
Optimization and Validation of a High Throughput UHPLC-MS/MS Method for Determination of the EU Regulated Lipophilic Marine Toxins and Occurrence in Fresh and Processed Shellfish



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Working Standard Solutions
2.2. LC-MS/MS Analysis
2.3. Sample Preparation
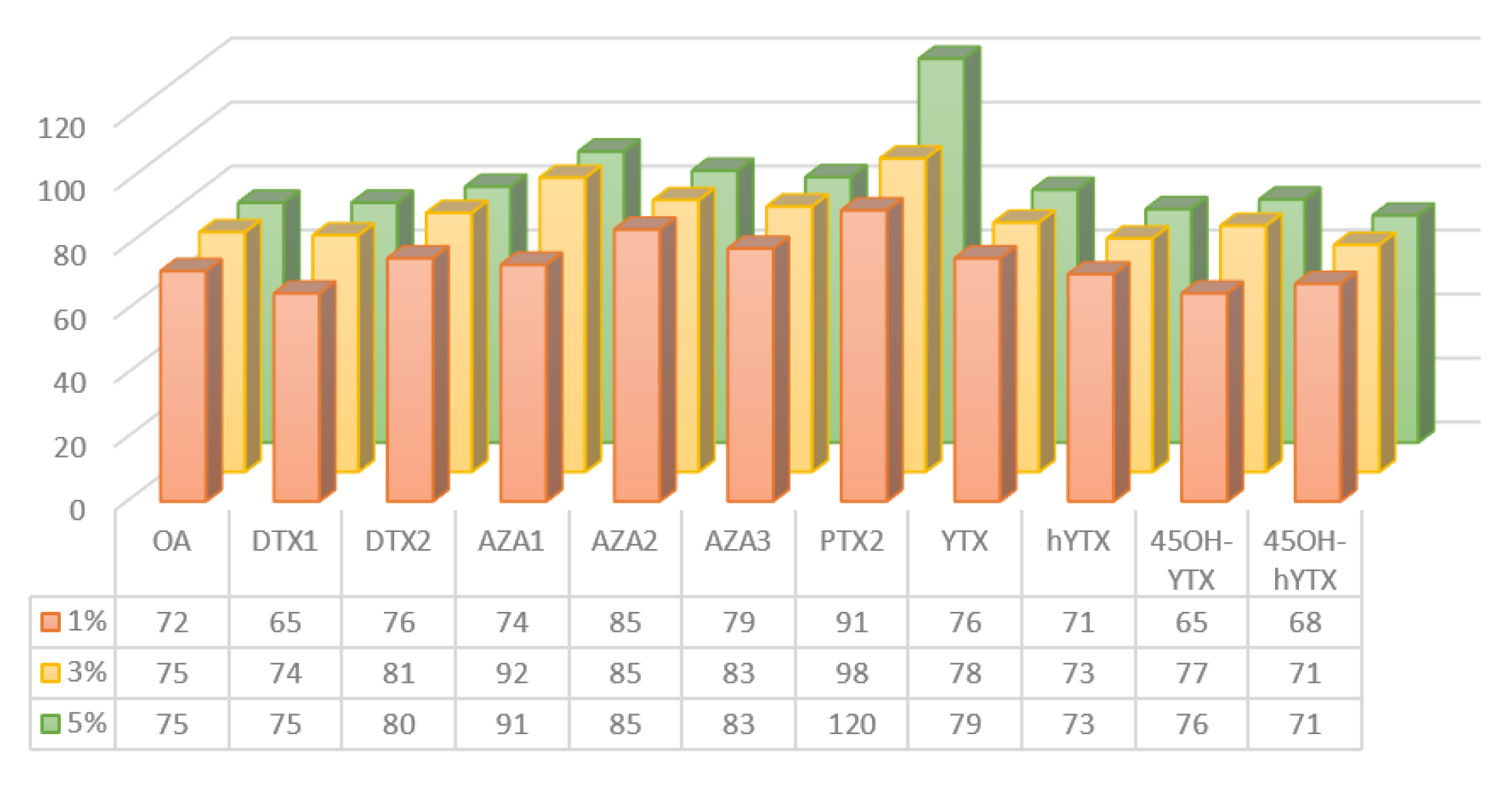
SPE Clean-Up
2.4. Validation Study
2.5. Interlaboratory Comparison: Proficiency Test Round
2.6. Software and Statistical Analysis
2.7. Sample Collection
3. Results and Discussion
3.1. Sample Preparation Optimization
3.2. Chromatographic Separation and Gradient Optimization
3.3. MS Parameters Optimization
3.3.1. Acquisition Mode
3.3.2. SRM Method and Vaporization Temperature Optimization
3.4. Method Validation
3.5. Application to Naturally Contaminated Samples
3.6. Comparison with Other Methods
3.7. Interlaboratory Comparison: Proficiency Test Round
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Food Safety Authority (EFSA). Marine Biotoxins in Shellfish—Okadaic Acid and Analogues—Scientific Opinion of the Panel on Contaminants in the Food Chain. EFSA J. 2008, 589, 23. [Google Scholar] [CrossRef]
- Gerssen, A.; Pol-Hofstad, I.E.; Poelman, M.; Mulder, P.P.; Van den Top, H.J.; De Boer, J. Marine Toxins: Chemistry, Toxicity, Occurrence and Detection, with Special Reference to the Dutch Situation. Toxins 2010, 2, 878–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Food Safety Authority (EFSA). Marine Biotoxins in Shellfish—Summary on Regulated Marine Biotoxins. EFSA J. 2009, 7, 1–23. [Google Scholar] [CrossRef]
- Morabito, S.; Silvestro, S.; Faggio, C. How the Marine Biotoxins Affect Human Health. Nat. Prod. Res. 2018, 32, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Gerssen, A.; Klijnstra, M.D. The Determination of marine biotoxins in seafood. In Analysis of Food Toxins and Toxicants; Wong, Y., Lewis, R.J., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 319–362. ISBN 978-1-118-99268-5. [Google Scholar]
- FAO—Food and Agriculture Organization of the United Nations; WHO—World Health Organization. Technical Paper on Toxicity Equivalency Factors for Marine Biotoxins Associated with Bivalve Molluscs; FAO/WHO: Rome, Italy, 2016; ISBN 978-92-5-109345-0. [Google Scholar]
- Blanco, J. Accumulation of Dinophysis Toxins in Bivalve Molluscs. Toxins 2018, 10, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FAO—Food and Agriculture Organization of the United Nations. Food and Nutrition Paper on Marine Biotoxins; FAO: Rome, Italy, 2004; Volume 80, ISBN 95-5-105129-1. [Google Scholar]
- Corriere, M.; Soliño, L.; Costa, P.R. Effects of the Marine Biotoxins Okadaic Acid and Dinophysistoxins on Fish. J. Mar. Sci. Eng. 2021, 9, 293. [Google Scholar] [CrossRef]
- Estevez, P.; Castro, D.; Pequeño-Valtierra, A.; Giraldez, J.; Gago-Martinez, A. Emerging Marine Biotoxins in Seafood from European Coasts: Incidence and Analytical Challenges. Foods 2019, 8, 149. [Google Scholar] [CrossRef] [Green Version]
- Visciano, P.; Schirone, M.; Berti, M.; Milandri, A.; Tofalo, R.; Suzzi, G. Marine Biotoxins: Occurrence, Toxicity, Regulatory Limits and Reference Methods. Front. Microbiol. 2016, 7, 1051. [Google Scholar] [CrossRef] [Green Version]
- European Food Safety Authority (EFSA). Marine Biotoxins in Shellfish—Yessotoxin Group-Scientific Opinion of the Panel on Contaminants in the Food Chain. EFSA J. 2009, 907, 1–62. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Marine Biotoxins in Shellfish—Azaspiracid Group-Scientific Opinion of the Panel on Contaminants in the Food Chain. EFSA J. 2008, 6, 723. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Marine Biotoxins in Shellfish—Pectenotoxin Group. EFSA J. 2009, 7, 1109. [Google Scholar] [CrossRef]
