), 0.89 (t, J = 7.0 Hz, 3H, H-18); 13C NMR (500 MHz, CDCl3) δ ppm 135.7 (C-7), 133.3 (C-10), 127.7 (C-9), 126.1 (C-8), 72.9 (C-6), 72.6 (C-1’), 71.8 (C-1), 70.6 (C-2’), 64.4 (C-3’), 37.3 (C-5), 32.0 (C-16), 29.8 (C-13), 29.6 (C-14), 29.5 (C-2), 29.4 (C-15, C-12), 27.9 (C-11), 26.2 (C-4), 25.3 (C-3), 22.8 (C-17), 14.2 (C-18); HRESIMS m/z 379.2827 [M + Na]+ (calcd for C21H40O4Na 379.2824).
The two diastereoisomers were separated by chiral chromatography on an amylose tris(3-chlorophenylcarbamate) immobilized on 5 μm silica column (analytical 4.6 × 250 mm, 1 mL/min and preparative 10 × 250 mm, 4.2 mL/min; isocratic heptane/isopropanol 90:10) to give Pricharaxin A (1a) (RT 16.9 min) and B (1b) (RT 19.4 min).
2’(S)-6(R)-Pericharaxin A (1a): + 5.4 (c 0.95, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ ppm 6.50 (dd, J = 15.1, 11.0 Hz, 1H, H-8), 5.97 (t, J = 10.9 Hz, 1H, H-9), 5.66 (dd, J = 15.2, 6.9 Hz, 1H, H-7), 5.46 (dt, J = 10.9, 7.6 Hz, 1H, H-10), 4.17 (dt, J = 6.6, 6.6 Hz, 1H, H-6), 3.86 (m, 1H, H-2’), 3.72 (dd, J = 11.4, 3.9 Hz, 1H, H-3’a), 3.65 (dd, J = 11.4, 5.1 Hz, 1H, H-3’b), 3.54 (dd, J = 9.5, 3.7 Hz, 1H, H-1’a), 3.50 (dd, J = 9.5, 6.1 Hz, 1H, H-1’b), 3.47 (t, J = 6.2 Hz, 2H, H-1), 2.18 (q, J = 7.2 Hz, 2H, H-11), 1.58–1.63 (m, 2H, H-2), 1.49–1.58 (m, 2H, H-5), 1.35–1.42 (m, 6H, H3, H-4 and H-12), 1.23–1.33 (m, 10H, H-13 to H-17), 0.89 (t, J = 7.0 Hz, 3H, H-18); 13C NMR (500 MHz, CDCl3) δ ppm 135.8 (C-7), 133.4 (C-10), 127.7 (C-9), 126.1 (C-8), 72.9 (C-6), 72.7 (C-1’), 71.8 (C-1), 70.6 (C-2’), 64.4 (C-3’), 37.3 (C-5), 32.0 (C-16), 29.8 (C-13), 29.6 (C-2, C-14), 29.4 (C-12, C-15), 27.9 (C-11), 26.2 (C-4), 25.3 (C-3), 22.8 (C-17), 14.2 (C-18).
2’(S)-6(S)-Pericharaxin B (1b): [α]D20 −0.8 (c 0.90, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ ppm 6.50 (dd, J = 15.1, 11.0 Hz, 1H, H-8), 5.97 (t, J = 10.9 Hz, 1H, H-9), 5.66 (dd, J = 15.2, 6.9 Hz, 1H, H-7), 5.46 (dt, J = 10.9, 7.6 Hz, 1H, H-10), 4.17 (dt, J = 6.6, 6.6 Hz, 1H, H-6), 3.86 (m, 1H, H-2’), 3.72 (dd, J = 11.4, 3.9 Hz, 1H, H-3’a), 3.65 (dd, J = 11.4, 5.1 Hz, 1H, H-3’b), 3.54 (dd, J = 9.5, 3.7 Hz, 1H, H-1’a), 3.50 (dd, J = 9.5, 6.1 Hz, 1H, H-1’b), 3.47 (t, J = 6.2 Hz, 2H, H-1), 2.18 (q, J = 7.2 Hz, 2H, H-11), 1.58–1.63 (m, 2H, H-2), 1.49–1.58 (m, 2H, H-5), 1.35–1.42 (m, 6H, H3, H-4 and H-12), 1.23–1.33 (m, 10H, H-13 to H-17), 0.89 (t, J = 7.0 Hz, 3H, H-18); 13C NMR (500 MHz, CDCl3) δ ppm 135.8 (C-7), 133.4 (C-10), 127.7 (C-9), 126.1 (C-8), 72.9 (C-6), 72.7 (C-1’), 71.8 (C-1), 70.6 (C-2’), 64.4 (C-3’), 37.3 (C-5), 32.0 (C-16), 29.8 (C-13), 29.6 (C-2, C-14), 29.4 (C-12, C-15), 27.9 (C-11), 26.2 (C-4), 25.3 (C-3), 22.8 (C-17), 14.2 (C-18).
Mosher determination of the C-6 configuration for epimer (1b) [
15,
16]: To a stirred solution of compound
1b (1 mg, 0.003 mmol) in anhydrous pyridine (80 μL) at room temperature, DMAP (1 crystal) was added under an inert atmosphere. After 5 min,
R-(-)-MTPA-Cl (4.3 mg, 0.017 mmol, 6 equiv.) was added. The reaction progress was monitored by TLC on silica gel (100% EtOAc). After 12 h, Pericharaxin (
1b) was totally consumed. The pyridine solvent was evaporated, the crude product was dissolved in CH
2Cl
2, washed with water, and concentrated, to give the ®-MTPA-triesterified product (
18).
1H NMR (500 MHz, CDCl
3): See
Table S1.
To a stirred solution of Pericharaxin B (
1b) (1 mg, 0.003 mmol) in anhydrous pyridine (80 μL) at room temperature, DMAP (1 crystal) was added under an inert atmosphere. After 5 min,
S-(-)-MTPA-Cl (4.3 mg, 0.017 mmol, 6 equiv.) was added. The reaction progress was monitored by TLC on silica gel (100% EtOAc). After 12 h, compound
1b was totally consumed. The pyridine solvent was evaporated, the crude product was dissolved in dichloromethane, washed with water, and concentrated, to give the (
S)-MTPA-triesterified product
19.
1H NMR (500 MHz, CDCl
3):See
Table S1.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}