2. Results and Discussion
Marinobazzanan (
1) was obtained as a pale-yellow amorphous powder. The molecular formula of
1 was determined as C
15H
2235ClNO
2 based on a protonated adduct at
m/z 284.1415 [M + H]
+ (calculated for C
15H
2335ClNO
2, 284.1417) in a high-resolution electrospray ionization mass spectrum (HR-ESI-MS), which indicated five degrees of unsaturation. The
1H NMR spectrum of
1 displayed one olefinic (
δH 6.66), two exo-methylene (
δH 5.44, 5.21), eight methylene (
δH 1.54, 1.65, 1.79, 1.87, 2.05, 2.21, 2.21, 2.39), two methine (
δH 3.86, 4.29), and two methyl singlet (
δH 1.20, 0.92) hydrogens. The
13C and HSQC NMR spectroscopic data revealed one carbonyl (
δC 172.2), four quaternary (
δC 153.7, 131.3, 48.5, 36.8), three methine (
δC 134.0, 75.6, 69.2), five methylene (
δC 111.9, 40.5, 33.0, 27.4, 21.8), and two methyl singlets (
δC 24.5, 17.1) carbons (
Table 1,
Figures S1 and S2). The molecular formula and the HSQC NMR spectroscopic data of
1 suggested that compound
1 possessed a bicyclic ring system.
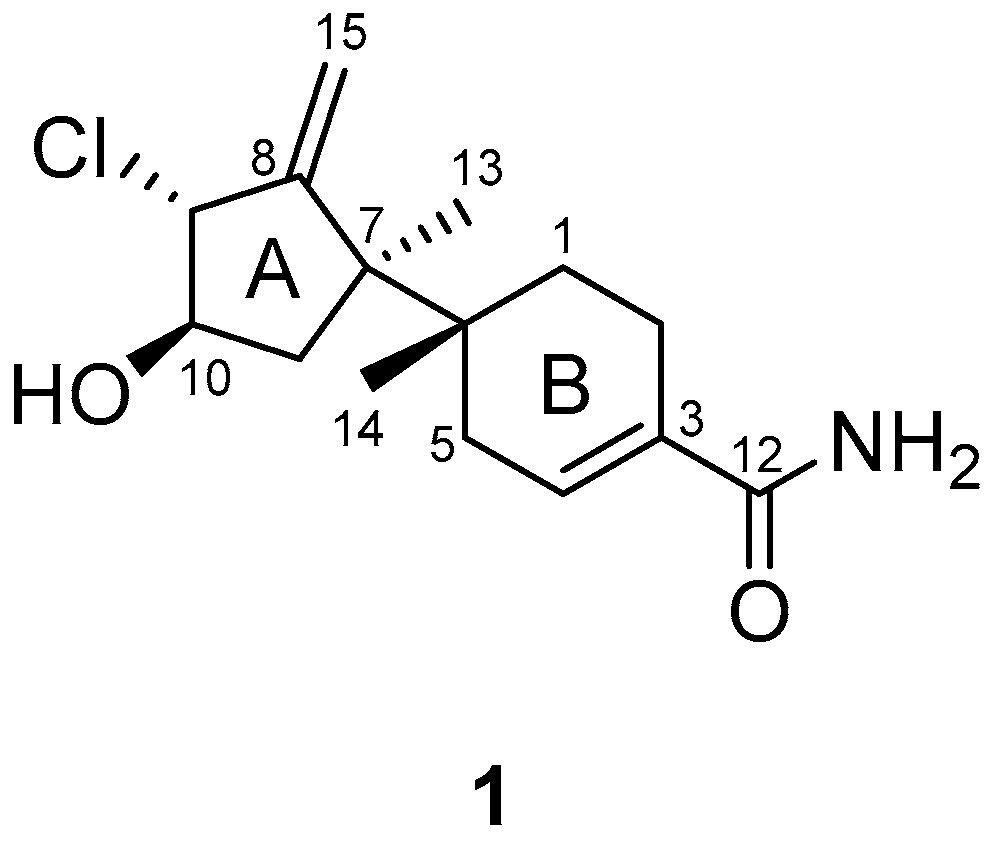
The interpretation of 2D NMR spectra (
Figures S3–S5) allowed the construction of a bazzanane-type sesquiterpenoid framework for
1. The COSY NMR crosspeaks [H-9 (
δH 4.29)/H-10 (
δH 3.86)/H-11 (
δH 1.87, 1.79)] and the long-range HMBC correlations from H-9 (
δH 4.29) to C-8 (
δC 153.7), C-15 (
δC 111.9), from exo-methylene H-15 (
δH 5.44, 5.21) to C-7 (
δC 48.5), C-8 (
δC 153.7), and from the methyl singlet H-13 (
δH 1.20) to C-7 (
δC 48.5), C-8 (
δC 153.7), and C-11 (
δC 40.5) allowed the construction of the five-membered ring (ring A) of bazzanane. The six-membered ring (ring B) was established by analyzing the COSY crosspeaks [H-1 (
δH 1.65, 1.54)/H-2 (
δH 2.39, 2.21) and H-4 (
δH 6.66)/H-5 (
δH 2.21, 2.05)], along with the long-range HMBC correlations from H-5 (
δH 2.21, 2.05) to C-1 (
δC 27.4), C-3 (
δC 131.3), C-4 (
δC 134.0), C-6 (
δC 36.8), and C-14 (
δC 17.1). In addition, the HMBC NMR correlations from H-4 (
δH 6.66) to the carbonyl carbon C-12 (
δC 172.2) allowed the connection of the amide group to ring B. Meanwhile, the connection of C-6/C-7 was secured by observing the HMBC correlations from H-13 (
δH 1.20) to C-6 (
δC 36.8), C-7 (
δC 48.5) and from H-14 (
δH 0.92) to the same two carbons. Furthermore, the attachment of a hydroxy group at C-10 was assigned through the presence of the carbon chemical shifts at
δC 75.6. In addition, a chlorine atom at C-9 was established by considering the isotope ratio (3:1) of the two protonated adduct [M + H]
+ and [M + H+2]
+ in the low-resolution electrospray ionization mass spectrum (LR-ESI-MS) and the chemical shift at
δC 69.2 in the chlorinated methine, which completed the planar structure of
1, as shown in
Figure 2.
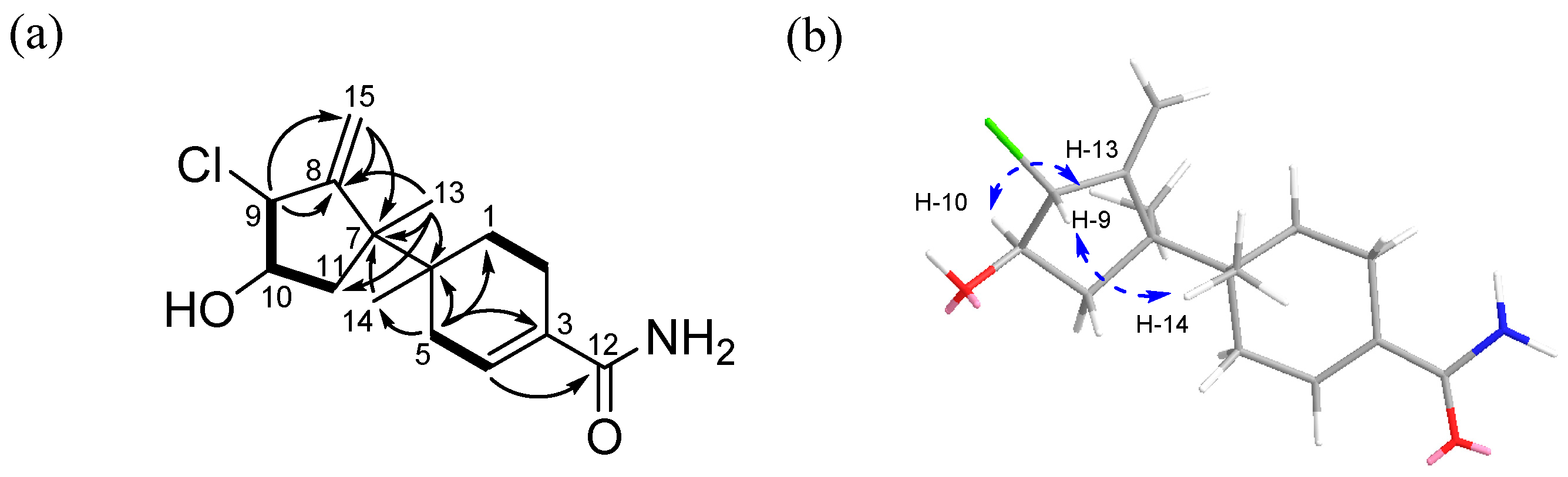
The relative stereochemistry of
1 was determined by analysis of the NOESY NMR spectroscopic data. A NOESY correlation (
Figure S6) between H-9 (
δH 4.29) and H-14 (
δH 0.92) indicated that these protons should be located on the same face of the molecule. Meanwhile, a NOESY crosspeak between H-10 (
δH 3.86) and H-13 (
δH 1.20) as well as its coupling constant (
3JH−H = 10.0 Hz) established their
syn relationship. Therefore, the relative configuration of
1 was assigned as 6
S*, 7
S*, 9
S*, and 10
S* (
Figure 2b).