- O’Mahony, M. EU Regulatory Risk Management of Marine Biotoxins in the Marine Bivalve Mollusc Food-Chain. Toxins 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- European Commission. Regulation (EC). No 853/2004 of the European Parliament and of the Council of 29 April 2004 Laying down Specific Hygiene Rules for Food of Animal Origin. Official Journal of the European. Union 2004, L. 139/55. Available online: https://data.europa.eu/eli/reg/2004/853/2021-10-28 (accessed on 9 December 2021).
- European Commission. Regulation (EU). 627/2019 of 15 March 2019—Laying down Uniform Practical Arrangements for the Performance of Official Controls on Products of Animal Origin Intended for Human Consumption in Accordance with Regulation (EU) 625/2017 of the European Parliament and of the Council and Amending Commission Regulation (EC) No 2074/2005 as Regards Official Controls. Official Journal of the European Union 2019, L131/50. Available online: https://data.europa.eu/eli/reg_impl/2019/627/2021-10-14 (accessed on 9 December 2021).
- Schirone, M.; Berti, M.; Visciano, P.; Chiumiento, F.; Migliorati, G.; Tofalo, R.; Suzzi, G.; Di Giacinto, F.; Ferri, N. Determination of Lipophilic Marine Biotoxins in Mussels Harvested from the Adriatic Sea by LC-MS/MS. Front. Microbiol. 2018, 9, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.; Fong, F.; Ho, K.-C.; Lee, F. The Mechanism of Diarrhetic Shellfish Poisoning Toxin Production in Prorocentrum Spp.: Physiological and Molecular Perspectives. Toxins 2016, 8, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, B.; Daranas, A.; Norte, M.; Riobó, P.; Franco, J.; Fernández, J. Yessotoxins, a Group of Marine Polyether Toxins: An Overview. Mar. Drugs 2008, 6, 73–102. [Google Scholar] [CrossRef]
- Alfonso, A.; Vieytes, M.; Botana, L. Yessotoxin, a Promising Therapeutic Tool. Mar. Drugs 2016, 14, 30. [Google Scholar] [CrossRef] [Green Version]
- Ferreiro, S.F.; Vilariño, N.; Carrera, C.; Louzao, M.C.; Santamarina, G.; Cantalapiedra, A.G.; Cifuentes, J.M.; Vieira, A.C.; Botana, L.M. Subacute Immunotoxicity of the Marine Phycotoxin Yessotoxin in Rats. Toxicon 2017, 129, 74–80. [Google Scholar] [CrossRef]
- Hu, W.; Xu, J.; Sinkkonen, J.; Wu, J. Polyketides from Marine Dinoflagellates of the Genus Prorocentrum, Biosynthetic Origin and Bioactivity of Their Okadaic Acid Analogues. Mini Rev. Med. Chem. 2010, 10, 51–61. [Google Scholar] [CrossRef]
- Valdiglesias, V.; Prego-Faraldo, M.; Pásaro, E.; Méndez, J.; Laffon, B. Okadaic Acid: More than a Diarrheic Toxin. Mar. Drugs 2013, 11, 4328–4349. [Google Scholar] [CrossRef] [Green Version]
- Ferron, P.-J.; Dumazeau, K.; Beaulieu, J.-F.; Le Hégarat, L.; Fessard, V. Combined Effects of Lipophilic Phycotoxins (Okadaic Acid, Azaspiracid-1 and Yessotoxin) on Human Intestinal Cells Models. Toxins 2016, 8, 50. [Google Scholar] [CrossRef] [Green Version]
- Toyofuku, H. Joint FAO/WHO/IOC Activities to Provide Scientific Advice on Marine Biotoxins (Research Report). Mar. Pollut. Bull. 2006, 52, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Regulation (EU) 1374/2021 of 12 April 2021 Amending Annex III to Regulation (EC) No 853/2004 of the European Parliament and of the Council on Specific Hygiene Requirements for Food of Animal Origin. Official Journal of the European Union 2021, L 297/1. Available online: https://data.europa.eu/eli/reg_del/2021/1374/oj (accessed on 10 December 2021).
