
Marine Puupehenone and Puupehedione: Synthesis and Future Perspectives


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
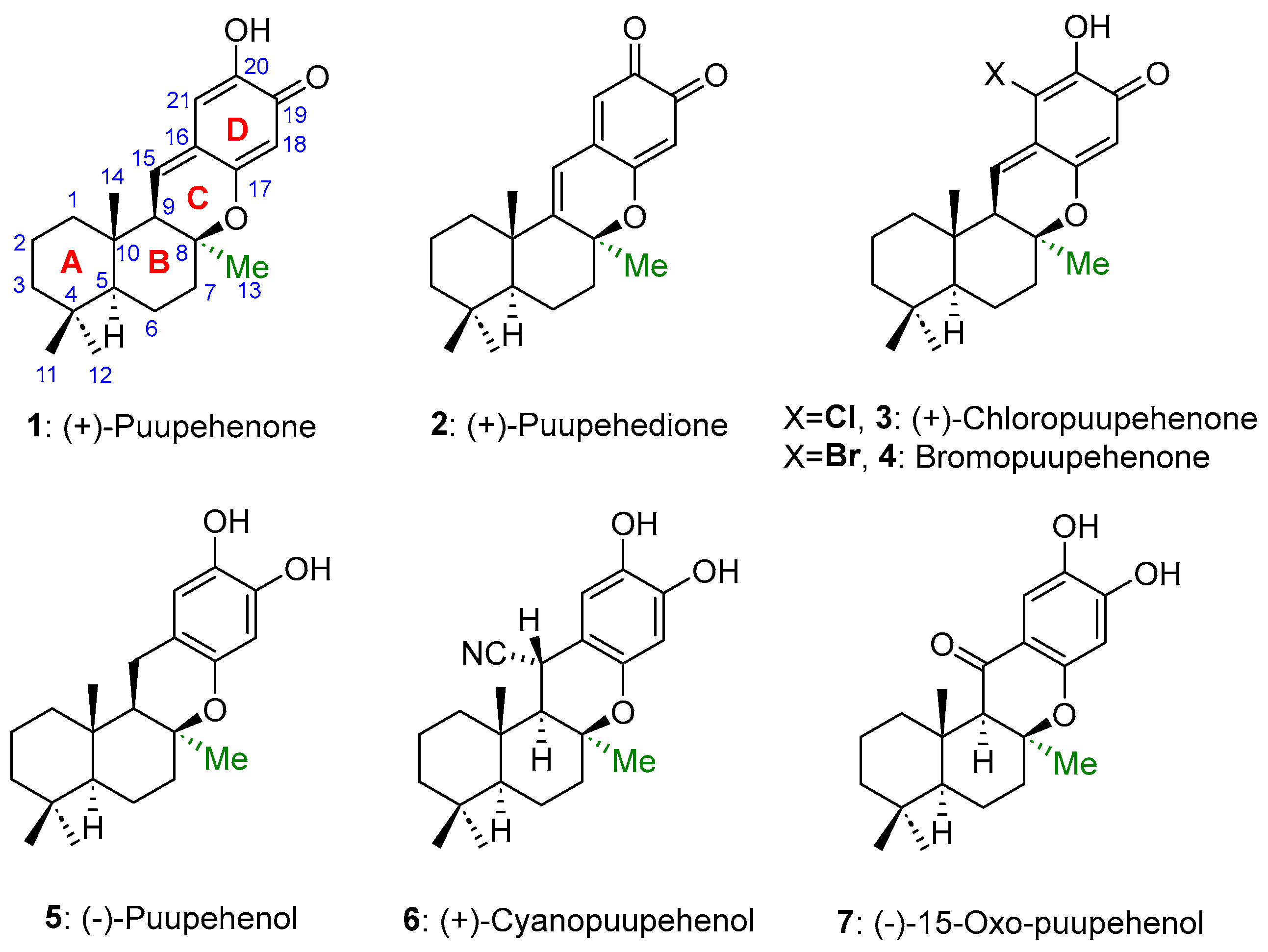
:1. Introduction
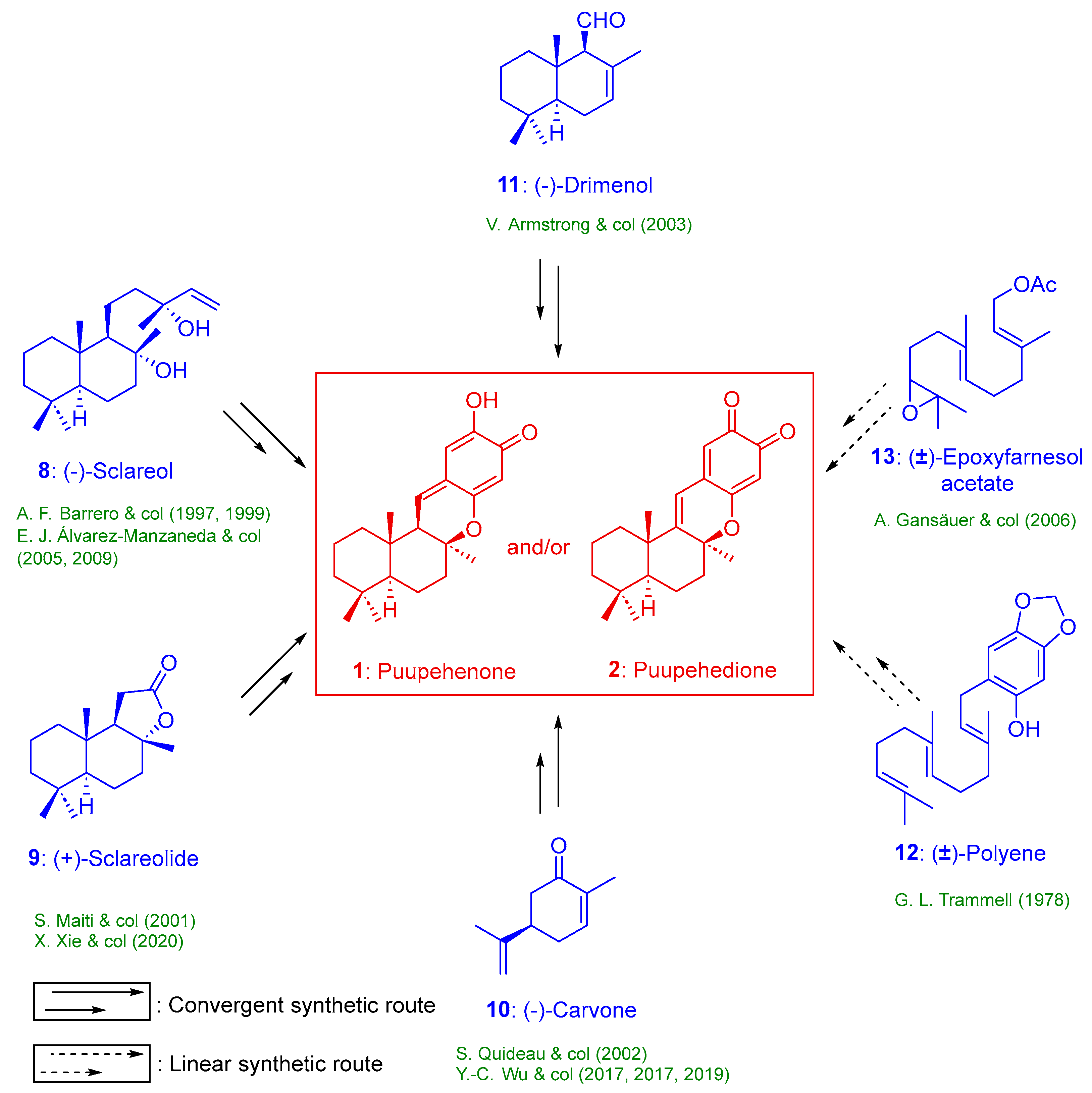
2. Results
2.1. Synthesis of Puupehenone (1) and Puupehedione (2) from Chiral Starting Materials
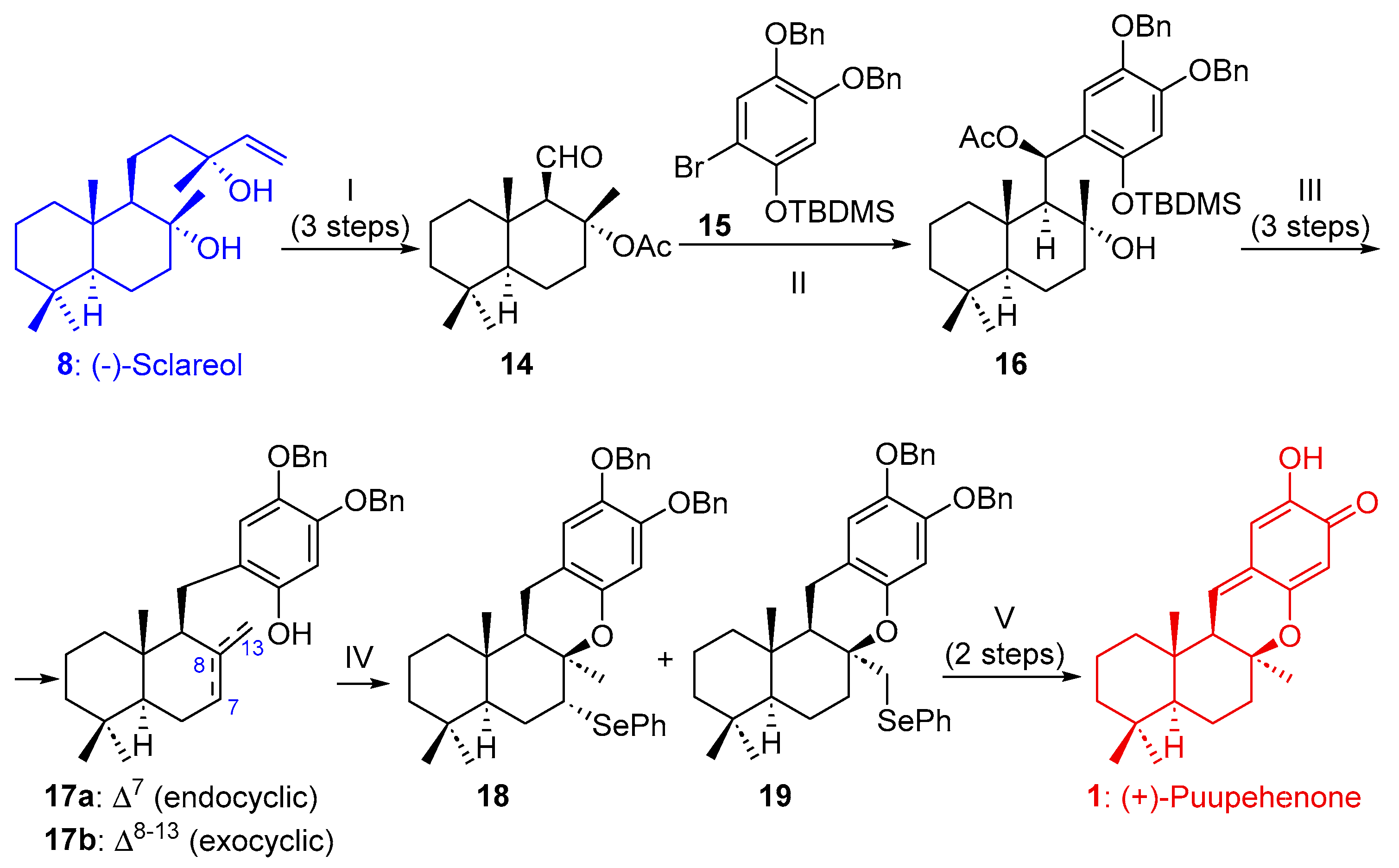
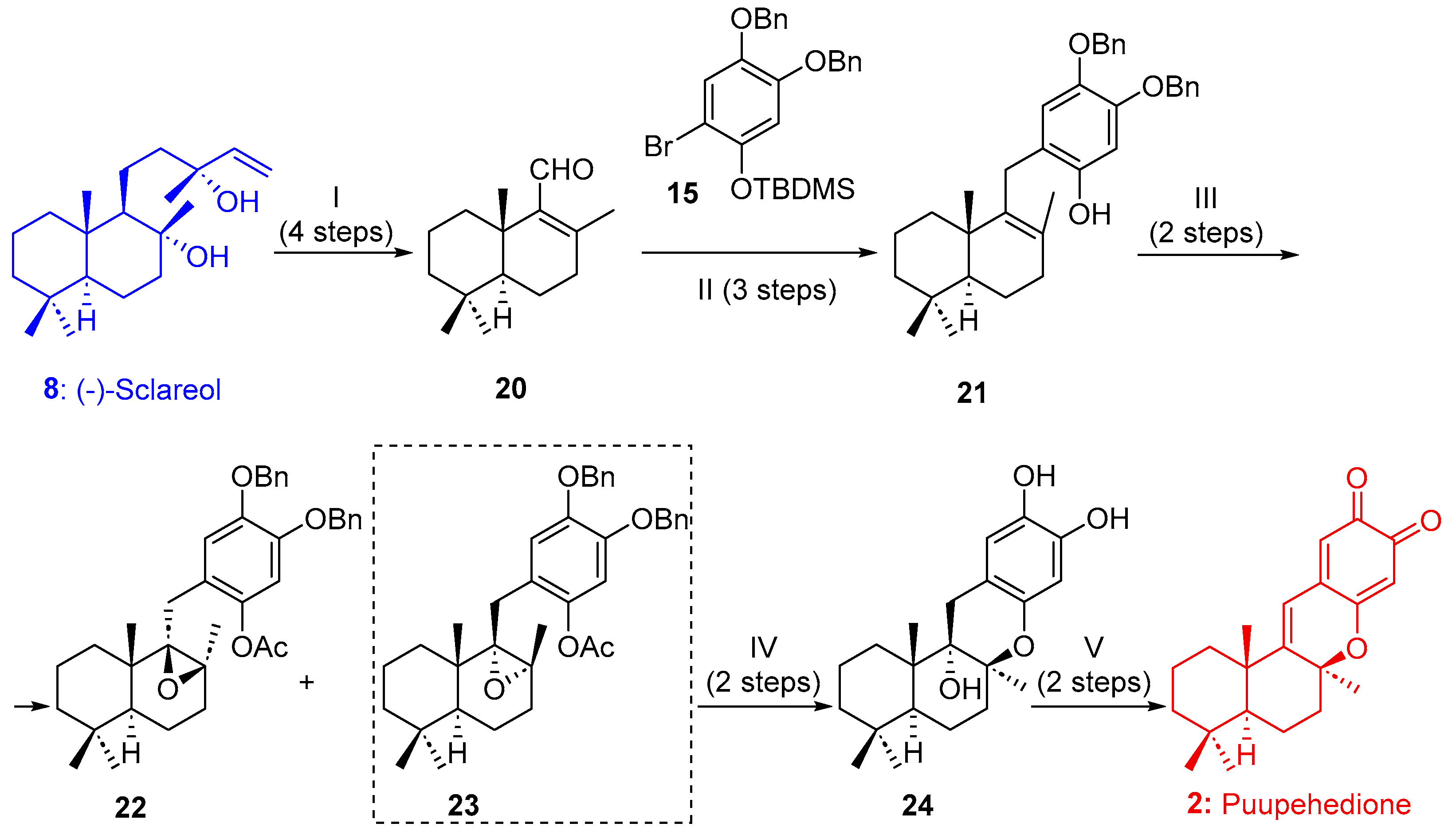
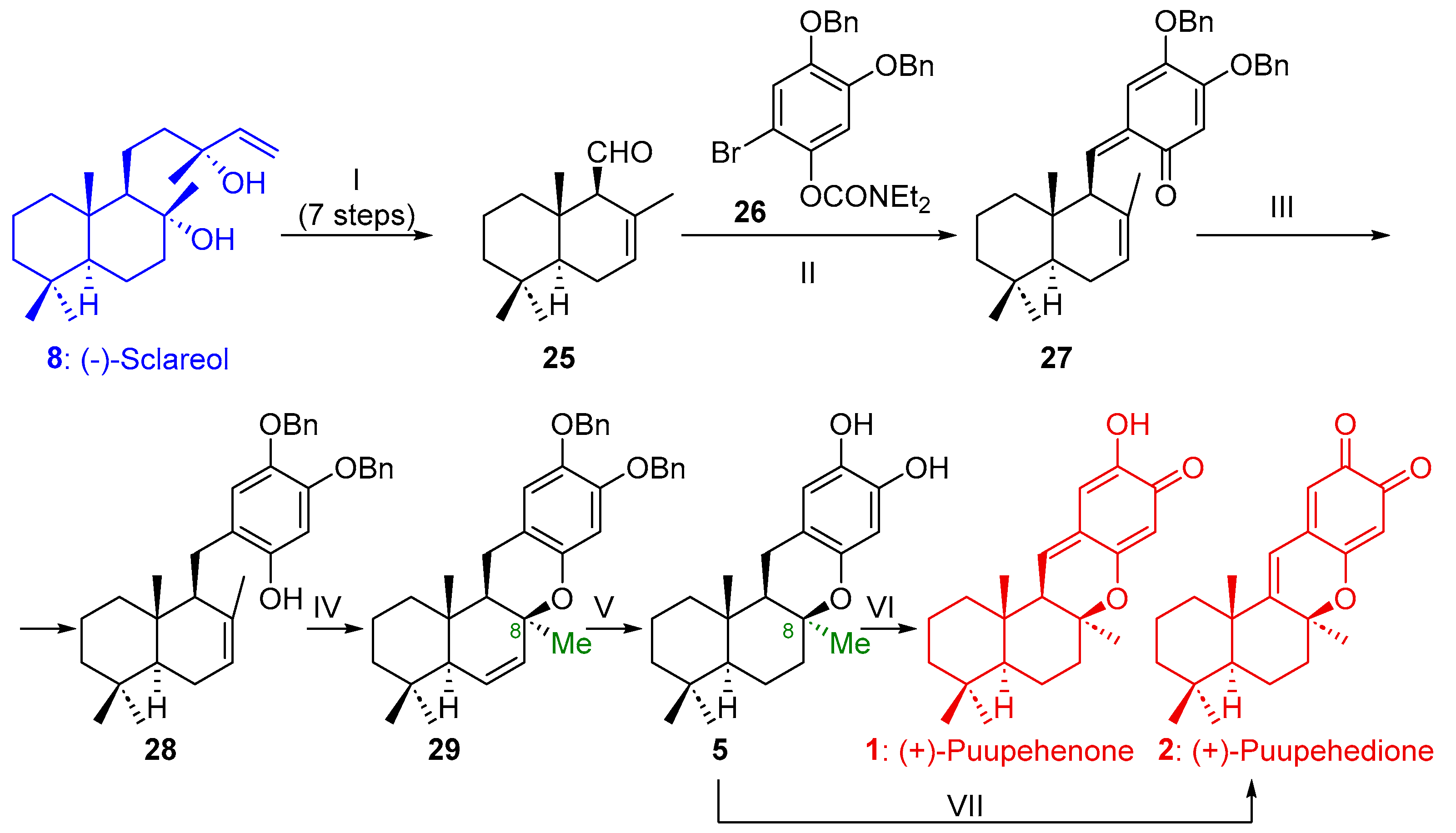
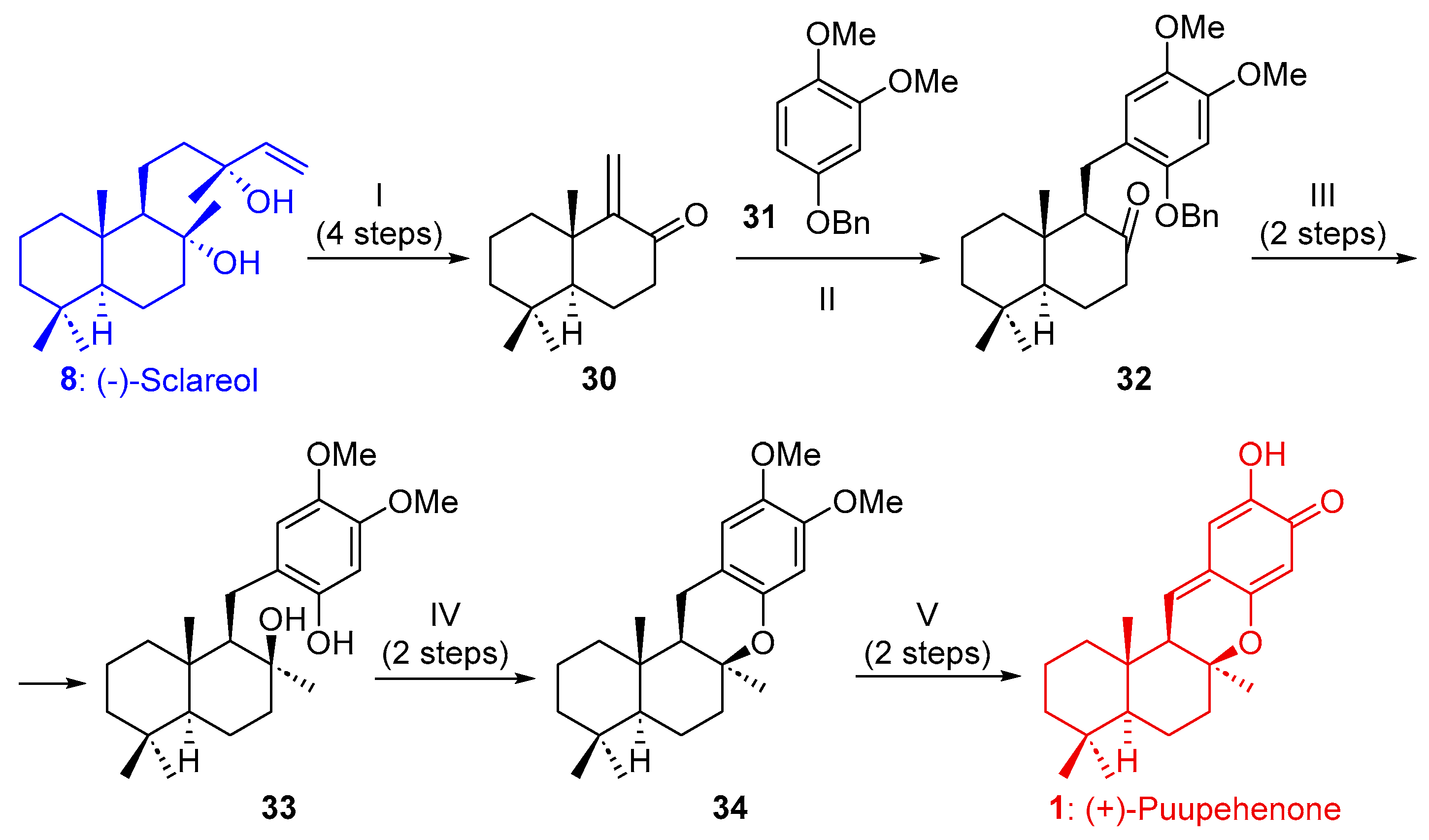
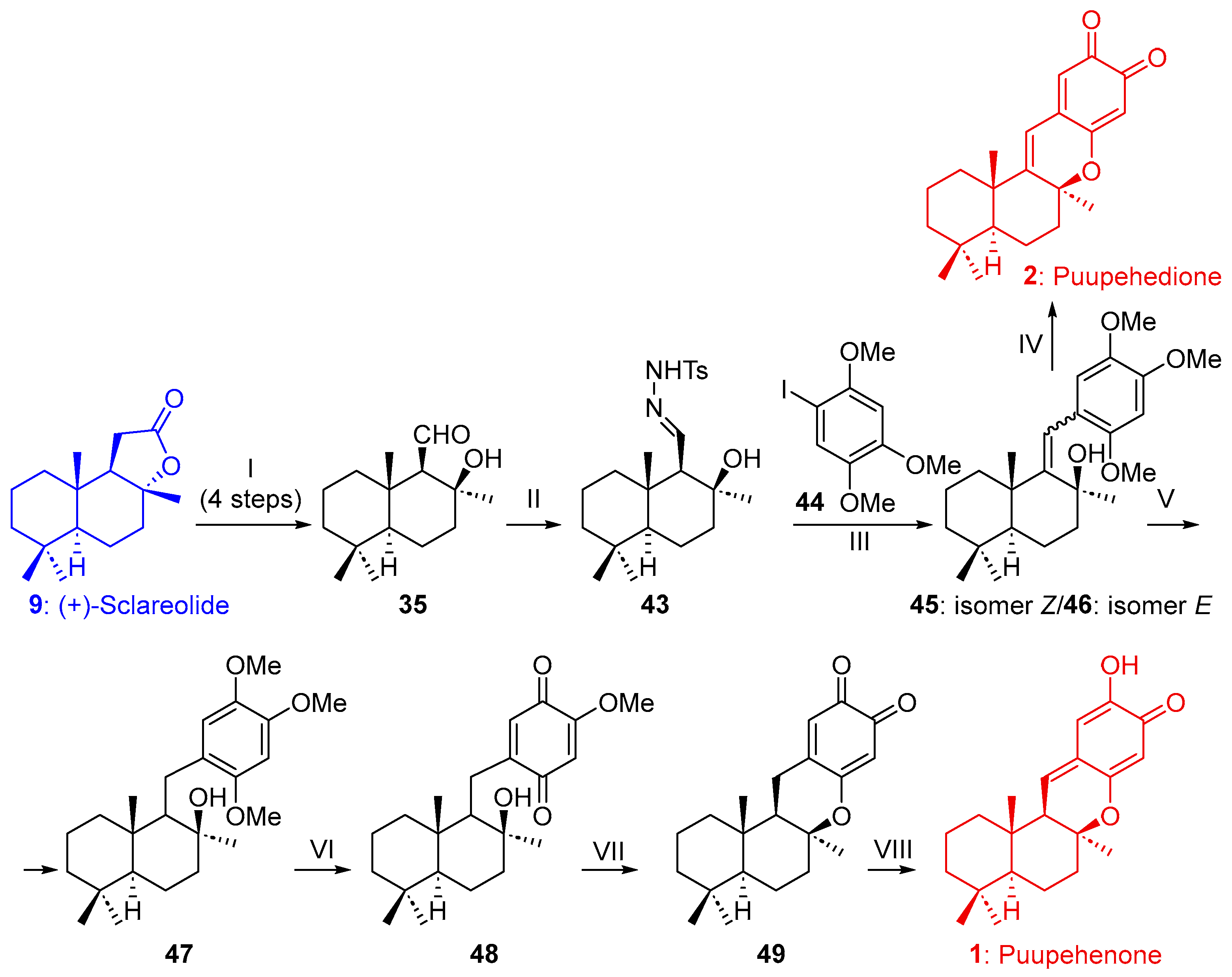
2.1.1. Synthesis of Puupehenone (1) and Puupehedione (2) from (-)-Sclareol (8)
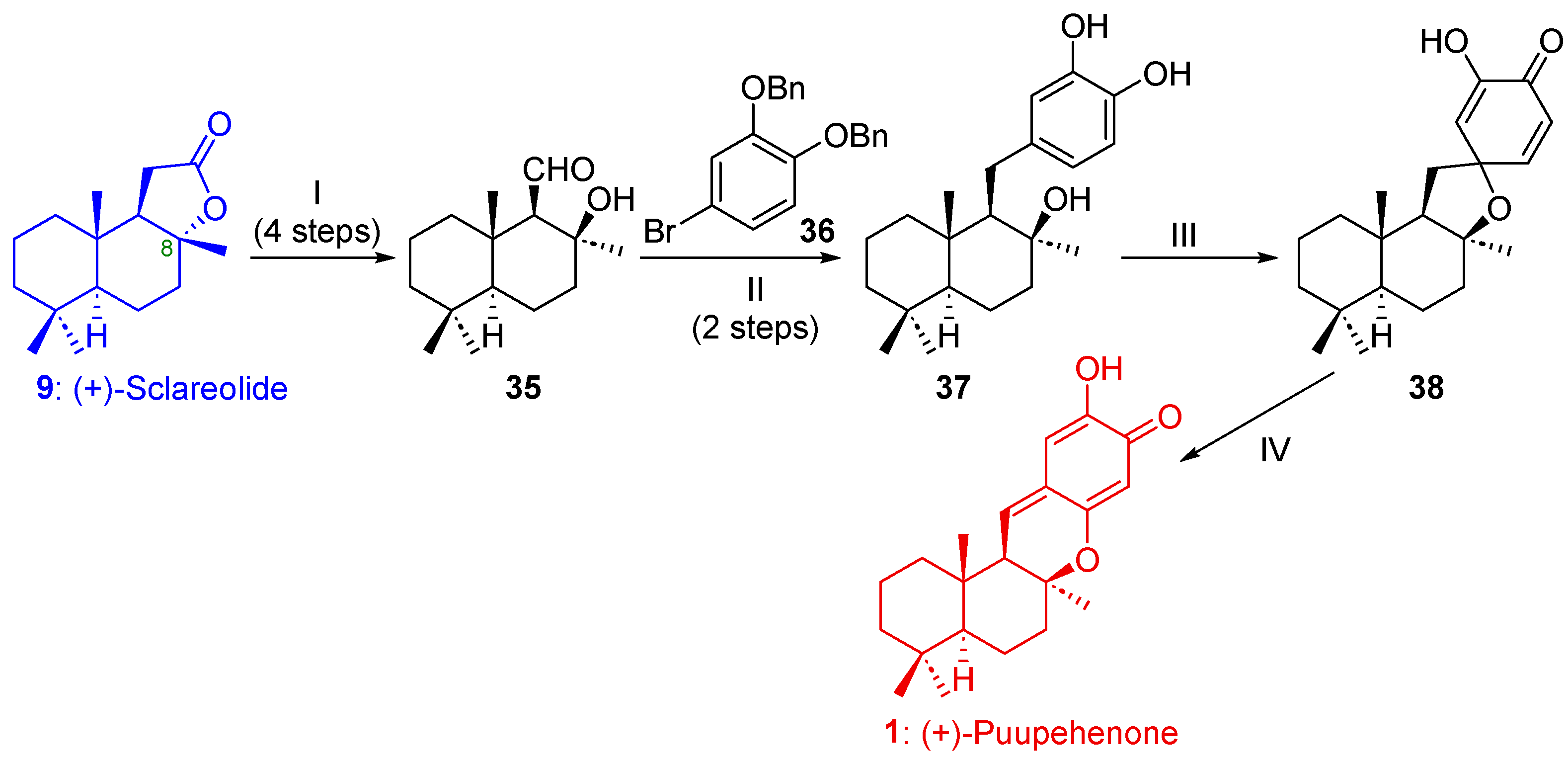
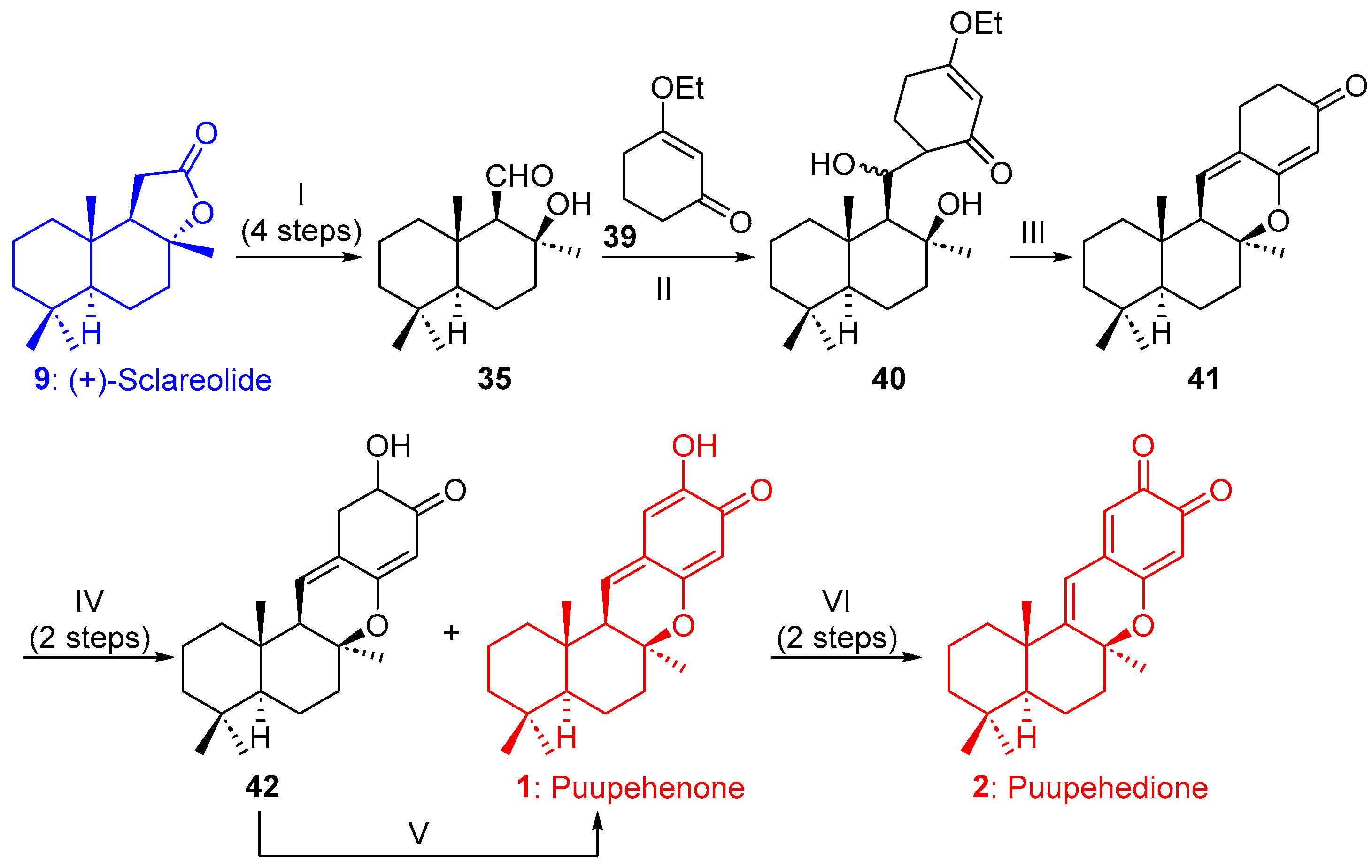
2.1.2. Synthesis of Puupehenone (1) and/or Puupehedione (2) from (+)-Sclareolide (9)
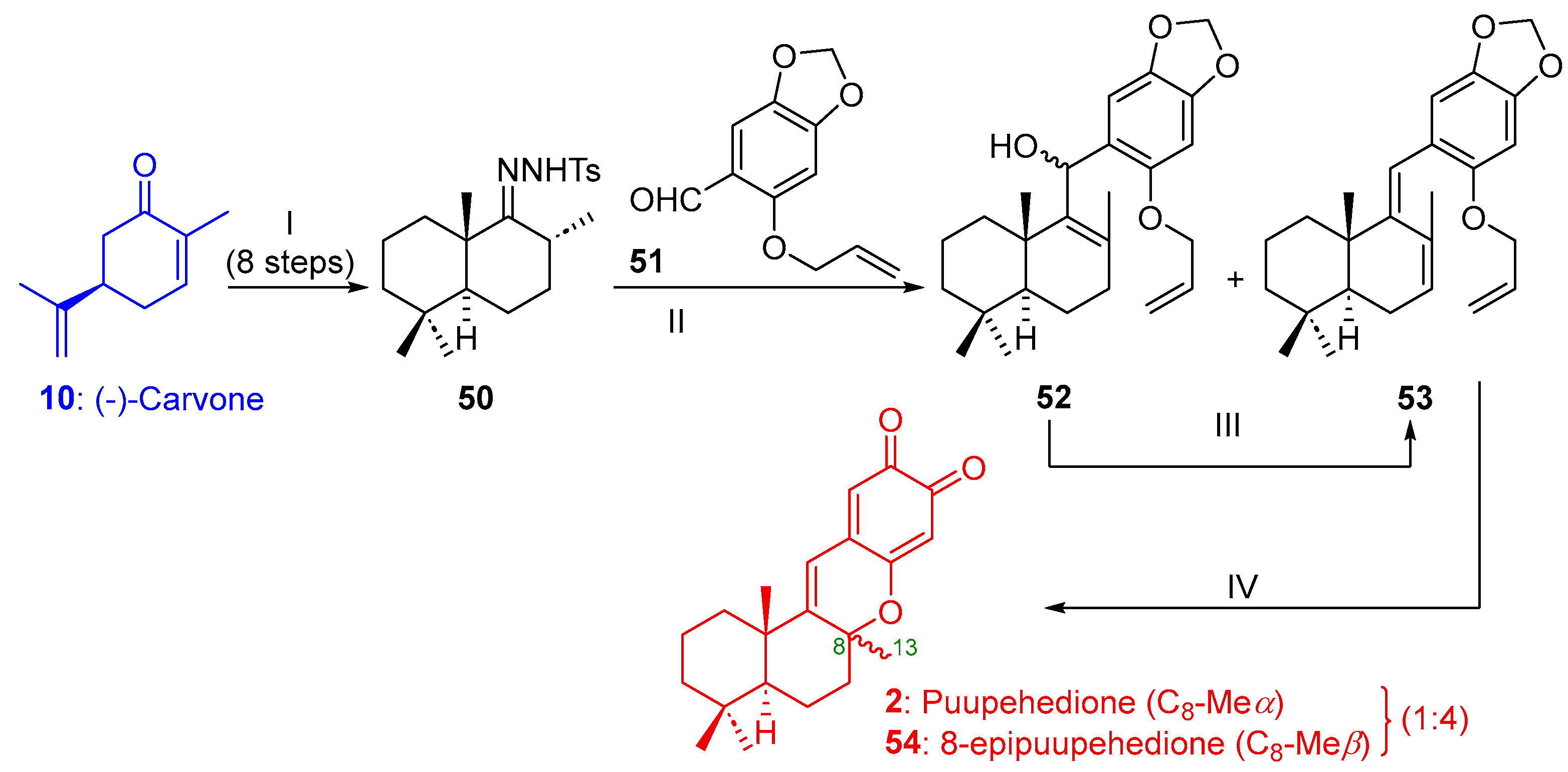
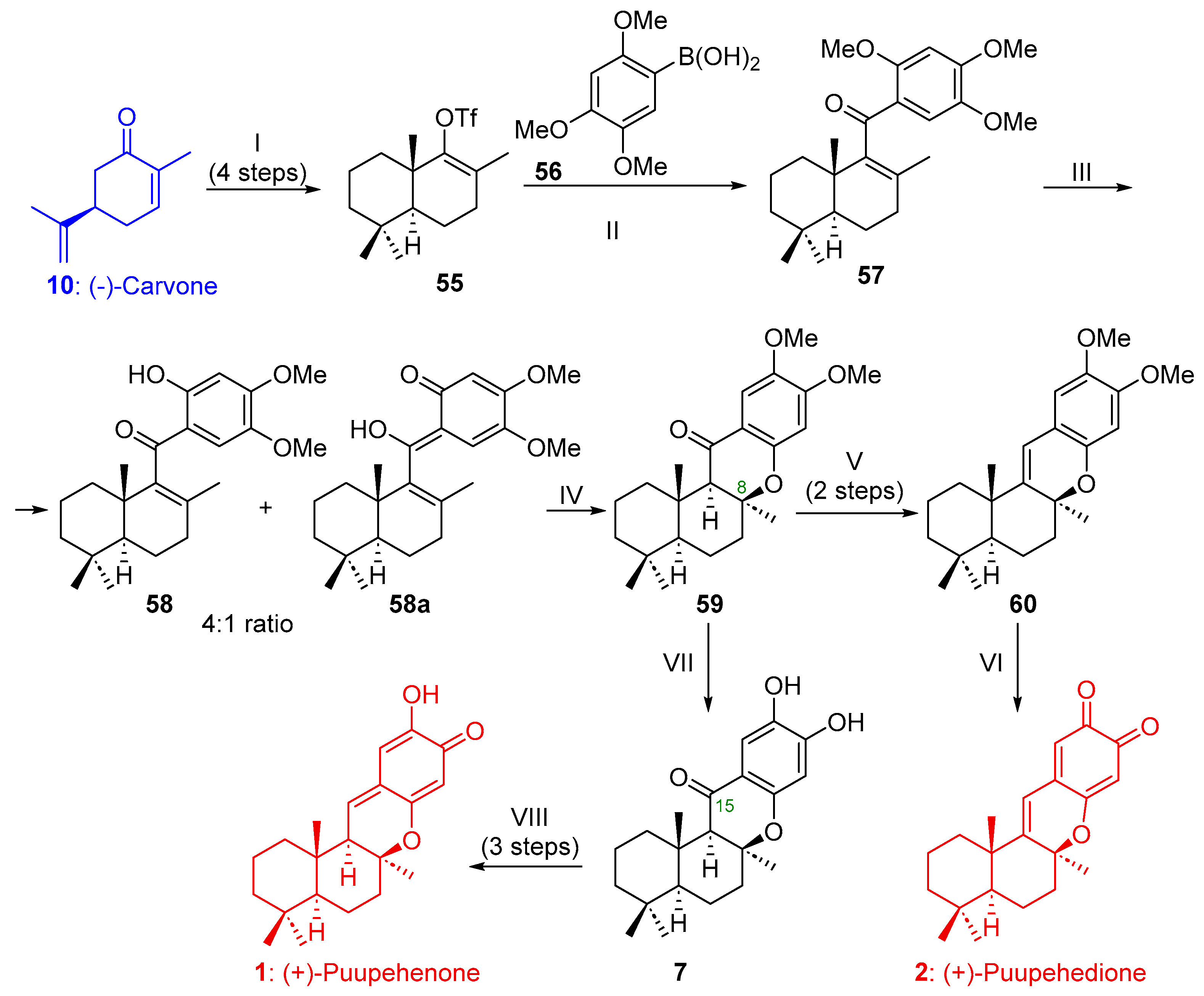
2.1.3. Synthesis of (+)-Puupehenone (1) and/or (+)-Puupehedione (2) from (-)-Carvone (10)
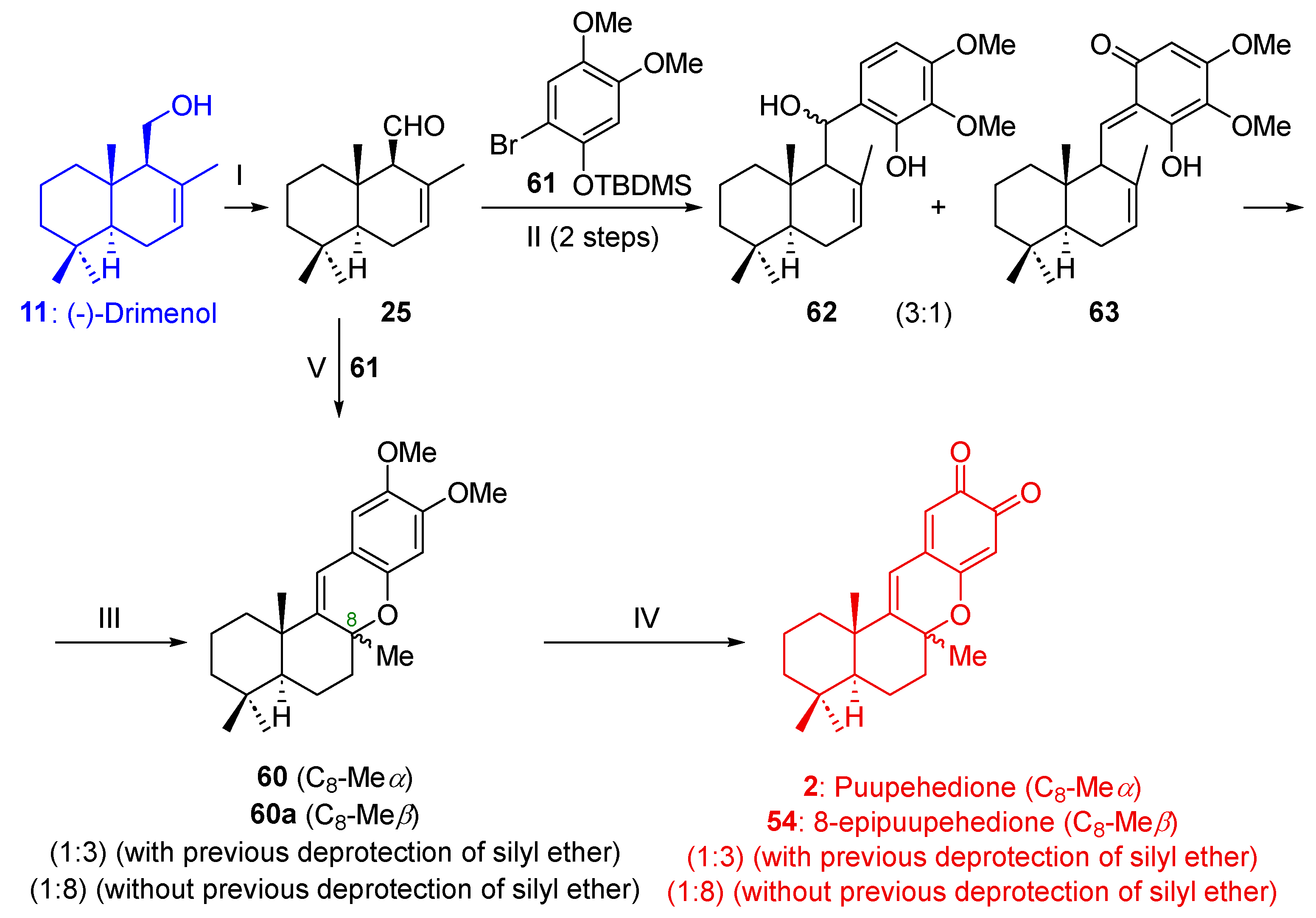
2.1.4. Synthesis of Puupehedione (2) from (-)-Drimenol (11)
2.2. Synthesis of Puupehenone (1) and Puupehedione (2) from Non-Chiral Starting Material
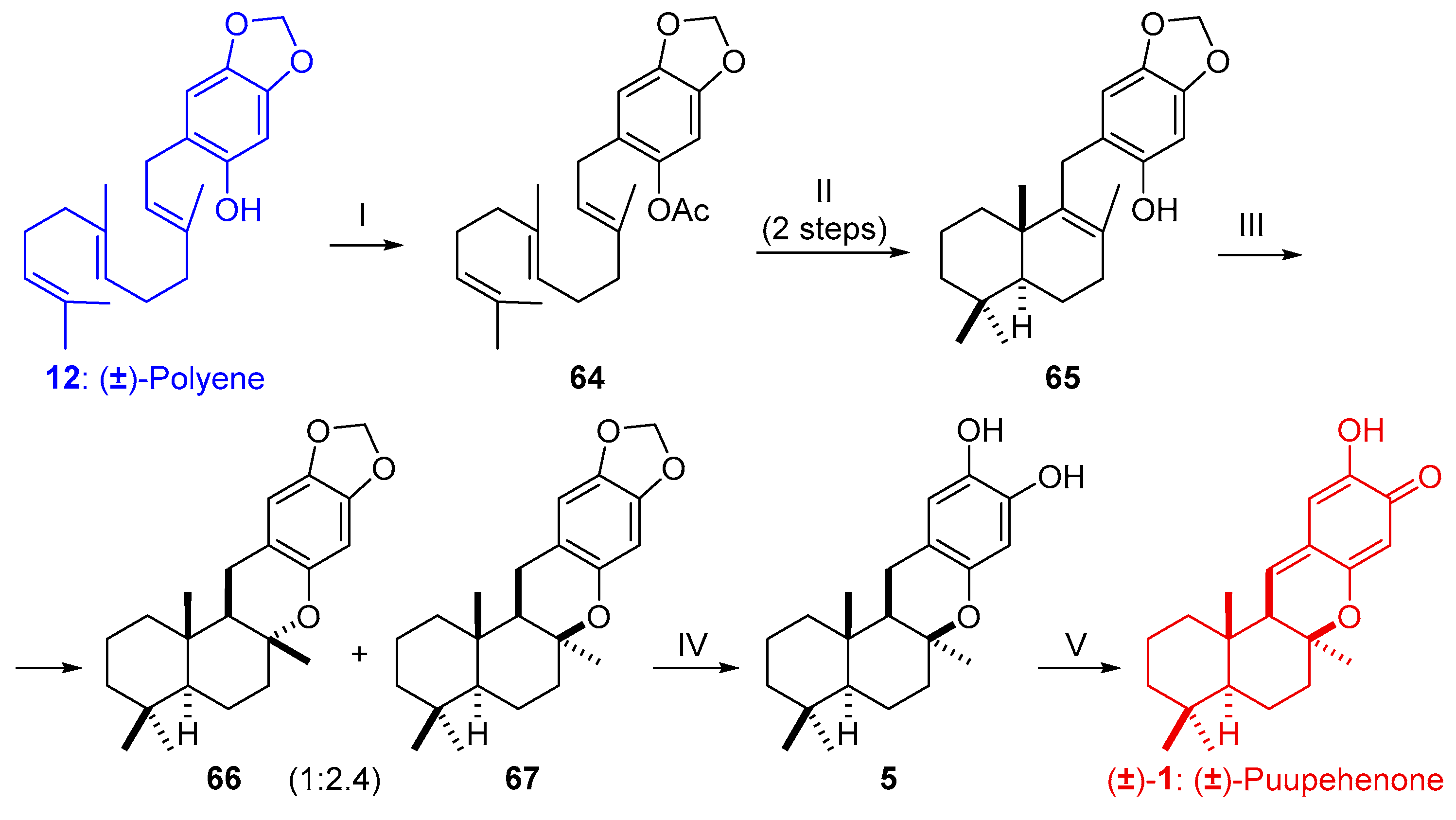
2.2.1. Synthesis of (±)-Puupehenone (2) from Farnesyl Bromide
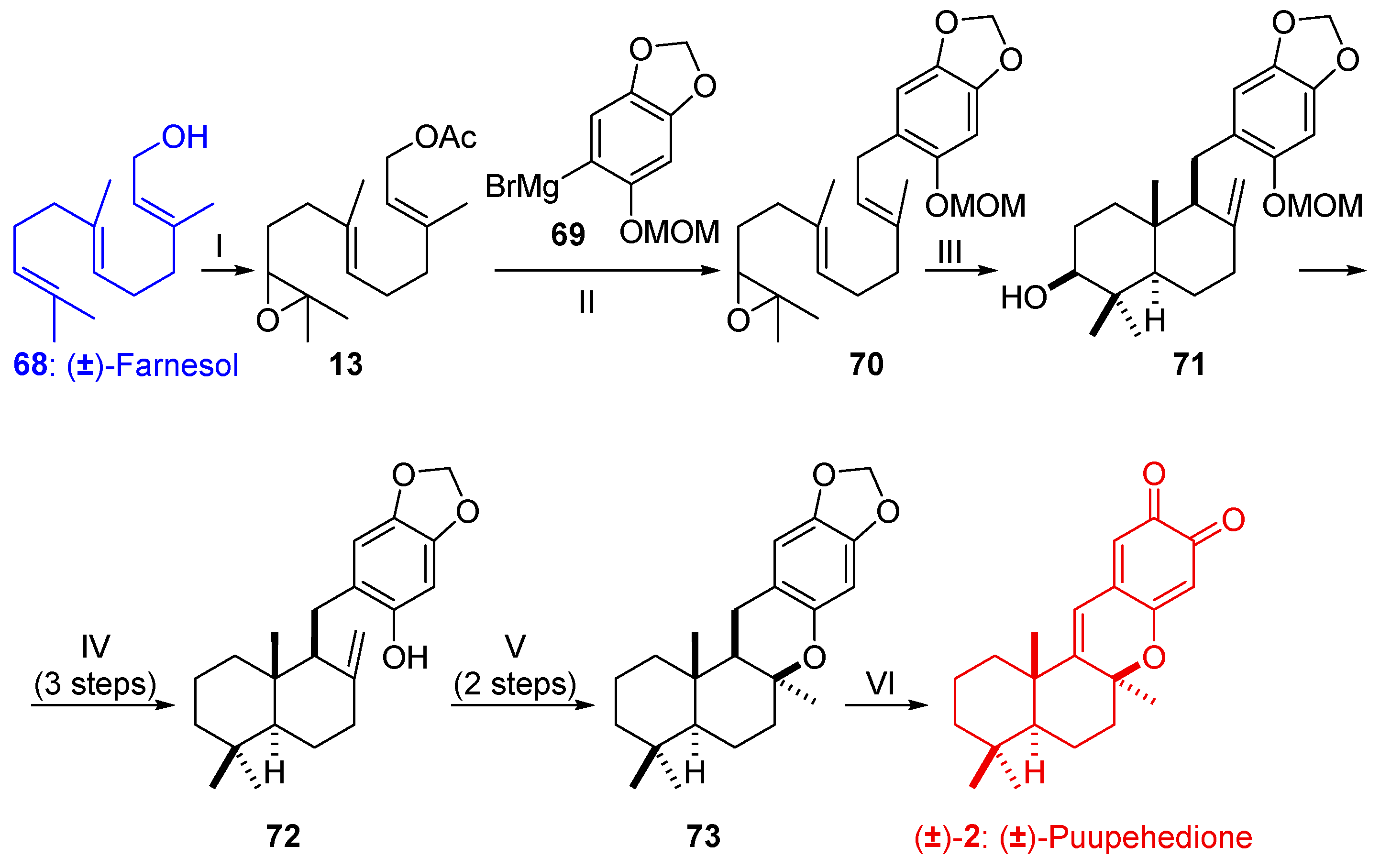
2.2.2. Synthesis of (±)-Puupehedione ((±)2) from Farnesol
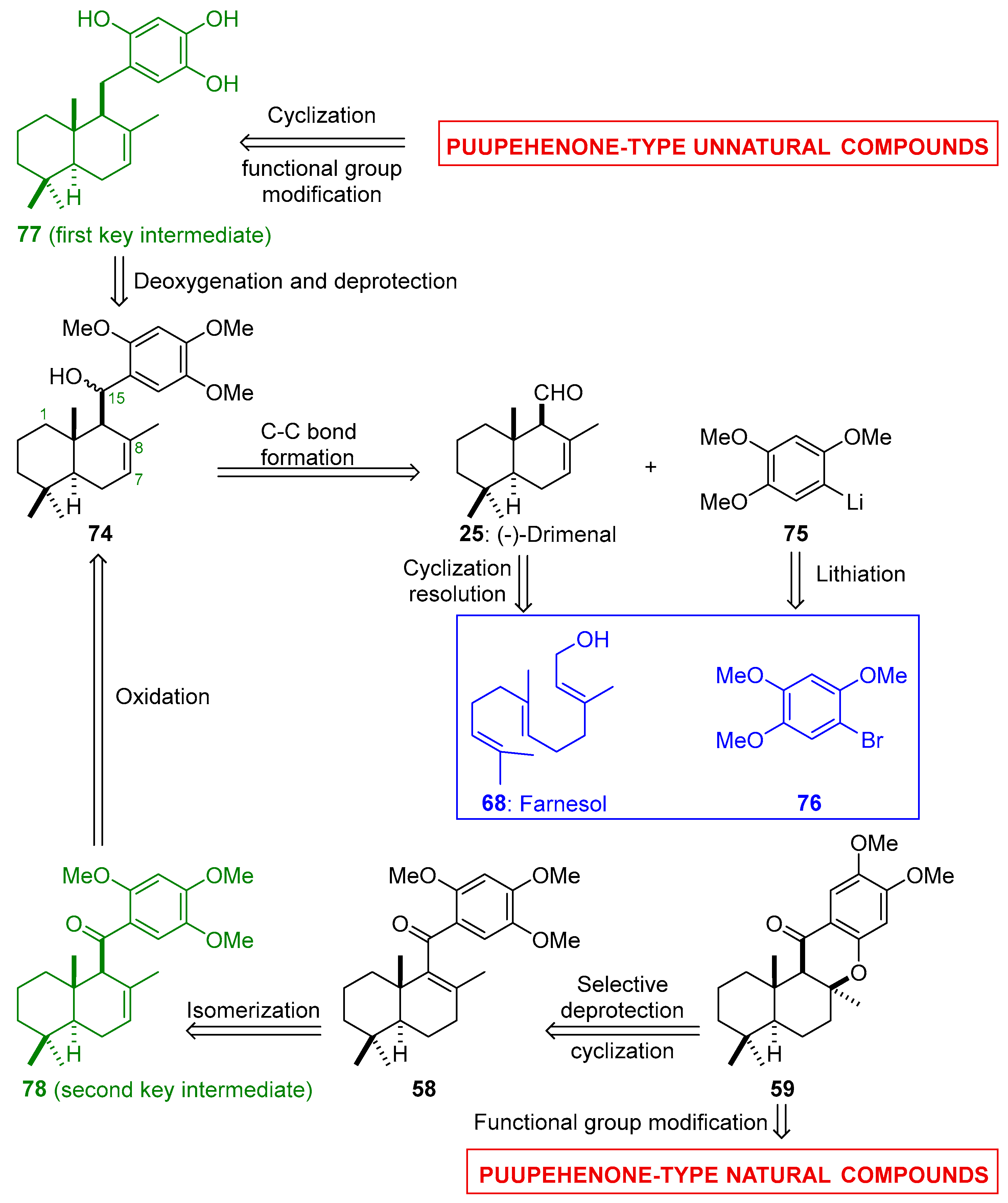
3. Future Perspectives
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marcos, I.S.; Conde, A.; Moro, R.F.; Basabe, P.; Diez, D.; Urones, J.G. Quinone/Hydroquinone Sesquiterpenes. Mini-Rev. Org. Chem. 2010, 7, 230. [Google Scholar] [CrossRef]
- Djura, P.; Stierle, D.B.; Sullivan, B.; Faulkner, D.J.; Arnold, E.; Clardy, J. Some metabolites of the marine sponges Smenospongia aurea and Smenospongia (polyfibrospongia) echina. J. Org. Chem. 1980, 45, 1435–1441. [Google Scholar] [CrossRef]