The absolute configuration of
1 was established using the modified Mosher’s method [
19,
20], combined with a comparison between the measured and calculated vibrational circular dichroism (VCD) spectra [
21]. First, esterification of
1 with (
R)- and (
S)-MTPA-Cl (
α-methoxy-
α-(trifluoromethyl) phenylacetyl chloride) yielded the (
S)- and (
R)-MTPA esters of
1 (
1a and
1b, respectively). Analysis of the
1H NMR (
Figures S7 and S8) Δ
δ(S−R) values revealed a consistent sign distribution, thus verifying the
R configuration at C-10 (
Figure 3). Therefore, the absolute stereochemistry of the three chiral centers of ring A in
1 was determined as 7
R, 9
R, and 10
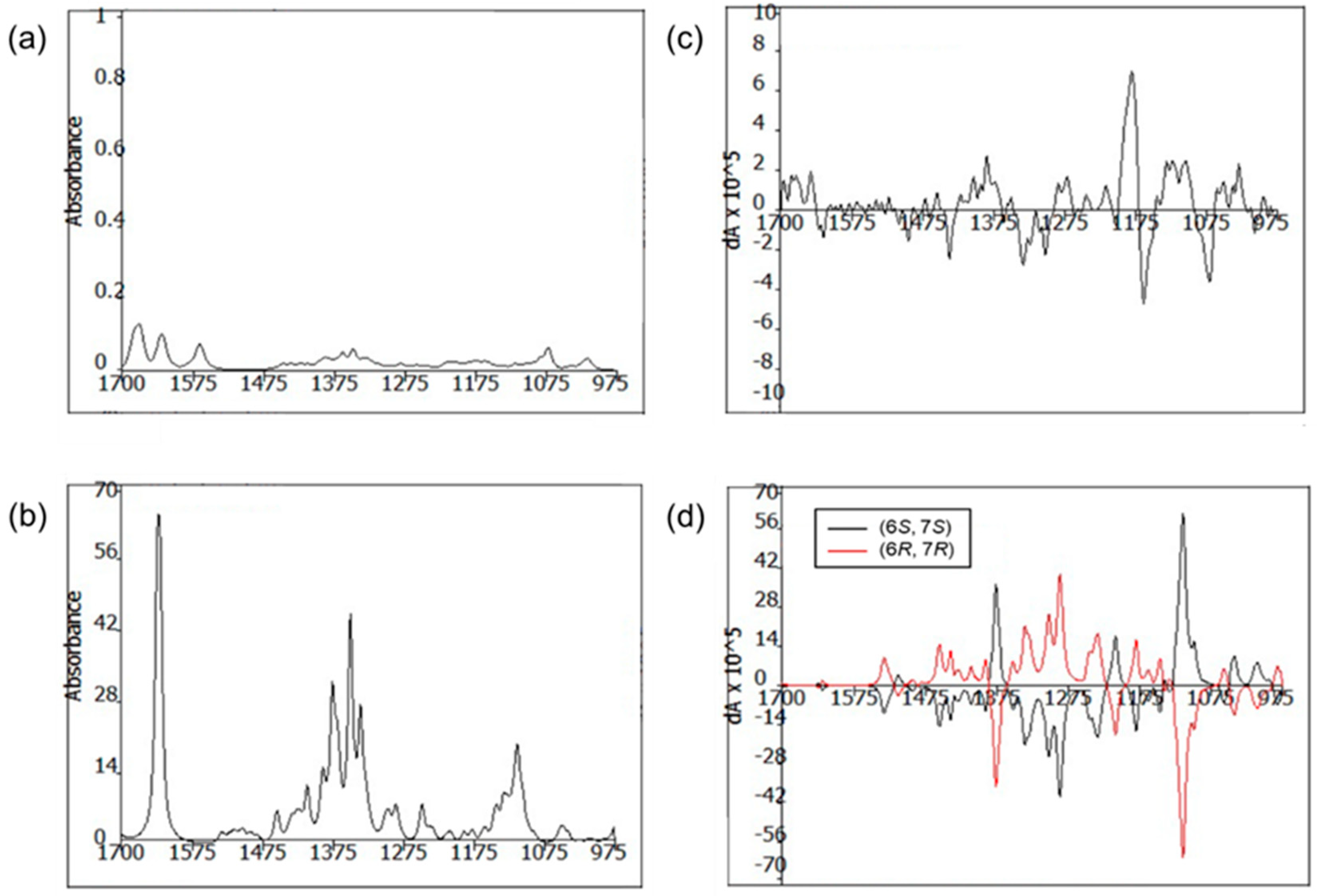
R. The absolute configurations of C-6 were also confirmed by comparing the experimental and calculated VCD spectra. The VCD spectra were calculated using density functional theory (DFT) at the B3LYP/6-31+G(d) level using the Gaussian 09 software (Gaussian, Inc., Wallingford, CT 06492, United States) and the calculated VCD spectrum of the (6
R, 7
R) configuration showed good agreement with the experimental spectrum of
1, with a confidence level of 87% (
Figure 4).
Bazzanane-type sesquiterpenes have been mainly found in liverworts, including the
Bazzania genus. After the first reported isolation of bazzanene from
Bazzania pompeana (Lac.) Mitt. in 1969 [
22], only a few bazzanane-type sesquiterpenes have been reported, indicating that they are very rare in nature [
23,
24]. The most similar compound to
1 was isolated from the New Zealand liverwort
Frullania falciloba [
25]. However, previously reported bazzanane-type sesquiterpenes are neither chlorinated nor possess an amide group in the molecule, as in
1. Furthermore,
1 is the first bazzanane-type sesquiterpenoid to be isolated from a strain of the genus
Acremonium and the first reported chlorinated bazzanane-type sesquiterpene with an amide group in its structure.
A methyl thiazolyl tetrazolium (MTT) cytotoxicity bioassay was used to evaluate the effect of
1 treatment in various concentrations (10, 25, 50, and 100 μM) on the viability of AGS (gastric cancer), A549 (lung cancer), and Caco-2 (colorectal cancer) cells. The cell viability of A549 was unaffected during the treatment with 10–50 μM of
1 for 48 h; however, the viability decreased marginally during the treatment with 100 μM of
1, as shown in
Figure 5. Similarly, the viability of AGS and Caco-2 did not decrease with treatment of 10–25 μM of
1 but decreased significantly at concentrations of 50–100 μM. Thus, these observations demonstrate that treatment with
1 is relatively non-toxic toward A549, AGS, and Caco-2 cells at concentrations less than 25 μM.