- Otero, P.; Rodríguez, P.; Botana, A.M.; Alfonso, A.; Botana, L.M. Analysis of natural toxins. In Liquid Chromatography; Elsevier: Amsterdam, The Netherlands, 2013; pp. 411–430. ISBN 978-0-12-415806-1. [Google Scholar]
- EU-RLMB—European Union Reference Laboratory for Marine Biotoxins. EU-Harmonised Standard Operating Procedure for Determination of Lipophilic Marine Biotoxins in Molluscs by LC-MS/MS, Version 5. 2015. Available online: http://www.aesan.gob.es/AECOSAN/docs/documentos/laboratorios/LNRBM/ARCHIVO2EU-Harmonised-SOP-LIPO-LCMSMS_Version5.pdf (accessed on 10 December 2021).
- European Commission. Commission Decision 657/2002 of 14 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results. Official Journal of the European Union 2002, L 221/8. Available online: https://data.europa.eu/eli/dec/2002/657/2021-06-10 (accessed on 10 December 2021).
- European Commission. Regulation (EU) 2017/625 of the European Parliament and of the Council of 15 March 2017 on Official Controls and Other Official Activities Performed to Ensure the Application of Food and Feed Law, Rules on Animal Health and Welfare, Plant Health and Plant Protection Products, Amending Regulations (EC) No 999/2001, (EC) No 396/2005, (EC) No 1069/2009, (EC) No 1107/2009, (EU) No 1151/2012, (EU) No 652/2014, (EU) 2016/429 and (EU) 2016/2031 of the European Parliament and of the Council, Council Regulations (EC) No 1/2005 and (EC) No 1099/2009 and Council Directives 98/58/EC, 1999/74/EC, 2007/43/EC, 2008/119/EC and 2008/120/EC, and Repealing Regulations (EC) No 854/2004 and (EC) No 882/2004 of the European Parliament and of the Council, Council Directives 89/608/EEC, 89/662/EEC, 90/425/EEC, 91/496/EEC, 96/23/EC, 96/93/EC and 97/78/EC and Council Decision 92/438/EEC (Official Controls Regulation). Official Journal of the European Union 2017, L 95/1. Available online: https://data.europa.eu/eli/reg/2017/625/2021-10-28 (accessed on 10 December 2021).
- ICH Expert Working Group. ICH Harmonised Guideline. Validation of Analytical Procedures: Text and Methodology; Somatek Inc.: San Diego, CA, USA, 2006; p. 15. [Google Scholar]
- EURACHEM. Eurachem Guideline—The Fitness for Purpose of Analytical Methods. A Laboratory Guide to Method Validation and Related Topics; EURACHEM: Lisboa, Portugal, 2014; ISBN 978-91-87461-59-0. [Google Scholar]
- ISO/IEC 17025:2017; General Requirements for the Competence of Testing and Calibration Laboratories. International Organization for Standardization ISO: Geneva, Switzerland, 2017.
- ISO 5725-2:2019; Accuracy (Trueness and Precision) of Measurement Methods and Results—Part 2: Basic Method for the Determination of Repeatability and Reproducibility of a Standard Measurement Method. International Organization for Standardadization ISO: Geneva, Switzerland, 2019.
- ISO/IEC 17043:2010; Conformity Assessment—General Requirements for Proficiency Testing. International Organization for Standardadization ISO: Geneva, Switzerland, 2010.