- Kohmoto, S.; McConnell, O.J.; Wright, A. Puupehenone, a cytotoxic metabolite from a deep water marine sponge, Stronglyophora hartman. J. Nat. Prod. 1987, 50, 336. [Google Scholar] [CrossRef]
- Pina, I.C.; Sanders, M.L.; Crews, P. Puupehenone congeners from an indoPacific Hyrtiossponge. J. Nat. Prod. 2003, 66, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 1998, 15, 113–158. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.T.; Scheuer, P.J. Cyanopuupehenol, an antiviral metabolite of a sponge of the order Verongida. Tetrahedron Lett. 1991, 32, 5671–5672. [Google Scholar] [CrossRef]
- Nasu, S.S.; Yeung, B.K.; Hamann, M.T. Puupehenone-related metabolites from two Hawaiian sponges, Hyrtios spp. J. Org. Chem. 1995, 60, 7290–7292. [Google Scholar] [CrossRef]
- Navi, B.N.; Perzanovski, H.P.; Ross, R.A.; Erdman, T.R.; Scheuer, P.J.; Finar, J.; Clardy, J. Recent research in marine natural products: The puupehenones. Pure Appl. Chem. 1979, 51, 1893–1898. [Google Scholar]
- Urban, S.; Capon, R.J. Absolute Stereochemistry of Puupehenone and Related Metabolites. J. Nat. Prod. 1996, 59, 900–901. [Google Scholar] [CrossRef]
- Martínez-Poveda, B.; Quesada, A.R.; Medina, M.A. Pleiotropic Role of Puupehenones in Biomedical Research. Mar. Drugs 2017, 15, 325. [Google Scholar] [CrossRef] [PubMed]
- Barrero, A.F.; Álvarez-Manzaneda, E.J.; Chahboun, R. Enantiospecific Synthesis of (+)-Puupehenone from (-)-Sclareol and Protocatechualdehyde. Tetrahedron Lett. 1997, 38, 2325–2328. [Google Scholar] [CrossRef]
- Barrero, A.F.; Álvarez-Manzaneda, E.E.; Chahboun, R.; Cortés, M.; Armstrong, V. Synthesis and Antitumor Activity of Puupehedione and related Compounds. Tetrahedron 1999, 55, 15181–15208. [Google Scholar] [CrossRef]