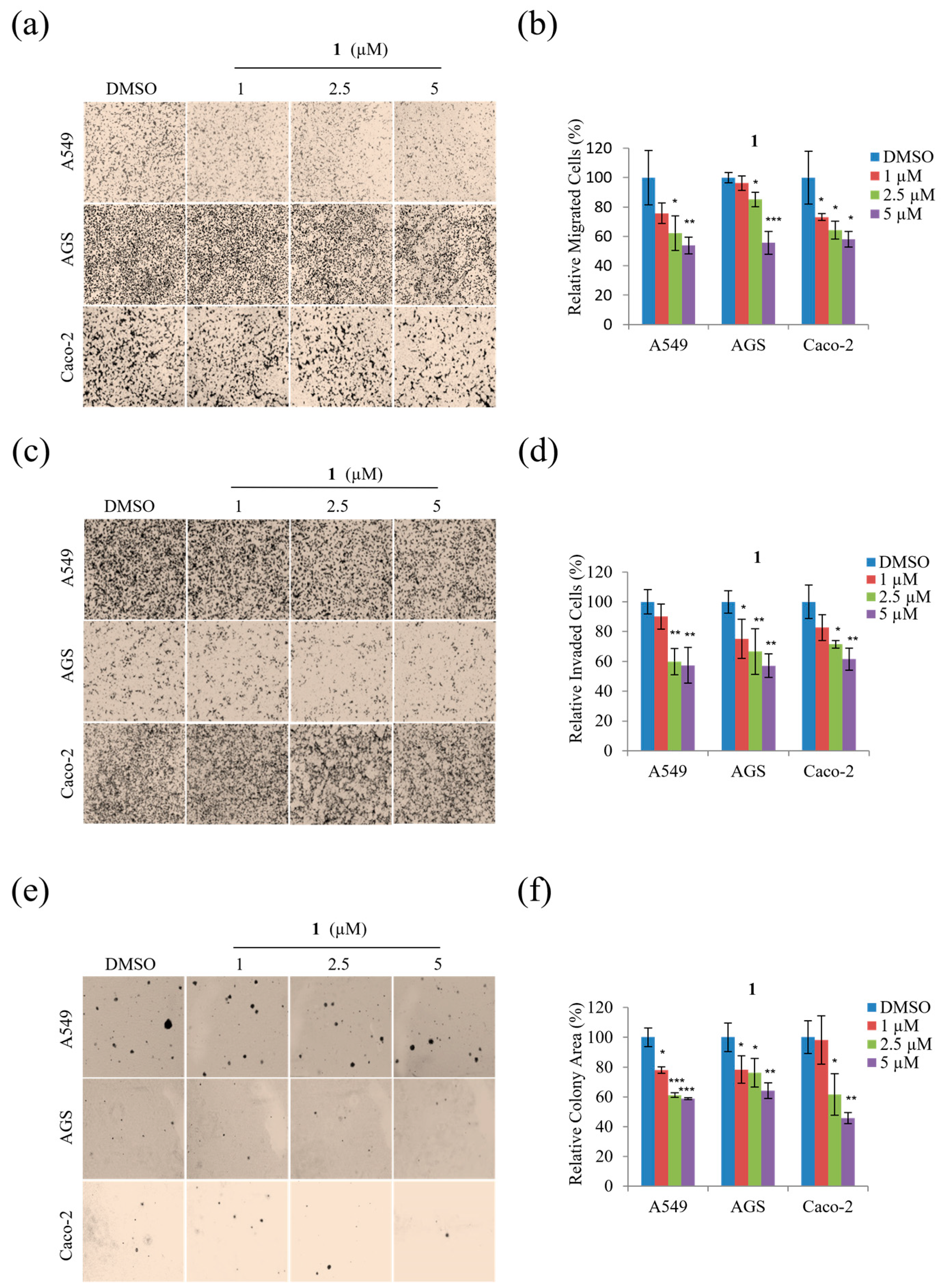
Next, migration and invasion assays were performed using non-toxic concentrations (1, 2.5, and 5 μM) to determine whether
1 inhibits the motility of A549, AGS, and Caco-2 cells. As shown in
Figure 6,
1 displayed a dose-dependent inhibitory effect on the migration of all three cell types at concentrations from 1 to 5 µM (
Figure 6a,b) alongside dose-dependent inhibition of invasion by each cell type by up to ~45% at 5 µM concentrations (
Figure 6c,d).
In addition, the potential anti-tumorigenic activity of
1 was evaluated by examining the soft agar colony formation of A549, AGS, and Caco-2 cells exposed to non-toxic concentrations (1, 2.5, and 5 µM). As shown in
Figure 6e,f, compound
1 dose-dependently decreased the colony formation of A549, AGS, and Caco-2 cells. Moreover,
Figure 6f reveals that treatment with 5 µM of
1 significantly decreased the tumorigenicity of A549, AGS, and Caco-2 cells. Overall, these results demonstrate that treatment with 1, 2.5, and 5 µM concentrations of
1 significantly suppressed the motility and tumorigenicity of A549, AGS, and Caco-2 cells.
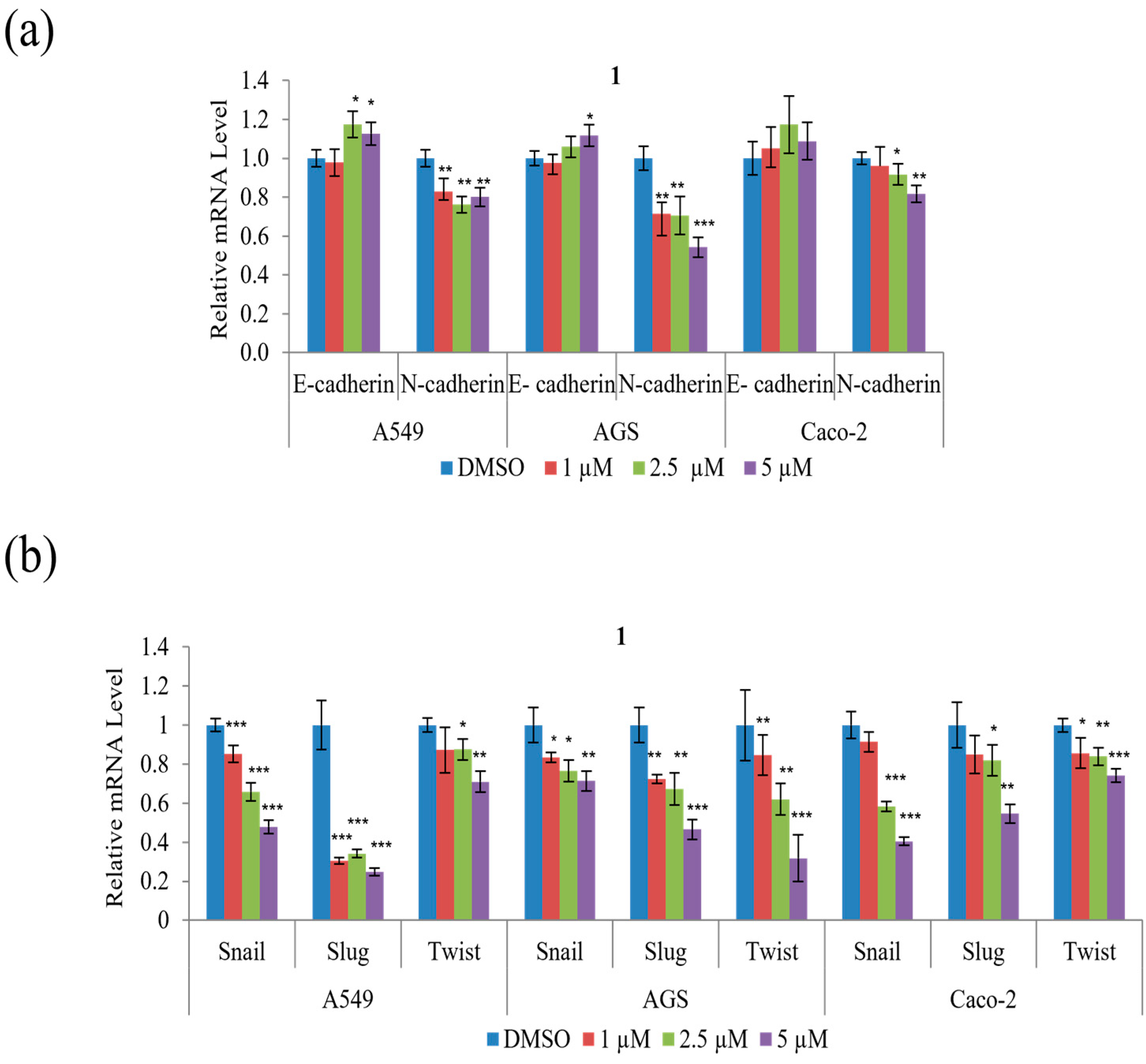
To determine whether the suppression of A549, AGS, and Caco-2 cell motility and tumorigenicity in the presence of
1 involves the epithelial–mesenchymal transition (EMT), the expression of EMT effectors and transcription factors were examined. As shown in
Figure 7a,
1 decreased mRNA expression of the mesenchymal marker N-cadherin but increased that of the epithelial marker E-cadherin in all three cell types. Further,
1 significantly decreased the mRNA expression of the EMT transcription factors, Snail, Slug, and Twist, in all three cell types, as shown in
Figure 7b. In summary, these results indicate that
1 modulates the expression of the EMT effector N-cadherin by downregulating the transcription factors Snail, Slug, and Twist.