- European Food Safety Authority (EFSA). Management of Left-Censored Data in Dietary Exposure Assessment of Chemical Substances. EFSA J. 2010, 8, 1557. [Google Scholar] [CrossRef] [Green Version]
- These, A.; Scholz, J.; Preiss-Weigert, A. Sensitive Method for the Determination of Lipophilic Marine Biotoxins in Extracts of Mussels and Processed Shellfish by High-Performance Liquid Chromatography–Tandem Mass Spectrometry Based on Enrichment by Solid-Phase Extraction. J. Chromatogr. A 2009, 1216, 4529–4538. [Google Scholar] [CrossRef]
- Wang, L.; Shi, X.; Zhao, Q.; Sun, A.; Li, D.; Zhao, J. Determination of Lipophilic Marine Toxins in Fresh and Processed Shellfish Using Modified QuEChERS and Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry. Food Chem. 2019, 272, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Gerssen, A.; McElhinney, M.A.; Mulder, P.P.J.; Bire, R.; Hess, P.; de Boer, J. Solid Phase Extraction for Removal of Matrix Effects in Lipophilic Marine Toxin Analysis by Liquid Chromatography-Tandem Mass Spectrometry. Anal. Bioanal. Chem. 2009, 394, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fux, E.; Rode, D.; Bire, R.; Hess, P. Approaches to the Evaluation of Matrix Effects in the Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis of Three Regulated Lipophilic Toxin Groups in Mussel Matrix (Mytilus edulis). Food Addit. Contam. Part A 2008, 25, 1024–1032. [Google Scholar] [CrossRef]
- Fang, L.; Yao, X.; Wang, L.; Li, J. Solid-Phase Extraction-Based Ultra-Sensitive Detection of Four Lipophilic Marine Biotoxins in Bivalves by High-Performance Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. Sci. 2015, 53, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Regueiro, J.; Rossignoli, A.E.; Álvarez, G.; Blanco, J. Automated On-Line Solid-Phase Extraction Coupled to Liquid Chromatography–Tandem Mass Spectrometry for Determination of Lipophilic Marine Toxins in Shellfish. Food Chem. 2011, 129, 533–540. [Google Scholar] [CrossRef]
- Gerssen, A.; Mulder, P.P.J.; McElhinney, M.A.; de Boer, J. Liquid Chromatography–Tandem Mass Spectrometry Method for the Detection of Marine Lipophilic Toxins under Alkaline Conditions. J. Chromatogr. A 2009, 1216, 1421–1430. [Google Scholar] [CrossRef]
- García-Altares, M.; Diogène, J.; de la Iglesia, P. The Implementation of Liquid Chromatography Tandem Mass Spectrometry for the Official Control of Lipophilic Toxins in Seafood: Single-Laboratory Validation under Four Chromatographic Conditions. J. Chromatogr. A 2013, 1275, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Stobo, L.A.; Lacaze, J.-P.C.L.; Scott, A.C.; Gallacher, S.; Smith, E.A.; Quilliam, M.A. Liquid Chromatography with Mass Spectrometry—Detection of Lipophilic Shellfish Toxins. J. AOAC Int. 2005, 88, 1371–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Commission. Commission Regulation (EU) No 15/2011 of 10 January 2011 Amending Regulation (EC) No 2074/2005 as Regards Recognised Testing Methods for Detecting Marine Biotoxins in Live Bivalve Molluscs. Official Journal of the European Union 2011, L 6/3. Available online: https://data.europa.eu/eli/reg/2011/15/oj (accessed on 10 December 2021).
- Rúbies, A.; Muñoz, E.; Gibert, D.; Cortés-Francisco, N.; Granados, M.; Caixach, J.; Centrich, F. New Method for the Analysis of Lipophilic Marine Biotoxins in Fresh and Canned Bivalves by Liquid Chromatography Coupled to High Resolution Mass Spectrometry: A Quick, Easy, Cheap, Efficient, Rugged, Safe Approach. J. Chromatogr. A 2015, 1386, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Domènech, A.; Cortés-Francisco, N.; Palacios, O.; Franco, J.M.; Riobó, P.; Llerena, J.J.; Vichi, S.; Caixach, J. Determination of Lipophilic Marine Toxins in Mussels. Quantification and Confirmation Criteria Using High Resolution Mass Spectrometry. J. Chromatogr. A 2014, 1328, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Blay, P.; Hui, J.P.M.; Chang, J.; Melanson, J.E. Screening for Multiple Classes of Marine Biotoxins by Liquid Chromatography–High-Resolution Mass Spectrometry. Anal. Bioanal. Chem. 2011, 400, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Bacchiocchi, S.; Siracusa, M.; Ruzzi, A.; Gorbi, S.; Ercolessi, M.; Cosentino, M.A.; Ammazzalorso, P.; Orletti, R. Two-Year Study of Lipophilic Marine Toxin Profile in Mussels of the North-Central Adriatic Sea: First Report of Azaspiracids in Mediterranean Seafood. Toxicon 2015, 108, 115–125. [Google Scholar] [CrossRef]