- Álvarez-Manzaneda, E.J.; Chahboun, R.; Barranco Pérez, I.; Cabrera, E.; Álvarez, E.; Álvarez-Manzaneda, R. First Enantiospecific Synthesis of the Antitumor Marine Sponge Metabolite (−)-15-Oxopuupehenol from (−)-Sclareol. Org. Lett. 2005, 7, 1477–1480. [Google Scholar] [CrossRef]
- Álvarez-Manzaneda, E.; Chahboun, R.; Cabrera, E.; Álvarez, E.; Haidour, A.; Ramos, J.M.; Álvarez-Manzaneda, R.; Tapia, R.; Es-Samti, H.; Fernández, A.; et al. A Convenient Enantiospecific Route towards Bioactive Merosesquiterpenes by Cationic-Resin-Promoted Friedel–Crafts Alkylation with α,β-Enones. Eur. J. Org. Chem. 2009, 2009, 1139–1143. [Google Scholar] [CrossRef]
- Quideau, S.; Lebon, M.; Lamidey, A.-M. Enantiospecific Synthesis of the Antituberculosis Marine Sponge Metabolite (+)-Puupehenone. The Arenol Oxidative Activation Route. Org. Lett. 2002, 4, 3975–3978. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-S.; Li, H.-J.; Wang, J.-L.; Wu, Y.-C. Protecting-group-free synthesis of haterumadienone- and puupehenone-type marine natural products. Green Chem. 2017, 19, 2140–2144. [Google Scholar] [CrossRef]
- Wang, H.-S.; Li, H.-J.; Nan, X.; Luo, Y.-Y.; Wu, Y.-C. Enantiospecific Semisynthesis of Puupehedione-Type Marine Natural Products. J. Org. Chem. 2017, 82, 12914–12919. [Google Scholar] [CrossRef]
- Maiti, S.; Sengupta, S.; Giri, C.; Achari, B.; Banerjee, A.K. Enantiospecific synthesis of 8-epipuupehedione from (R)-(−)-carvone. Tetrahedron Lett. 2001, 42, 2389–2391. [Google Scholar] [CrossRef]
- Song, H.; Liu, L.; Yang, M.; Wu, G.; Chen, P.; Xie, X.; She, X. Total syntheses of (−)-15-oxopuupehenol and (+)-puupehenone and formal syntheses of (−)-puupehenol and (+)-puupehedione. Org. Chem. Front. 2020, 7, 35–42. [Google Scholar] [CrossRef]