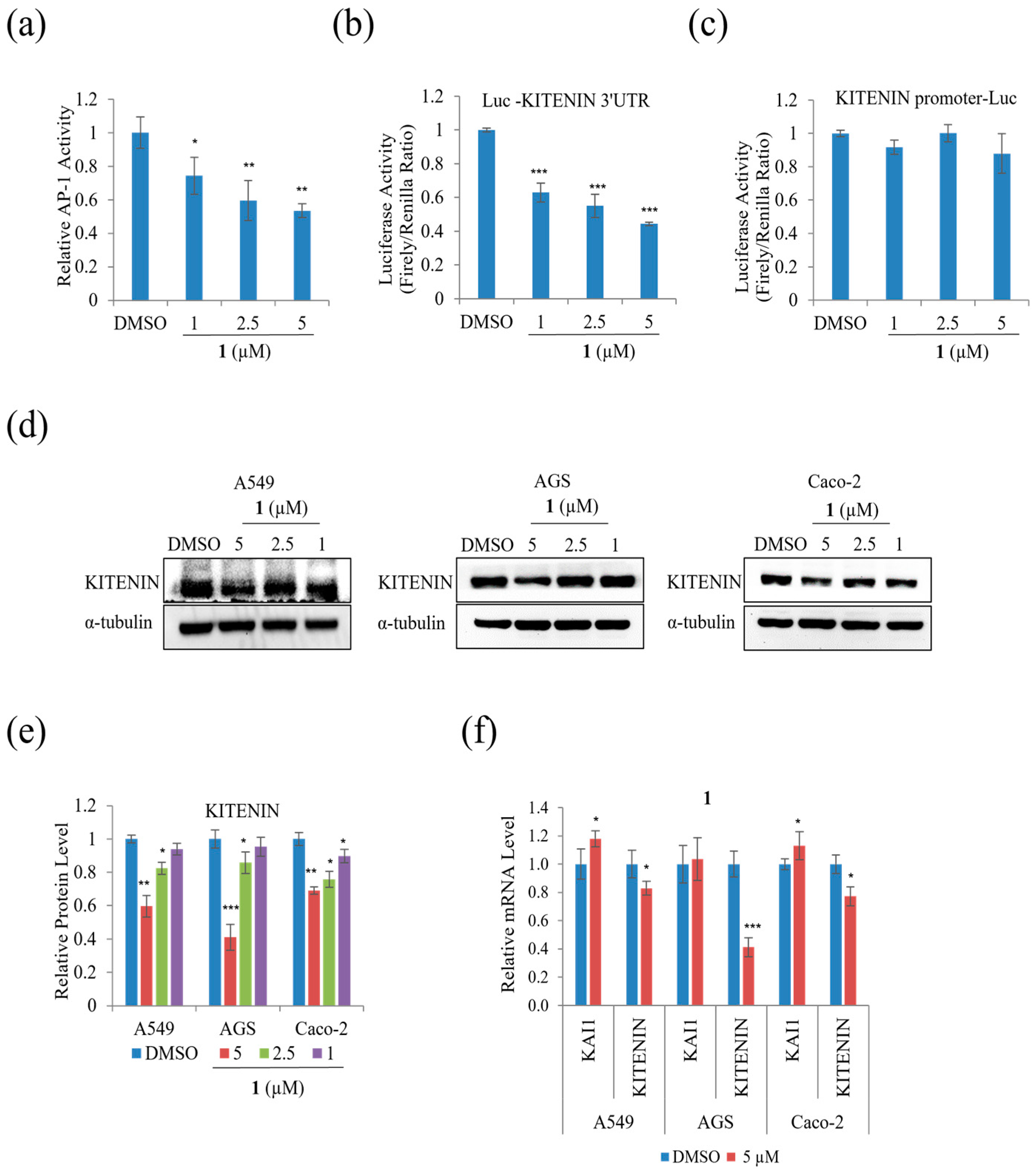
To examine whether the suppression of A549, AGS, and Caco-2 cell motility and tumorigenicity in the presence of
1 involves KITENIN and AP-1, the protein and mRNA expression levels of KITENIN and AP-1, including their activities, were examined. Epidermal growth factor increases KITENIN-mediated AP-1 activity, and there is an inverse relationship between KAI and KITENIN [
26]. As shown in
Figure 8a, at a concentration of 5 μM,
1 suppressed AP-1 activity.
Figure 8b,c show that
1 suppressed the activity of the KITENIN 3′-UTR, while the KITENIN promoter did not show significant change.
Figure 8d,e indicates that the protein level of KITENIN decreased in A549, AGS, and Caco-2 during treatment. The mRNA expression level of KITENIN was also suppressed in A549, AGS, and Caco-2, whereas the mRNA level of KAI1 was increased by
1 (
Figure 8f). As a result,
1 decreased cell motility by downregulating the expression level of KITENIN while upregulating that of KAI1.
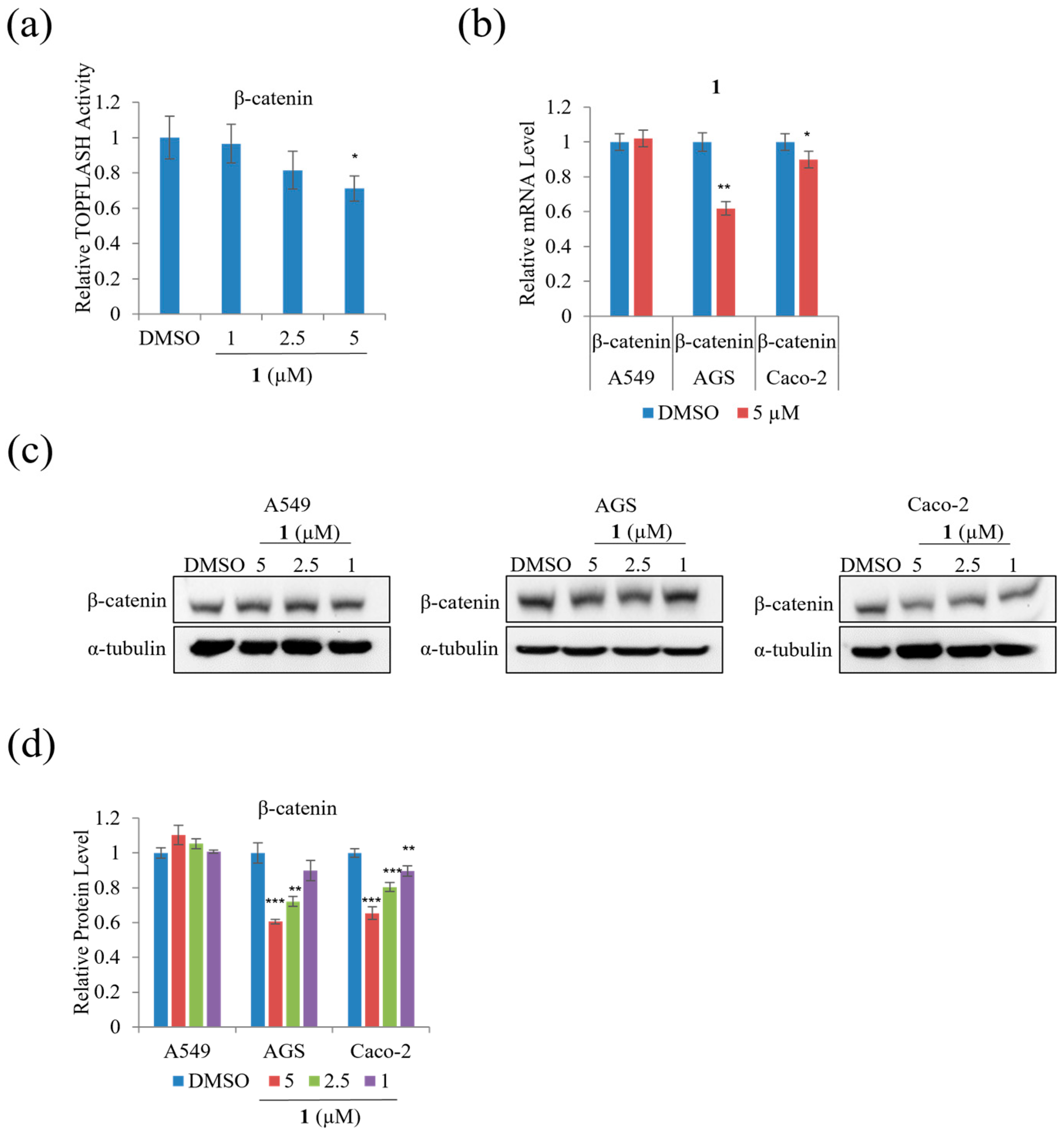
We performed TOPFLASH reporter assays to assess whether treatment with
1 modulates β-catenin-mediated and/or KITENIN-mediated signaling activity. Treatment with 5 µM of
1 significantly decreased β-catenin-mediated TOPFLASH activity on HEK293T by 30% (
Figure 9a). In addition,
Figure 9b–d indicates that treatment with
1 decreased mRNA expression and the protein level of β-catenin on AGS and Caco-2 cells; however, treatment with
1 did not significantly affect the mRNA expression and protein level of β-catenin in A549 cells.