- Gerssen, A.; van Olst, E.H.W.; Mulder, P.P.J.; de Boer, J. In-House Validation of a Liquid Chromatography Tandem Mass Spectrometry Method for the Analysis of Lipophilic Marine Toxins in Shellfish Using Matrix-Matched Calibration. Anal. Bioanal. Chem. 2010, 397, 3079–3088. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, I.; Alfonso, A.; González-Jartín, J.M.; Vieytes, M.R.; Botana, L.M. A Single Run UPLC-MS/MS Method for Detection of All EU-Regulated Marine Toxins. Talanta 2018, 189, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Orea, C.; Sanchís, J.; Farré, M.; Barceló, D. Analysis of Lipophilic Marine Biotoxins by Liquid Chromatography Coupled with High-Resolution Mass Spectrometry in Seawater from the Catalan Coast. Anal. Bioanal. Chem. 2017, 409, 5451–5462. [Google Scholar] [CrossRef]
- Oller-Ruiz, A.; Campillo, N.; Hernández-Córdoba, M.; Gilabert, J.; Viñas, P. Monitoring Lipophilic Toxins in Seawater Using Dispersive Liquid—Liquid Microextraction and Liquid Chromatography with Triple Quadrupole Mass Spectrometry. Toxins 2021, 13, 57. [Google Scholar] [CrossRef]
- van den Top, H.J.; Gerssen, A.; McCarron, P.; van Egmond, H.P. Quantitative Determination of Marine Lipophilic Toxins in Mussels, Oysters and Cockles Using Liquid Chromatography-Mass Spectrometry: Inter-Laboratory Validation Study. Food Addit. Contam. Part A 2011, 28, 1745–1747. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Broadwater, M.H.; Ramsdell, J.S. Analysis of Diarrhetic Shellfish Poisoning Toxins and Pectenotoxin-2 in the Bottlenose Dolphin (Tursiops Truncatus) by Liquid Chromatography–Tandem Mass Spectrometry. J. Chromatogr. A 2015, 1416, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Gałuszka, A.; Migaszewski, Z.; Namieśnik, J. The 12 Principles of Green Analytical Chemistry and the SIGNIFICANCE Mnemonic of Green Analytical Practices. TrAC Trends Anal. Chem. 2013, 50, 78–84. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Biotoxin Group | Relevant and Regulated Compounds | Molecular Formula | Number of Identified Analogues | Legal Limit in Molluscs | TEF | Acute Reference Dose | Chemical Properties | Algae Producing Species | Mechanism of Toxicity | Main Symptomatology | Refs. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| OA group | OA Okadaic acid | C44H68O13 | >30 | 160 μg kg−1 of OA equivalents | 1 | 0.3 μg eq. kg−1 b.w. | polyketide structure; C1 carboxyl group; polycyclic ethers and poly alkoxy with 3 spiro-keto rings; derivate from of a C38 fatty acid | Dinophysis fortii, Dinophysis acuta, Dinophysis ovum, Dinophysis acuminate, Dinophysis norvegica, Prorocentrum lima Prorocentrum belizeanum, Phalacroma rotundata, Phalacroma mitra | inhibition of serine/threonine protein phosphatases (PP) 2A, 1B, 2B | acute exposure: diarrhea, nausea, abdominal pain chronic exposure: mucosal damages of the intestinal tract, gastrointestinal cancer (tumor promoting, studies ongoing) | [1,2,3,6,7,8,9,11,19,23,24] |
| DTX1 Dinophysistoxin-1 | C45H70O13 | 1 | |||||||||
| DTX2 Dinophysistoxin-2 | C44H68O13 | 0.6 | |||||||||
| DTX3 Dinophysistoxin-3 | acylated derivatives of OA analogues (C length: C14–C22; most common is C16-palmitic) number of unsaturation: 0–6 | 1 | |||||||||
| PTX group | PTX1 Pectenotoxin-1 | C47H70O15 | 15 | 160 μg kg−1 of PTX equivalents | 1 | 0.8 μg eq. kg−1 b.w. | macrolactonic structure; poly hydroxyl polycyclic ethers | Dinophysis fortii, Dinophysis acuta, Dinophysis acuminate | alteration of actin-based cytoskeleton, induction of apoptosis and subsequent cell death | liver necrosis, cardiac muscle damage (in vitro and in vivo: mice) | [2,3,6,8,11,14] |
| PTX2 Pectenotoxin-2 | C47H70O14 | 1 | |||||||||
