
Continuous Flow Chemistry: A Novel Technology for the Synthesis of Marine Drugs

 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
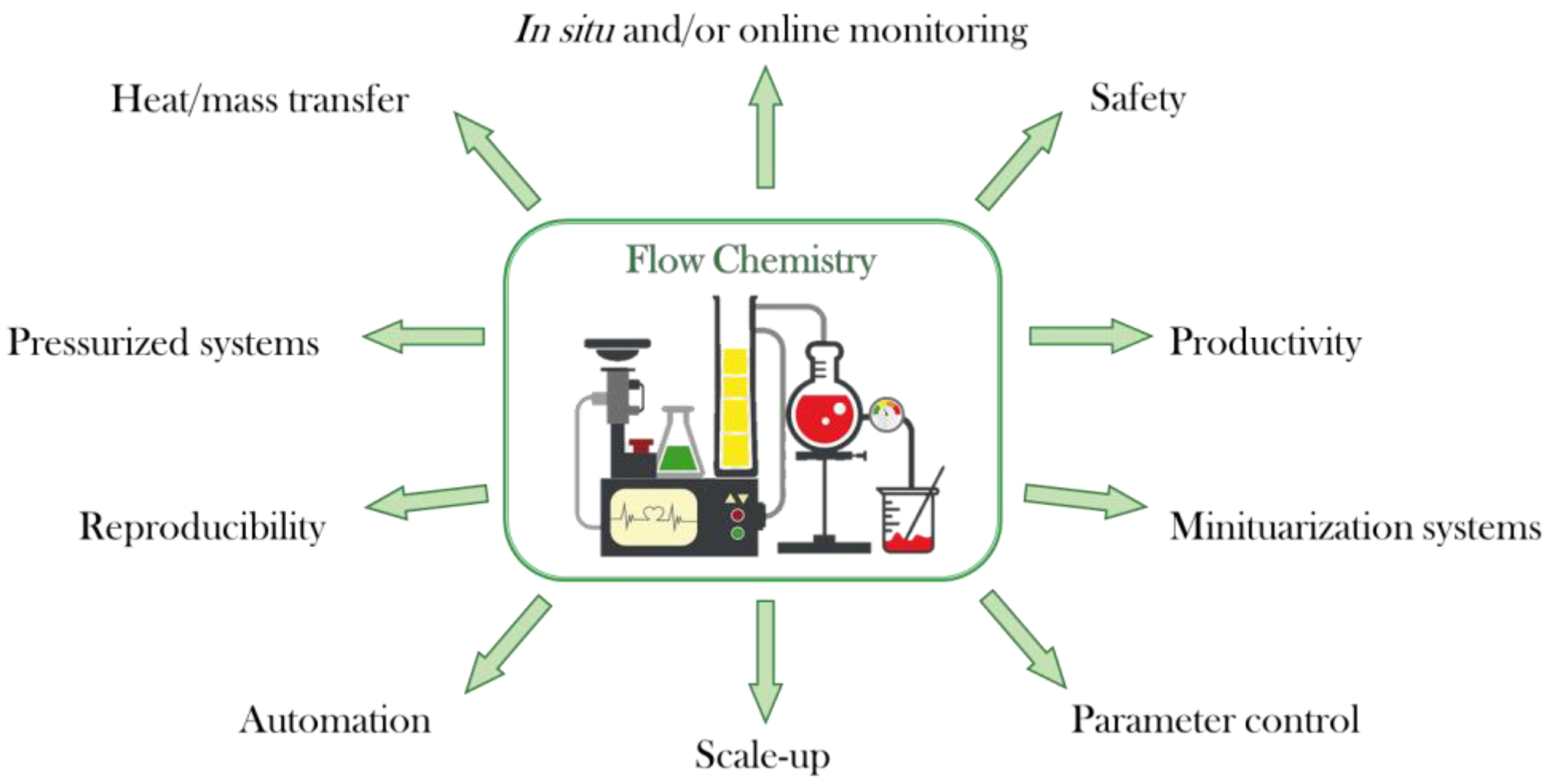
1. Introduction