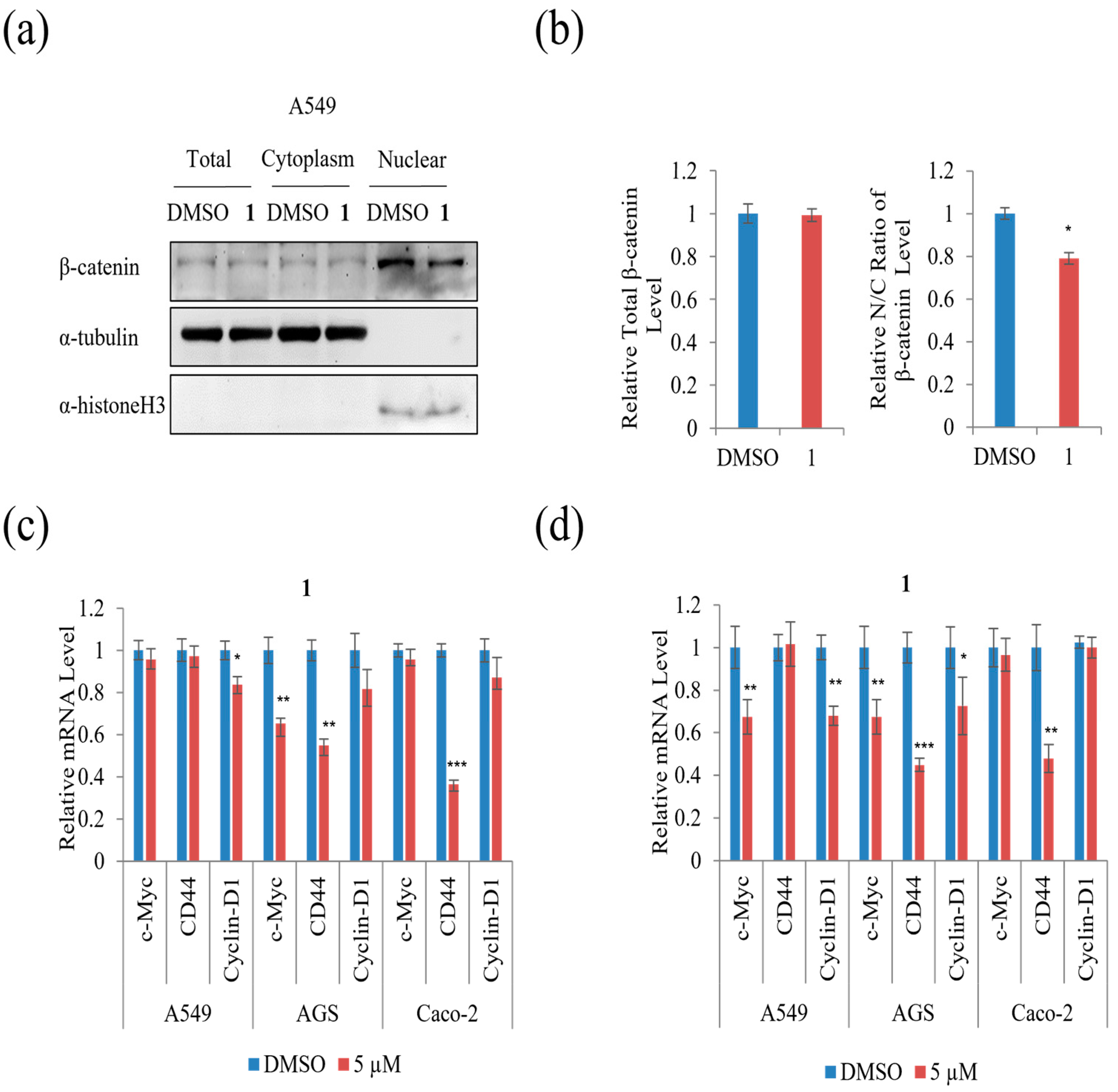
The protein level of total, cytoplasmic, and nuclear β-catenin was examined to test whether
1 affected the nuclear/cytoplasmic distribution of β-catenin in A549 cells. As shown in
Figure 10a,b, treatment with 5 µM of
1 did not affect the level of total β-catenin, whereas it decreased the β-catenin nuclear to cytoplasmic ratio remarkably in A549 cells compared to DMSO. To further test the effect of
1 on the levels of downstream target genes of β-catenin, qRT-PCR analysis was performed. As shown in
Figure 10c, treatment with 5 µM of
1 suppressed the mRNA expression of cyclin-D1 in A549 and CD44 in Caco-2, while treatment with 5 µM of
1 suppressed the mRNA expression of c-Myc and CD44 in AGS cells for 24 h. Moreover, the β-catenin downstream target genes, including c-Myc, CD44, and cyclin-D1, were suppressed in AGS cells during treatment with 5 µM of
1 for 48 h (
Figure 10d). Treatment with 5 µM of
1 suppressed the mRNA expression of cyclin-D1 and CD44 in A549, while 5 µM of
1 suppressed the mRNA expression of CD44 in Caco-2 cells for 48 h (
Figure 10d). These results show that
1 decreased β-catenin-mediated TOPFLASH activity and its downstream targets in three cancer cells.