- Armstrong, V.; Barrero, A.F.; Álvarez-Manzaneda, J.E.; Cortés, M.; Sepúlveda, B. An Efficient Stereoselective Synthesis of Cytotoxic 8-Epipuupehedione. J. Nat. Prod. 2003, 66, 1382–1383. [Google Scholar] [CrossRef]
- Trammell, G.L. The total synthesis of (±)-puupehenone. Tetrahedron Lett. 1978, 18, 1525–1528. [Google Scholar] [CrossRef]
- Gansäuer, A.; Justicia, J.; Rosales, A.; Worgull, D.; Rinker, B.; Cuerva, J.M.; Oltra, J.E. Transition-Metal-Catalyzed Allylic Substitution and Titanocene-Catalyzed Epoxypolyene Cyclization as a Powerful Tool for the Preparation of Terpenoids. Eur. J. Org. Chem. 2006, 2006, 4115–4127. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Cheng, Y.-F.; Li, H.-J. Approaches to the Total Synthesis of Puupehenone-Type Marine Natural Products. In Organic Synthesis—A Nascent Relook; Nandeshwarappa, B.P., Ed.; IntechOpen: Rijeka, Croatia, 2019; Chapter 4. [Google Scholar] [CrossRef]
- Wu, Y.-B.; Ni, Z.-Y.; Shi, Q.-W.; Dong, M.; Kiyota, H.; Gu, Y.-C.; Cong, B. Constituents from Salvia Species and Their Biological Activities. Chem. Rev. 2012, 112, 5967–6026. [Google Scholar] [CrossRef] [PubMed]
- Schalk, M.; Pastore, L.; Mirata, M.A.; Khim, S.; Schouwey, M.; Deguerry, F.; Pineda, V.; Rocci, L.; Daviet, L. Toward a Biosynthetic Route to Sclareol and Amber Odorants. J. Am. Chem. Soc. 2012, 134, 18900–18903. [Google Scholar] [CrossRef]
- Frija, L.M.T.; Frade, R.F.M.; Afonso, C.A.M. Isolation, Chemical, and Biotransformation Routes of Labdane-type Diterpenes. Chem. Rev. 2011, 111, 4418–4452. [Google Scholar] [CrossRef] [PubMed]