| AZA group | AZA1 Azaspiracid-1 | C47H71NO12 | >40 | 160 μg kg−1 of AZA equivalents | 1 | 0.2 μg eq. kg−1 b.w. | poly hydroxyl polycyclic ethers; piperidine ring (amino group = aza group); C1 carboxyl group | Azadinium spinosum, Amphidoma languida, Azadinium poporum | cytotoxic effect by increasing of calcium and cAMP; alterations in cytoskeletal structures and the E-cadherin system, with disruption of cell-cell- and cell-matrix interactions, and perturbation of the intestinal barrier function | injury of lamina propria and epithelial cells in small intestine, liver and thymus necrosis (in vivo: mice) | [2,3,6,8,11,13,25] |
| AZA2 Azaspiracid-2 | C48H73NO12 | 1.8 | |||||||||
| AZA3 Azaspiracid-3 | C46H69NO12 | 1.4 | |||||||||
| YTX group | YTX Yessotoxin | C55H82O21S2 | >90 | 3.75 mg kg−1 of YTX equivalents | 1 | 25 μg eq. kg−1 b.w. | organosulfate structure (two sulfooxy groups); polycyclic ethers | Protoceratium reticulatum | modifications of intracellular levels of cAMP, calcium, PDEs, PKC and AKAP-149 (not well clarified) | immunotoxicity and immunosuppressive effects (in vitro and in vivo: rats) | [2,3,4,6,8,11,12,20,21,22] |
| hYTX homoyessotoxin | C56H84O21S2 | 1 | |||||||||
| 45-OH-YTX 45-hydroxy-yessotoxin | C55H82O22S2 | 1 | |||||||||
| 45-OH-hYTX 45-hydroxy-homoyessotoxin | C56H84O22S2 | 0.5 |
| Compound | Ion | Polarity | Precursor Ion (m/z) | Product ion (m/z) 1 Quantifier | Collision Energy 2 | Product Ion (m/z) 1 Qualifier | Collision Energy 2 | RF Lens Voltage 2 |
|---|---|---|---|---|---|---|---|---|
| OA | [M + Na]+ | + | 827.5 | 723.4 | 49 | 809.4 791.4 | 44 46 | 298 |
| DTX2 | [M + Na]+ | + | 827.5 | 723.4 | 49 | 809.4 791.4 | 44 46 | 218 |
| YTX | [M − 2H]2− | - | 570.4 | 467.2 | 30 | 502.2 386.2 | 23 31 | 298 |
| hYTX | [M − 2H]2− | - | 577.4 | 474.3 | 31 | 509.1 403.2 | 23 33 | 298 |
| 45-OH-YTX | [M − 2H]2− | - | 578.4 | 467.4 | 31 | 396.4 | 31 | 298 |
| 45-OH-hYTX | [M − 2H]2− | - | 585.4 | 474.0 | 31 | 403.4 | 31 | 298 |
| DTX1 | [M + Na]+ | + | 841.5 | 737.4 | 55 | 823.5 805.4 | 44 51 | 255 |
| AZA3 | [M + H]+ | + | 828.5 | 810.5 | 33 | 792.5 640.4 | 42 50 | 298 |
| AZA1 | [M + H]+ | + | 842.5 | 824.5 | 32 | 806.5 654.4 | 42 53 | 298 |
| AZA2 | [M + H]+ | + | 856.5 | 838.5 | 33 | 820.5 672.4 | 42 51 | 298 |
| PTX2 | [M + NH4]+ | + | 876.6 | 823.5 | 25 | 841.4 787.4 | 22 30 | 298 |
| Performance Characteristics | Evaluation/Measurement Approach |
|---|---|
| Linearity | Injection of LMTs standard solutions in methanol 2, 10, 20, 40, 60, 80 μg L−1 (three replicates at each concentration level) regression of calibration curve with the least square method. Mandel test to check linearity. Calculation of determination coefficient value (R2 > 0.98). |
| Selectivity | Analysis of 20 non-hydrolysed blank samples and 20 hydrolysed samples of fresh, frozen, precooked and canned mussels, for checking the absence of interfering peaks in the retention-time window of ± 3% of each analyte. |
| Limit of detection Limit of quantification | Gradual dilution (80, 40, 20, 10, 2, 1 μg L−1) of a matrix matched extract obtained by pooling the blank matrices used for selectivity study and spiking it with all the LMTs. The comparison of measured signals of quantifier ions with signals of blank samples, defined as signal-to-noise ratio (S/N), permitted the establishment of the minimum concentration at which the analyte could be reliably detected/quantified. A S/N of 3 and 10 for LoD and LoQ, respectively, was considered acceptable. |
| Precision and trueness | Analysis of a blank mussel sample spiked at 20 and 80 μg kg−1 with a mix LMTs standard solution (six replicates in two different working sessions with the same instrument, different days, operators and instrumental calibrations). The relative standard deviation for each analyte and recovery values were evaluated. Evaluation of method trueness by use of CRM-FDMT1: recovery values obtained on samples spiked at 80 μg kg−1 were used to correct the results of six independent tests obtained by using CRM-FDMT1. |
| Measurement uncertainty | Use of the maximum standard uncertainty approach: α = numeric factor depending on the value of C. |
| Matrix effect | Evaluation using calibration graph method: as the ratio between the slope of the curve obtained for the matrix-matched extracts (matrix: mussels) and the slope of the curve for the standard calibration curve minus 1, expressed in percentage.