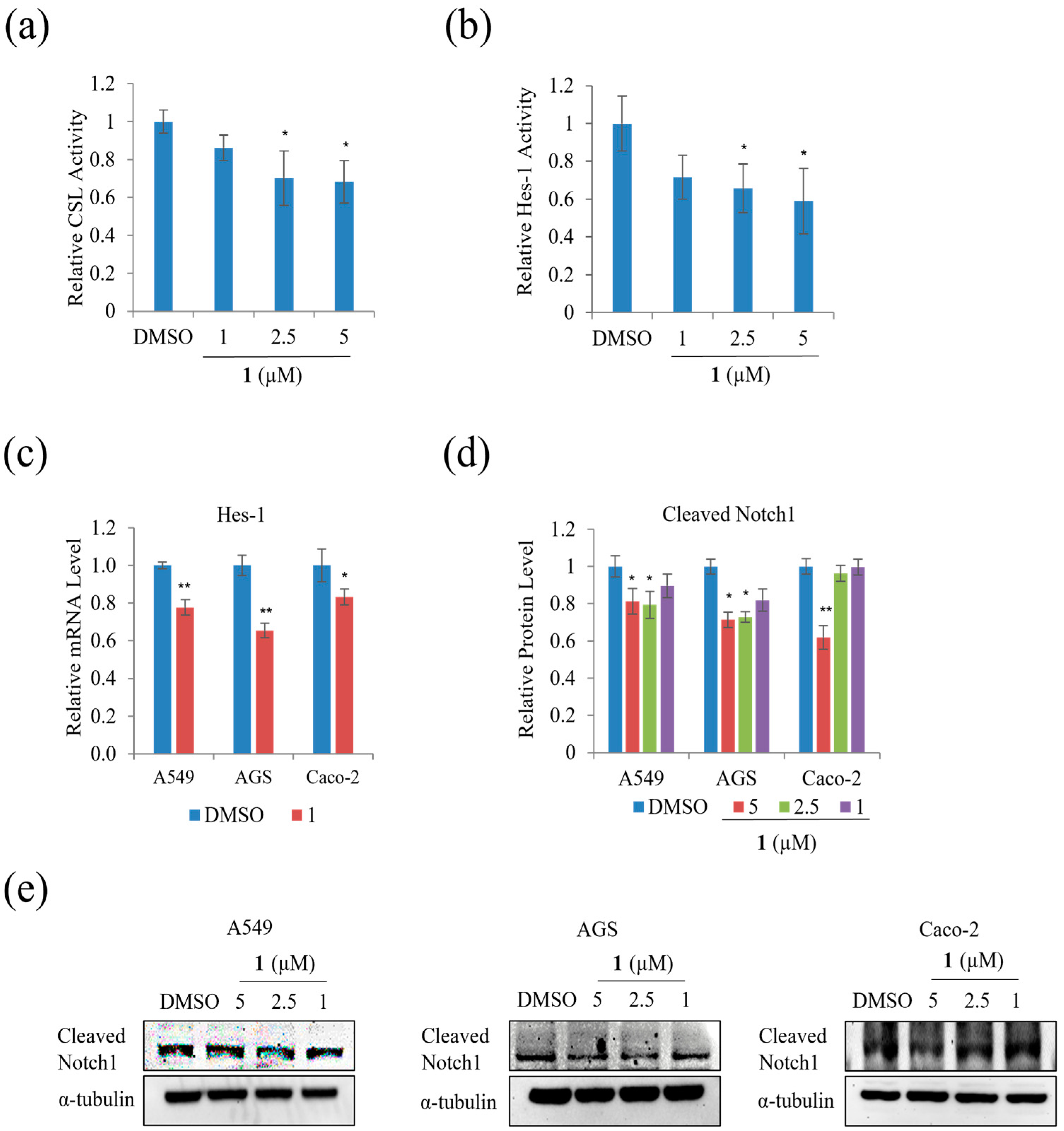
We examined the effect of
1 on the Notch signal pathway in A549, AGS, and Caco-2 cells and found that treatment with 5 µM of
1 significantly decreased the relative CSL activity in HEK293T cells by approximately 20% (
Figure 11a). In addition, the relative Hes-1 activity in HEK293T and mRNA expression of Hes-1 in AGS, A549, and Caco-2 cells decreased during treatment with 5 µM, as shown in
Figure 11b,c. The level of expression of HES correlated with NICD and CSL. Treatment with
1 decreased the level of Cleaved Notch1 in A549, AGS, and Caco-2 cells (
Figure 11d,e). This result indicates that
1 slightly suppressed the Notch signal pathway in A549, AGS, and Caco-2 cells.
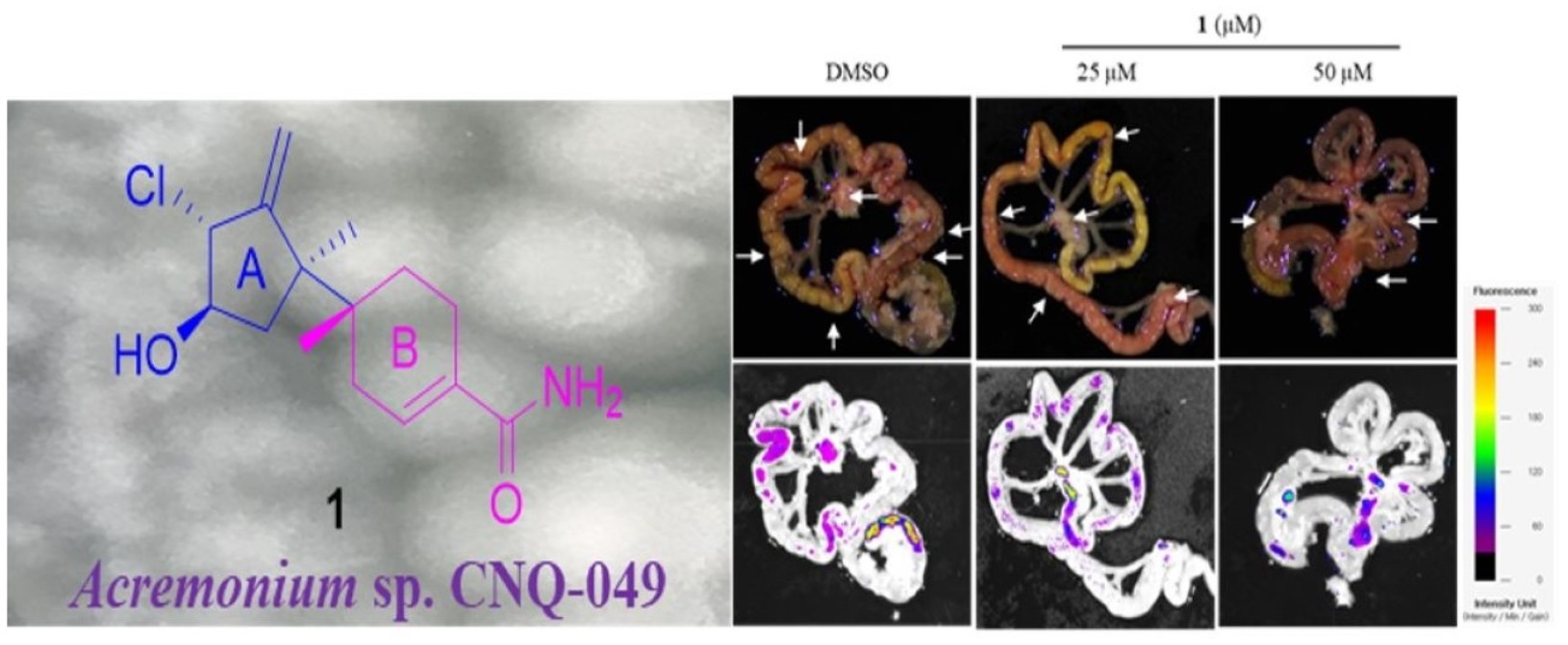
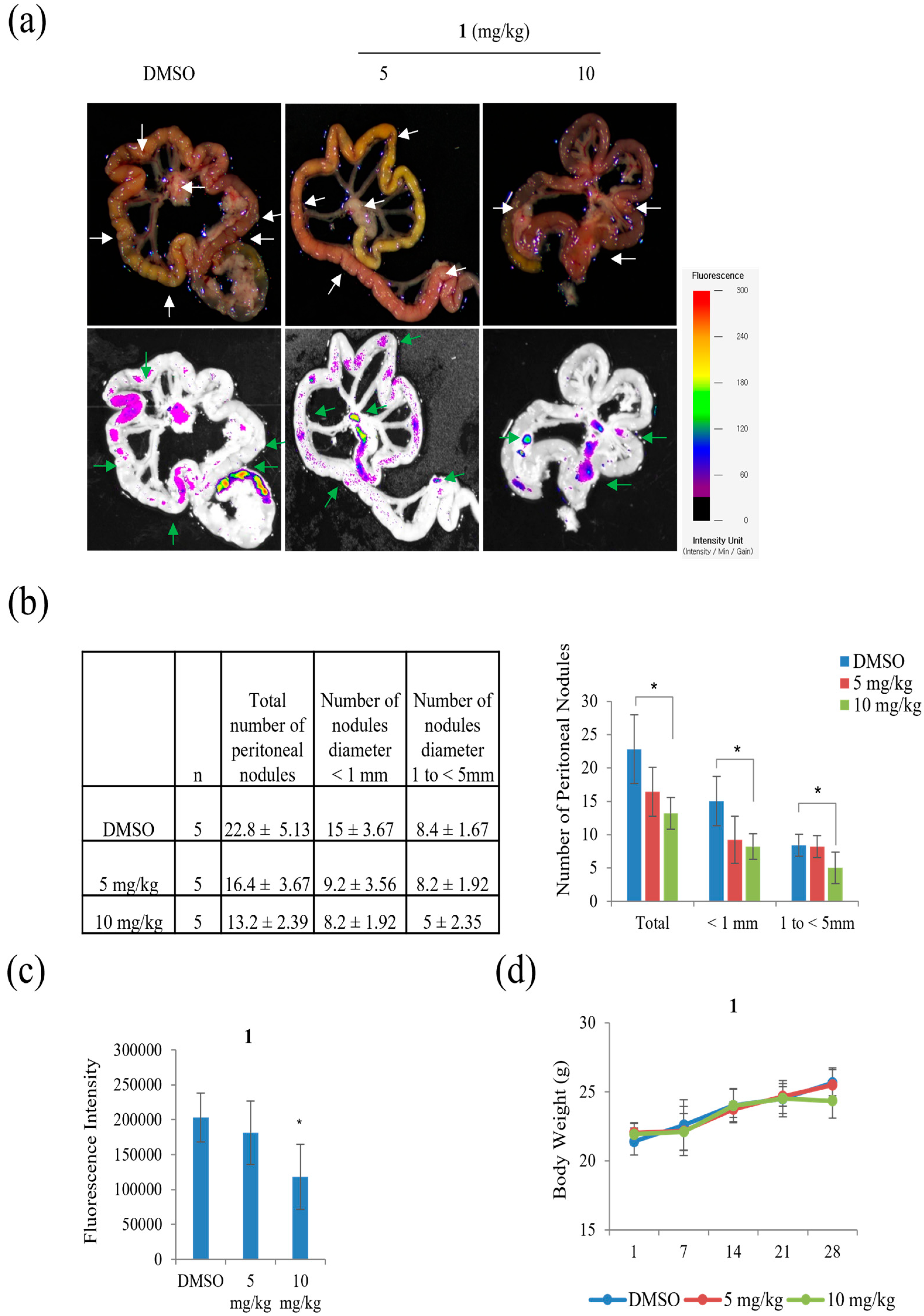
Peritoneal carcinomatosis occurs when gastric cancer metastasizes to the peritoneal cavity [
27]. Peritoneal carcinomatosis is a hallmark of advanced peritoneal tumor progression, and peritoneal recurrence from gastric cancer occurs due to resistance to chemotherapy. On day 28 after inoculation of AGS-iRFP, treatment of 10 mg/kg of
1 significantly reduced the three different categories of the number of tumor nodules in the mesentery compared to the control (
Figure 12). As shown in
Figure 12b, treatment with 10 mg/kg of
1 reduced the number of nodules with diameters >1 compared to other groups (control group, 8.4 ± 1.67 and 5 mg/kg of
1 group, 8.2 ± 1.92 versus 10 mg/kg of
1, 5 ± 2.35,
p < 0.05). The number of nodules with diameters ranging from 1 to < 5 mm in the group with 10 mg/kg of
1 was also lower than those in the other groups (control group, 8.4 ± 1.67 and 5 mg/kg of
1 group, 8.2 ± 1.92 versus 10 mg/kg of
1, 5 ± 2.35,
p < 0.05). As a result, the quantitative data showed that the total number of metastatic nodules was significantly reduced in the mice treated with 10 mg/kg of
1 compared to those in the control group (
Figure 12c).
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotation was acquired using a Kruss Optronic P-8000 polarimeter with a 5-cm cell. The UV spectrum was recorded in methanol (MeOH) on a Scinco UVS2100, and the VCD spectra were measured using a BioTools dualPEM ChiralIR spectrophotometer. The IR spectrum was collected on a Varian Scimitar Series. The 1H and 2D NMR spectra were recorded at 400 and 500 MHz in CD3OD, containing Me4Si as the internal standard on Varian Inova spectrometers. The 13C NMR spectra were acquired at 75 MHz on a Varian Inova spectrometer. The high-resolution mass spectrum was obtained on a JMS-700 (JEOL) mass spectrometer, and the low-resolution LC-MS data were measured using the Agilent Technologies 1260 quadrupole and Waters Micromass ZQ LC/MS system with a reversed-phase column (Phenomenex Luna C18 (2), 100 Å, 50 mm × 4.6 mm, 5 µm), at a flow rate of 1.0 mL/min, at the National Research Facilities and Equipment Center (NanoBioEnergy Materials Center) at Ewha Womans University. Medium-pressure liquid chromatography (MPLC) was performed on a Biotage Isolera One system (SE-751 03 Uppsala, Sweden), using Biotage SNAP KP-Sil, with a step gradient solvent of dichloromethane (DCM) and methanol (MeOH). The fractions were purified by reversed-phase high-performance liquid chromatography (HPLC) (Phenomenex Luna C18 (2), 100 Å, 250 nm × 10 mm, 5 μm).