- Barrero, A.F.; Manzaneda, E.A.; Altajeros, J.; Salido, S.; Ramos, J.M.; Simmonds, M.S.J.; Blaney, W.M. Synthesis of Biologically Active Drimanes and Homodrimanes from (-)-Sclareol. Tetrahedron 1995, 51, 7435–7450. [Google Scholar] [CrossRef]
- Barrero, A.F.; Álvarez-Manzanda, J.E.; Chahboun, R. Synthesis of Wiedendiol-A and Wiedendiol-B from Labdane Diterpenes. Tetrahedron 1998, 54, 5635–5650. [Google Scholar] [CrossRef]
- Topcu, G.; Ulubelen, A.; Tam, T.C.-M.; Che, C.-T. Norditerpenes and Norsesterterpenes from Salvia yosgadensis. J. Nat. Prod. 1996, 59, 113–116. [Google Scholar] [CrossRef]
- Hajime, H. Aroma of cigar tobacco. II. Isolation of norambreinolide from cigar tobacco. Agric. Biol. Chem. 1971, 35, 1461–1462. [Google Scholar]
- Dixon, D.; Lockner, J.W.; Zhou, Q.; Baran, P.S. Scalable, Divergent Synthesis of Meroterpenoids via “Borono-sclareolide”. J. Am. Chem. Soc. 2012, 134, 8432–8435. [Google Scholar] [CrossRef]
- Cheng, Y.; Lyu, X.; Liu, C.; Wang, X.; Cheng, J.; Zhang, D.; Meng, X.; Zhao, Y. Synthesis and Biological Evaluation of Sclareolide-Indole Conjugates and Their Derivatives. Molecules 2023, 28, 1737. [Google Scholar] [CrossRef] [PubMed]
- Ravid, U.; Bassat, M.; Putievsky, E.; Weistein, V.; Ikan, R. Isolation and determination of optically pure carvone enantiomers from caraway (Carum carvi L.), dill (Anethum graveolens L.), spearmint (Mentha spicata L.) and Mentha longifolia (L.) Huds. Flavour Fragance J. 1987, 2, 95–97. [Google Scholar] [CrossRef]
- Ncube, E.N.; Steenkamp, L.; Dubery, I.A. Ambrafuran (AmbroxTM) Synthesis from Natural Plant Product Precursors. Molecules 2020, 25, 3851. [Google Scholar] [CrossRef]
- Quideau, S.; Pouységu, C.; Deffieux, D. Oxidative Dearomatization of Phenols: Why, How and What For? Synlett 2008, 4, 467–495. [Google Scholar] [CrossRef]