|
| Matrix Ruggedness | Conditions of major changes (matrix to analyze). Six additional experiments for each new matrix spiked at 80 μg kg−1 (oysters, clams, cockles, scallops and cephalopod molluscs). Comparison of precision and recovery data with the results obtained for validation matrix. |
| Compound | LoQ µg kg−1 | Precision (Mean) RSD% | Recovery (Mean) % | Identification Criteria (Ion Ratio % Qualifier 1/Quantifier) ± 35% | Matrix Effect % | Selectivity | Matrix RUGGEDNESS |
|---|---|---|---|---|---|---|---|
| OA | 8 | 7.8 | 75.4 | 59 ± 35 | −9 | verified for fresh, frozen, precooked and canned mussels | oysters, cockles, clams, scallops and cephalopod molluscs (a,b) |
| DTX2 | 7 | 8.2 | 81.6 | 59 ± 35 | −6 | ||
| YTX | 8 | 8.1 | 73.8 | 31 ± 35 | −7 | ||
| hYTX | 5 | 8.9 | 73.1 | 31 ± 35 | −3 | ||
| 45-OH-YTX | 4 | 8.1 | 73.8 | 31 ± 35 | / | ||
| 45-OH-hYTX | 5 | 8.9 | 73.1 | 31 ± 35 | / | ||
| DTX1 | 7 | 8.2 | 76.4 | 54 ± 35 | −2 | ||
| AZA3 | 8 | 3.6 | 82.3 | 23 ± 35 | 6 | ||
| AZA1 | 4 | 4.4 | 87.4 | 23 ± 35 | −8 | ||
| AZA2 | 3 | 3.4 | 81.8 | 23 ± 35 | 11 | ||
| PTX2 | 3 | 11.8 | 101.3 | 71 ± 35 | 19 |
| N | Median | Mean | SD 1 | IQR 2 | Min | Max | 25th Percentile | 75th Percentile | |
|---|---|---|---|---|---|---|---|---|---|
| OA group | 203 | 19.0 | 35.3 | 57.8 | 35.0 | 8.00 | 620 | 8.00 | 43.0 |
| YTX group | 203 | 30.0 | 60.0 | 109 | 62.0 | 8.00 | 1220 | 8.00 | 70.0 |
| PTX group | 203 | 8.00 | 8.19 | 1.65 | 0.00 | 8.00 | 27.0 | 8.00 | 8.00 |
| AZA group | 203 | 8.00 | 8.05 | 0.84 | 0.00 | 8.00 | 20.0 | 8.00 | 8.00 |
| OA Group per Year | |||||||||
| 2019 | 101 | 18.0 | 28.9 | 62.4 | 24.0 | 8.00 | 620 | 8.00 | 32.0 |
| 2020 | 77 | 31.0 | 50.9 | 57.1 | 40.0 | 8.00 | 278 | 17.0 | 57.0 |
| 2021 | 25 | 8.00 | 12.9 | 13.2 | 0.00 | 8.00 | 58.0 | 8.00 | 8.0 |
| YTX Group per Year | |||||||||
| 2019 | 101 | 40.0 | 72.0 | 144 | 66.0 | 8.00 | 1220 | 8.00 | 74.0 |
| 2020 | 77 | 8.00 | 36.0 | 50.0 | 42.0 | 8.00 | 260 | 8.00 | 50.0 |
| 2021 | 25 | 90.0 | 82.0 | 49.0 | 40.0 | 8.00 | 180 | 60.0 | 100 |
| OA Group per Family | |||||||||
| Mytilidae | 197 | 19.0 | 35.3 | 58.6 | 35.0 | 8.00 | 620 | 8.00 | 43.0 |
| Ostreidae | 6 | 30.0 | 35.9 | 19.19 | 24.8 | 18.0 | 66.0 | 30.0 | 46.5 |
| YTX Group per Family | |||||||||
| Mytilidae | 197 | 30.0 | 61.0 | 0.11 | 66.0 | 8.00 | 1220 | 8.00 | 74.0 |
| Ostreidae | 6 | 8.00 | 8.00 | 0.0 | 0.0 | 8.00 | 8.00 | 8.00 | 8.00 |
| References | Extraction and Clean-Up | Detection | Analytes | Matrices | Recovery (%) Range | LoQ Range | Validation Parameters Evaluated | Notes |
|---|---|---|---|---|---|---|---|---|
| Rùbies et al. (2015) | QuEChERS | UHPLC-ESI-Q-Orbitrap | AZA1, AZA2, AZA3, DTX1, DTX2, PTX1, PTX2, SPX1, OA, YTX, hYTX, 45OHYTX, 45OHhYTX | fresh and canned bivalve molluscs | 69–119 | 25 µg kg−1 | selectivity, linearity, trueness, precision | eprinomectin as internal standard |
| Blay et al. (2011) | SLE MeOH | LC-ESI-Orbitrap | AZA1, AZA2, AZA3, DTX1, DTX2, PTX2, PTX2, SPX1, OA, PSTs | shellfish | N/A | 10–30 µg kg−1 | linearity | screening |
| Regueiro et al. (2011) | SLE: MeOH/H2O online-SPE | HPLC-ESI-QqQ-MS/MS | AZA1, DTX1, DTX2, PTX2, SPX1, OA, YTX, GYM | mussels | 97–102 | 1.12–8 µg kg−1 | linearity, trueness, precision, matrix effect | |
| Fang et al. (2014) | SLE: MeOHSPE | UFLC-ESI-QqQ-MS/MS | AZA2, PTX2, SPX1, GYM | bivalve molluscs | 71–101 | 0.037–0.27 µg kg−1 | linearity, trueness, precision, matrix effect | |
| Rodríguez et al. (2018) | SLE: MeOH | HPLC-ESI-QqQ-MS/MS | AZA1, AZA2, AZA3, DTX1, DTX2, PTX2, SPX1, OA, YTX, hYTX, PSTs, TTX, DA | mussels | N/A | 0.047–40.15 µg kg−1 | linearity, precision, matrix effect | OA/DTX2 one peak; different extraction protocol for PSTs, TTX, DA |
| García-Altares et al. (2013) | SLE: MeOH | LC-QTRAP-ESI-MS/MS | AZA1, AZA2, AZA3, DTX1, DTX2, PTX2, SPX1, OA, YTX, hYTX, 45OHYTX, 45OHhYTX, GYM | bivalve molluscs | 28–150 | 1.5–377 µg kg−1 | linearity, precision, trueness, matrix effect | comparative study (different mobile phase pH) |