3.2. Collection and Phylogenetic Analysis of the Strain CNQ-049
The marine-derived Acremonium sp. CNQ-049 was isolated from marine sediment collected off the coast of Southern California. The strain CNQ-049 was identified as Acremonium sp. with 99.4% similarity to that of Acremonium fusidiodes, based on 18S rRNA gene sequence analysis (GenBank accession number KP131520.1).
3.3. Fermentation, Extraction, and Isolation
Strain CNQ-049 was cultured in 80 × 2.5 L Ultra Yield flasks, each containing 1 L of the medium (10 g/L soluble starch, 2 g/L yeast, 4 g/L peptone, and 34.75 g/L sea salt dissolved in distilled water), and shaken at 120 rpm and 27 °C. After seven days of cultivation, the broth was extracted with ethyl acetate (EtOAc) (80 L overall), and the soluble fraction was dried in vacuo to afford 5 g of the crude extract. This was then separated on a MPLC silica-gel column (Biotage® SNAP Cartridge, KP-SIL), with step gradients of MeOH/DCM (0 to 100%), to obtain 10 fractions, which were labeled as Q049-1−Q049-10. Fraction Q049-6 (762 mg) was separated into six subfractions, labeled Q049-6-A−Q049-6-F, by C18 reversed-phase column chromatography with 37% aqueous acetonitrile. Subfraction Q049-6-B (221 mg) was further purified by reversed-phase HPLC (Phenomenex Luna C18 (2), 250 × 100 mm, 2.0 mL/min, 5 μm, 100 Å, UV = 210 nm) with 38% CH3CN to obtain 10.7 mg of marinobazzanan (1).
Marinobazzanan (
1): pale-yellow amorphous powder;
−29 (c 0.0625, MeOH); UV (MeOH) λ
max (log
ε) 205 (3.73) nm; IR (KBr) ν
max 3338, 2964, 2935, 2360, 1669, 1373, 1082 cm
−1;
1H NMR and 2D NMR (400 MHz and 500 MHz, CD
3OD) see
Table 1; HR-ESI-MS
m/z 284.1415 [M + H]
+ (calcd for C
15H
2235ClNO
2, 284.1417).
3.4. MTPA Esterification of Marinobazzanan
To obtain the (S)- and (R)-MTPA esters, 2.0 mg of 1 was completely dried under high vacuum for 12 h and dissolved in dried pyridine (0.6 mL). Catalytic amounts of crystalline 4-dimethylaminopyridine (DMAP) were added and, respectively, treated with (R)-and (S)-α-methoxy trifluoromethyl-phenylacetic acid (MTPA) chloride (6 μL). The mixtures were stirred for 12 h at 50 °C and purified by reversed-phase HPLC (Phenomenex Luna C18 (2), 250 × 100 mm, 2.0 mL/min, 5 μm, 100 Å), with 90% aqueous acetonitrile, to afford 0.8 mg and 0.7 mg of (S)- and (R)-MTPA esters of 1, respectively.
(S)-MTPA ester of 1: 1H NMR (500 MHz, in CDCl3) δH 0.88 (3H, s, H-14), 1.23 (3H, s, H-13), 1.41–1.61 (2H, overlapped, H-1), 1.86 (1H, dd, J = 13.0, 10.0 Hz, H-11a), 2.02 (1H, dd, J = 13.0, 7.5 Hz, H-11b), 1.99 (1H, m, H-5a), 2.12 (1H, m, H-5b), 2.21 (1H, m, H-2a), 2.29 (1H, m, H-2b), 4.46 (1H, dt, J = 10.0, 2.7 Hz, H-9), 5.22 (1H, d, J = 2.7 Hz, H-15a), 5.24 (1H, dt, J = 10.0, 7.5 Hz, H-10), 5.52 (1H, d, J = 2.7 Hz, H-15b), 6.55 (1H, br. s, H-4); LR-ESI-MS m/z 457.3 [M − CONH2 + H]+.