- Noble, A.; Roesner, S.; Aggarwal, V.K. Short Enantioselective Total Synthesis of Tatanan A and 3-epi-Tatanan A Using Assembly-Line Synthesis. Angew. Chem. Int. Ed. 2016, 55, 15920–15924. [Google Scholar] [CrossRef] [PubMed]
- Appel, H.H.; Brooks, C.J.W.; Overto, K.H. The Constitution and Stereochemistry of Drimenol, a Novel Bicyclic Xesquiterpenoid. J. Chem. Soc. 1959, 3322–3332. [Google Scholar] [CrossRef]
- Cortés, M.; Delgado, V.; Saitz, C.; Armstrong, V. Drimenol: A Versatile Synthon for Compounds with trans-Drimane Skeleton. Nat. Prod. Commun. 2011, 6, 477–490. [Google Scholar] [CrossRef]
- Chen, X.X.; Zhang, D.; Xu, D.; Zhou, H.; Xu, G. Remote C−H Activation Strategy Enables Total Syntheses of Nortriterpenoids (±)-Walsucochin B and (±)-Walsucochinoids M and N. Org. Lett. 2020, 17, 6993–6997. [Google Scholar] [CrossRef]
- Jung, Y.-Y.; Hwang, S.T.; Sethi, G.; Fan, L.; Arfuso, F.; Ahn, K.S. Potential Anti-Inflammatory and Anti-Cancer Properties of Farnesol. Molecules 2018, 23, 2827. [Google Scholar] [CrossRef]
- Rosales Martínez, A.; Pozo Morales, L.; Díaz Ojeda, E.; Castro Rodríguez, M.; Rodríguez-García, I. The Proven Versatility of Cp2TiCl. J. Org. Chem. 2021, 86, 1311–1329. [Google Scholar] [CrossRef]
- Ma, T.-K.; Parsons, P.J.; Barret, A.G.M. Meroterpenoid Synthesis via Sequential Polyketide Aromatization and Radical Anion Cascade Triene Cyclization: Biomimetic Total Syntheses of Austalide Natural Products. J. Org. Chem. 2019, 84, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- Gansäuer, A.; Rosales, A.; Justicia, J. Catalytic epoxypolyene cyclization via radicals: Highly diastereoselective formal synthesis of puupehedione and 8-epi-puupehedione. Synlett 2006, 6, 927–929. [Google Scholar] [CrossRef]