| These et al. (2009) | SLE: MeOH SPE | LC-QTRAP-ESI-MS/MS | AZA1, PTX2, OA, YTX, | bivalve molluscs and processed shellfish products | 86–147 | 1 µg kg−1 | linearity, trueness, precision | comparative study (different SPE cartridges) |
| Fux et al. (2009) | PEA | (1) HPLC-ESI-QqQ-MS/MS (2) HPLC-QTOF-MS/MS | AZA1, PTX2, OA | mussels | N/A | N/A | linearity, matrix effect | study of matrix effect evaluation |
| Wang et al. (2019) | QuEChERS dSPE | HPLC-ESI-QqQ-MS/MS | AZA1, AZA2, AZA3, DTX1, DTX2, SPX1, OA, YTX, hYTX | fresh and processed shellfish | 88–109 | 0.32–4.92 µg kg−1 | linearity, precision, trueness, matrix effect | comparative study (different sorbents) |
| Wang et al. (2015) | SLE: MeOH SPE | LC-QTRAP-ESI-MS/MS | DTX1, DTX2, PTX2, OA | bottlenose dolphin | 85–140 | 0.2–0.7 µg kg−1- | linearity, precision, trueness | |
| Domènech et al. (2014) | SLE: MeOH | UHPLC-ESI-Q-Orbitrap | AZA1, PTX2, SPX1, OA, YTX, GYM | mussels | 80–110 | 0.9–4.8 µg kg−1 | selectivity, linearity, trueness, precision, measurement uncertainty | robust validation study |
| Schirone et al. (2018) | SLE: MeOH | HPLC-ESI-QqQ-MS/MS | AZA1, AZA2, AZA3, DTX1, DTX2, PTX2, OA, YTX, hYTX | mussels | 85–104 | 40–60 µg kg−1 | selectivity, linearity, trueness, precision, measurement uncertainty | monitoring study |
| Gerssen et al. (2009) | SLE: MeOH SPE | HPLC-ESI-QqQ-MS/MS | OA, YTX, AZA1, PTX2, GYM, SPX1 | mussels, scallops and oysters | 63–117 | 9 µg kg−1 | linearity, trueness, precision, matrix effect | matrix effect study |
| Gerssen et al. (2010) | SLE: MeOH SPE | HPLC-ESI-QqQ-MS/MS | OA, YTX, AZA1, PTX2, SPX1 | mussels, oysters, cockles and clams | 102–111 | 16.4 µg kg−1 | linearity, trueness, precision | comparative study (with/without SPE) |
| Van den Top et al. (2011) | SLE: MeOH SPE | HPLC-ESI-QqQ-MS/MS | AZA1, AZA2, AZA3, DTX1, DTX2, PTX2, OA, YTX, 45OHYTX | mussels, oysters and cockles | 80–110 | 4–53 µg kg−1 | linearity, trueness, precision, matrix effect | inter-laboratory validation study |
| Oller-Ruiz et al. (2021) | DLLME | HPLC-ESI-QqQ-MS/MS | AZA1, AZA2, AZA3, AZA4, AZA5, DTX1, DTX2, PTX2, SPX1, OA, GYM | seawater | 82–123 | 0.7–19 ng L−1 | linearity, trueness, precision, matrix effect | monitoring study |
| This method | SLE: MeOH SPE | HPLC-ESI-QqQ-MS/MS | AZA1, AZA2, AZA3, DTX1, DTX2, PTX2, OA, YTX, hYTX, 45OHYTX, 45OHhYTX | fresh and processed mussels, oysters, scallops, clams, cockles and cephalopod molluscs | 73–101 | 3–8 µg kg−1 | selectivity, linearity, trueness, precision, matrix effect, measurement uncertainty, ruggedness |
| Compounds | Assigned Values (xa) µg kg−1 | Obtained Value µg kg−1 | Obtained Z-Score |
|---|---|---|---|
| OA | 302 | 382 | 1.20 |
| Total OA | 745 | 815 | 0.56 |
| OA + PTX group | 748 | 815 | 0.54 |
| YTX | 320 | 330 | 0.11 |
| hYTX | 2660 | 3550 | 2.05 |
| YTX group | 3940 | 3880 | −0.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amore, T.; Lo Magro, S.; Vita, V.; Di Taranto, A. Optimization and Validation of a High Throughput UHPLC-MS/MS Method for Determination of the EU Regulated Lipophilic Marine Toxins and Occurrence in Fresh and Processed Shellfish. Mar. Drugs 2022, 20, 173. https://doi.org/10.3390/md20030173
D’Amore T, Lo Magro S, Vita V, Di Taranto A. Optimization and Validation of a High Throughput UHPLC-MS/MS Method for Determination of the EU Regulated Lipophilic Marine Toxins and Occurrence in Fresh and Processed Shellfish. Marine Drugs. 2022; 20(3):173. https://doi.org/10.3390/md20030173
Chicago/Turabian StyleD’Amore, Teresa, Sonia Lo Magro, Valeria Vita, and Aurelia Di Taranto. 2022. "Optimization and Validation of a High Throughput UHPLC-MS/MS Method for Determination of the EU Regulated Lipophilic Marine Toxins and Occurrence in Fresh and Processed Shellfish" Marine Drugs 20, no. 3: 173. https://doi.org/10.3390/md20030173
APA StyleD’Amore, T., Lo Magro, S., Vita, V., & Di Taranto, A. (2022). Optimization and Validation of a High Throughput UHPLC-MS/MS Method for Determination of the EU Regulated Lipophilic Marine Toxins and Occurrence in Fresh and Processed Shellfish. Marine Drugs, 20(3), 173. https://doi.org/10.3390/md20030173