(R)-MTPA ester of 1: 1H NMR (500 MHz, in CDCl3) δH 0.84 (3H, s, H-14), 1.22 (3H, s, H-13), 1.38−1.60 (2H, overlapped, H-1), 1.67 (1H, dd, J = 13.0, 10.0 Hz, H-11a), 2.00 (1H, dd, J = 13.0, 7.5 Hz, H-11b), 1.95 (1H, m, H-5a), 2.07 (1H, m, H-5b), 2.18 (1H, m, H-2a), 2.26 (1H, m, H-2b), 4.50 (1H, dt, J = 10.0, 2.7 Hz, H-9), 5.20 (1H, dt, J = 10.0, 7.5 Hz, H-10), 5.22 (1H, d, J = 2.7 Hz, H-15a), 5.54 (1H, d, J = 2.7 Hz, H-15b), 6.52 (1H, br. s, H-4); LR-ESI-MS m/z 457.3 [M − CONH2 + H]+.
3.5. VCD Analysis and Calculations
The conformational assignments for the C-6 and C-7 positions of 1 were performed using the Macromodel software (Version 9.9, Schrodinger LLC.) with “Mixed torsional/Low-mode sampling” in the GAFF force field. The experiments were conducted in the gas phase with the 50 kJ/mol energy window limit and a maximum of 10,000 steps to thoroughly examine all low-energy conformers. The Polak–Ribière conjugate gradient method was utilized for the minimization processes with 10,000 maximum iterations and a 0.001 kJ (mol Å)−1 convergence threshold on the root mean square gradient. Conformers within 10 kJ/mol of each global minimum for the 6R,7R, and 6S,7S forms of 1 were used for calculating the gauge-independent atomic orbital shielding constant, without geometric optimization, by employing the TmoleX Version 4.2.1 software (COSMOlogic GmbH & Co. KG) at the B3LYP/6-31 + G(d) level in the gas phase. A sample of 1 (5.0 mg) was dissolved in CDCl3 (150 μL) and placed in a BaF2 cell with a path length of 100 μm, and data were acquired on a BioTools dualPEM ChiralIR spectrophotometer. The spectra were collected in 12 blocks, and each block was acquired for 3120 scans.
3.6. Cell Culture
The human cancer-cell lines A549 (lung cancer), AGS (gastric cancer), and Caco-2 (colorectal cancer) were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium or Dulbecco’s Modified Eagle Medium (DMEM) (Gen Depot, Barker, TX, USA), supplemented with 10% FBS and 1% penicillin–streptomycin solution in a humidified atmosphere containing 5% CO
2 at 37 °C [
28].
3.7. Methyl Thiazolyl Tetrazolium (MTT) Assay
Marinobazzanan (
1) was dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, St.Louis, MO, USA) and diluted to four concentrations (10, 25, 50, and 10 µM). The cells were seeded on 96-well plates (3 × 10
3 cells/well) for 12–16 h and then treated with 10, 25, 50, and 10 µM of compound
1 for 48 h [
29]. After 4 h of incubation with MTT in 5% CO
2 at 37 °C, the cells were lysed with 150 µL of DMSO (Sigma-Aldrich), and the absorbance was measured at 570 nm using a spectrophotometer (Bio Tek Instruments, Winooski, VT, USA).
3.8. Invasion Assay
The invasion of cancer cells was measured using Transwell chambers (Corning, New York, NY, USA) [
30] containing polycarbonate membranes with 8 µm pores coated with 1% gelatin. The AGS (3 × 10
5), A549 (3 × 10
5), and Caco-2 (2.5 × 10
5) cells were seeded in a culture medium containing 0.2% bovine serum albumin (BSA) and incubated with 1, 2.5, and 5 µM of compound
1 or DMSO control for 24 h. The lower chamber was filled with 600 μL DMEM/RPMI containing 0.2% BSA and 10 μg/mL fibronectin (EMD Millipore Corp., Billerica, MA, USA) as a chemoattractant. After 24 h of incubation, the invading cells were fixed using a Diff-Quik kit (Sysmex, Kobe, Japan). The number of cells was quantified using a Nikon Eclipse 400 fluorescence microscope (Nikon Instech, Co., Ltd., Kawasaki, Japan) and i-Solution FL Auto Software (IMT i-Solution Inc., Vancouver, QC, Canada; five fields/chamber).
3.9. Migration Assay
Migration assays were performed in non-coated Transwell chambers [
30]. The cells were seeded at a density of 2.5–3 × 10
5 cells/well in RPMI 1640/DMEM containing 0.2% BSA in the upper compartment of the chamber. The lower chamber was filled with 600 μL RPMI 1640/DMEM containing 0.2% BSA and fibronectin as a chemoattractant. The cells were cultured either in the absence or presence of compound
1 (1, 2.5, and 5 µM) for 24 h and were fixed using a Diff-Quick kit. The cells in the upper chamber were counted using a Nikon Eclipse 400 fluorescence microscope (Nikon Instech, Co., Ltd.) and i-Solution FL Auto Software (IMT i-Solution Inc.; five fields/chamber)
3.10. Soft Agar Colony Formation Assay
The cancer cells were suspended at a density of 2.5–3 × 10
3 cells/well in 1.0 mL of soft agar (0.35% soft-agar solution diluted 2-fold with 2 × DMEM/RPMI) and planted onto 1 mL of soft agar (0.5% agarose solution diluted 2-fold with 2 × DMEM/RPMI) in a 12-well plate and cultured for three weeks [
31]. The cells were fed twice per week with cell-culture media, compound
1 (1, 2.5, and 5 µM), or DMSO. The surface areas of the colonies in the five fields per well were estimated using a Nikon Eclipse 400 fluorescence microscope (Nikon Instech, Co., Ltd.) and i-Solution FL Auto Software (IMT i-Solution Inc.; five fields/chamber). Three replications were performed.
3.11. Quantitative Real-Time PCR
The total RNA was isolated from A549, AGS, and Caco-2 cells using RNAiso Plus (Takara, Otsu, Japan) according to the manufacturer’s instructions. Moloney murine leukemia virus reverse transcriptase (Invitrogen, Carlsbad, CA, USA) was used to convert 1 μg of RNA into cDNA. The dye SYBR Green (Enzynomics, Seoul, Republic of Korea) was used to analyze relative gene expression. Further, the qRT-PCR reaction and analysis were performed using CFX (Bio-Rad, Hercules, CA, USA).
3.12. Western Blotting
The A549, AGS, and Caco-2 cells were treated with 1 for 24 h, and washed twice with ice-cold phosphate-buffered saline (PBS). Lysis buffer was used for extraction. In some experiments, cytoplasmic and nuclear extracts were separated with the NE-PER nuclear and cytoplasmic extraction kit (Pierce Biotechnology, USA), and the extracted protein was separated using SDS-PAGE. The density of the bands was measured using the Multi Gauge 3.0 (Fujifilm, Tokyo, Japan) software, and the bands’ relative density was calculated based on the density of the control bands during loading in each sample.
3.13. Reporter Assay
HEK293T was seeded in 24-well plates. Following attachment, the cells were transfected with TOPFLASH, AP1, KITENIN, CSL, HES, NF-κb, KITENIN 3′-untranslated region (3′-UTR) reporters with renilla luciferase reporter plasmid (pRL-TK). After 12 h transfection of these reporters, the cells were treated with 1 µM, 2.5 µM, and 5 µM of compound 1. The Luciferase activity was calculated using the Dual-Luciferase Reporter Assay (Promega, Wisconsin, USA) and normalized to Renilla luciferase.
3.14. Lentiviral Transduction
The human gastric-cancer AGS cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium (Gen Depot, Barker, TX, USA), supplemented with puromycin solution. After 12 h, the lentiviral vector was transfected into the human gastric-cancer cell line by polybrene, and the returned cells were incubated. After 72 h, the single cells were then plated in individual wells of a 96-well plate and incubated for 7–10 days.
3.15. Animal Studies
The AGS-iRFP cell suspension (1 × 107cells in 0.1 mL PBS per mouse) was implanted into the abdominal cavity of male BALB/c nude mice (five weeks old), obtained from Central Lab. Animal Inc. (Seoul, Korea). All in-vivo experiments were performed according to the Guiding Principles for the Care and Use of Animals (DHEW publication, NIH 80-23) and were approved by the Sunchon National University Research Institutional Animal Care and Use Committee. The mice were randomly assigned to one of three groups: control, 5 mg/kg of 1, and 10 mg/kg of 1 by IP treatment. All animals were examined by measuring weight change. The treatment was initiated a week after the AGS-iRFP cells were injected. On day 28 after tumor inoculation, all the mice were sacrificed. The fluorescence area of the images alongside the representative images was then obtained through a fluorescence-labeled organism bioimaging instrument system.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}