- Snyder, S.A.; Corey, E.J. Concise Total Syntheses of Palominol, Dolabellatrienone, β-Araneosene, and Isoedunol via an Enantioselective Diels−Alder Macrobicyclization. J. Am. Chem. Soc. 2006, 128, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Rosales, A.; López-Sánchez, C.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Total Synthesis of (±)-Euryfuran Through Ti(III) Catalyzed Radical Cyclization. Lett. Org. Chem. 2007, 4, 553–555. [Google Scholar] [CrossRef]
- Rosales, A.; Muñoz-Bascón, J.; Moráles-Álcazar, V.M.; Castilla-Alcalá, J.A.; Oltra, J.E. Ti(III)-catalyzed, concise synthesis of marine furanospongian diterpenes. RSC Adv. 2012, 2, 12922–12925. [Google Scholar] [CrossRef]
- Rosales, A.; Muñoz-Bascón, J.; López-Sánchez, C.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I.; Oltra, J.E. Ti-Catalyzed Homolytic Opening of Ozonides: A sustainable C-C Bond-Forming Reaction. J. Org. Chem. 2012, 77, 4171–4176. [Google Scholar] [CrossRef]
- Rosales, A.; Foley, L.A.R.; Padial, N.M.; Muñoz-Bascón, J.; Sancho-Sanz, I.; Roldán-Molina, E.; Pozo-Morales, L.; Irías-Álvarez, A.; Rodríguez-Maecker, R.; Rodríguez-García, I.; et al. Diastereoselective Synthesis of (±)-ambroz by Titanium(III)-Catalyzed Radical Tandem Cyclization. Synlett 2016, 27, 369–374. [Google Scholar] [CrossRef]
- Rosales Martinez, A.; Pozo Morales, L.; Diaz Ojeda, E. Cp2TiCl-catalyzed, concise synthetic approach to marine natural product (±)-cyclozonarone. Synth. Commun. 2019, 49, 2554–2560. [Google Scholar] [CrossRef]
- Rosales Martínez, A.; Enríquez, L.; Jaraíz, M.; Pozo Morales, L.; Rodríguez-García, I.; Díaz Ojeda, E. A Concise Route for the Synthesis of Tetracyclic Meroterpenoids: (±)-Aureol Preparation and Mechanistic Interpretation. Mar. Drugs 2020, 18, 441. [Google Scholar] [CrossRef]
- Torres-García, I.; López-Martínez, J.L.; López-Domene, R.; Muñoz-Dorado, M.; Rodríguez-García, I.; Álvarez-Corral, M. Enantioselective total synthesis of putative dihydrorosefuran, a monoterpene with an unique 2,5-dihydrofuran structure. Beilstein J. Org. Chem. 2022, 18, 1264–1269. [Google Scholar] [CrossRef]
- López-Martínez, J.; Torres-García, I.; Moreno-Gutiérrez, I.; Oña-Burgos, P.; Rosales Martínez, A.; Muñoz-Dorado, M.; Álvarez-Corral, M.; Rodríguez-García, I. A Concise Diastereoselective Total Synthesis of α-Ambrinol. Mar. Drugs 2023, 21, 230. [Google Scholar] [CrossRef] [PubMed]
- Arjona, O.; Garranzo, M.; Mahugo, J.; Maroto, E.; Plumet, J.; Sáez, B. Total synthesis of both enantiomers of 15-oxopuupehenol methylendioxy derivatives. Tetrahedron Lett. 1997, 38, 7249–7252. [Google Scholar] [CrossRef]
- Jordine, G.; Bick, S.; Mgller, U.; Wezel, P.; Daucher, B.; Maas, G. Studies on forskolin ring C forming reactions. Tetrahedron 1994, 50, 139–160. [Google Scholar] [CrossRef]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosales Martínez, A.; Rodríguez-García, I. Marine Puupehenone and Puupehedione: Synthesis and Future Perspectives. Mar. Drugs 2023, 21, 322. https://doi.org/10.3390/md21060322
Rosales Martínez A, Rodríguez-García I. Marine Puupehenone and Puupehedione: Synthesis and Future Perspectives. Marine Drugs. 2023; 21(6):322. https://doi.org/10.3390/md21060322
Chicago/Turabian StyleRosales Martínez, Antonio, and Ignacio Rodríguez-García. 2023. "Marine Puupehenone and Puupehedione: Synthesis and Future Perspectives" Marine Drugs 21, no. 6: 322. https://doi.org/10.3390/md21060322
APA StyleRosales Martínez, A., & Rodríguez-García, I. (2023). Marine Puupehenone and Puupehedione: Synthesis and Future Perspectives. Marine Drugs, 21(6), 322. https://doi.org/10.3390/md21060322