2. Synthesis of Marine Drugs Using Flow Chemistry
2.1. Synthesis of Aplysamines
2.2. Synthesis of (−)-hennoxazole A
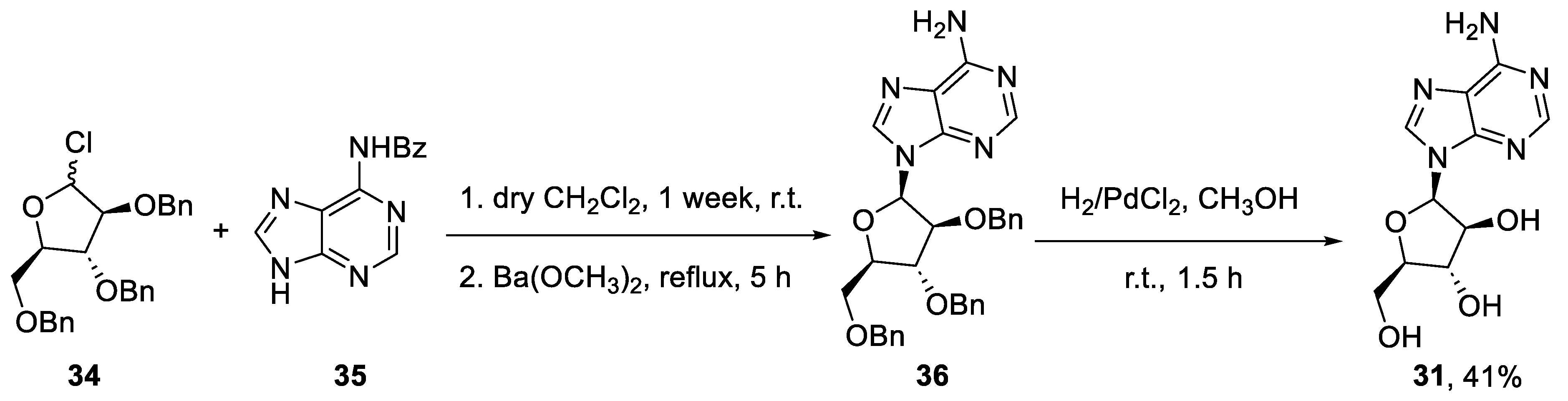
2.3. Synthesis of Vidarabine
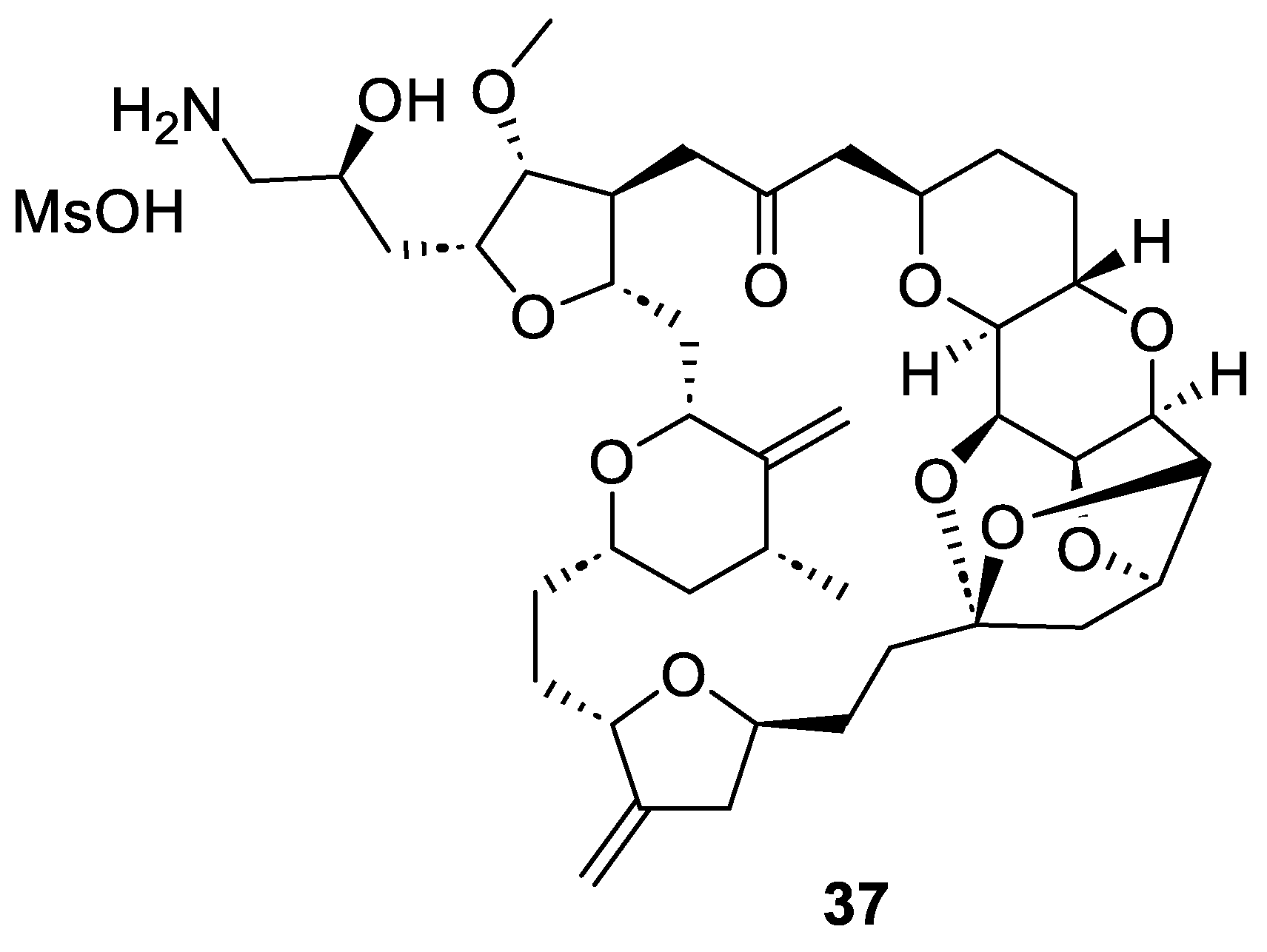
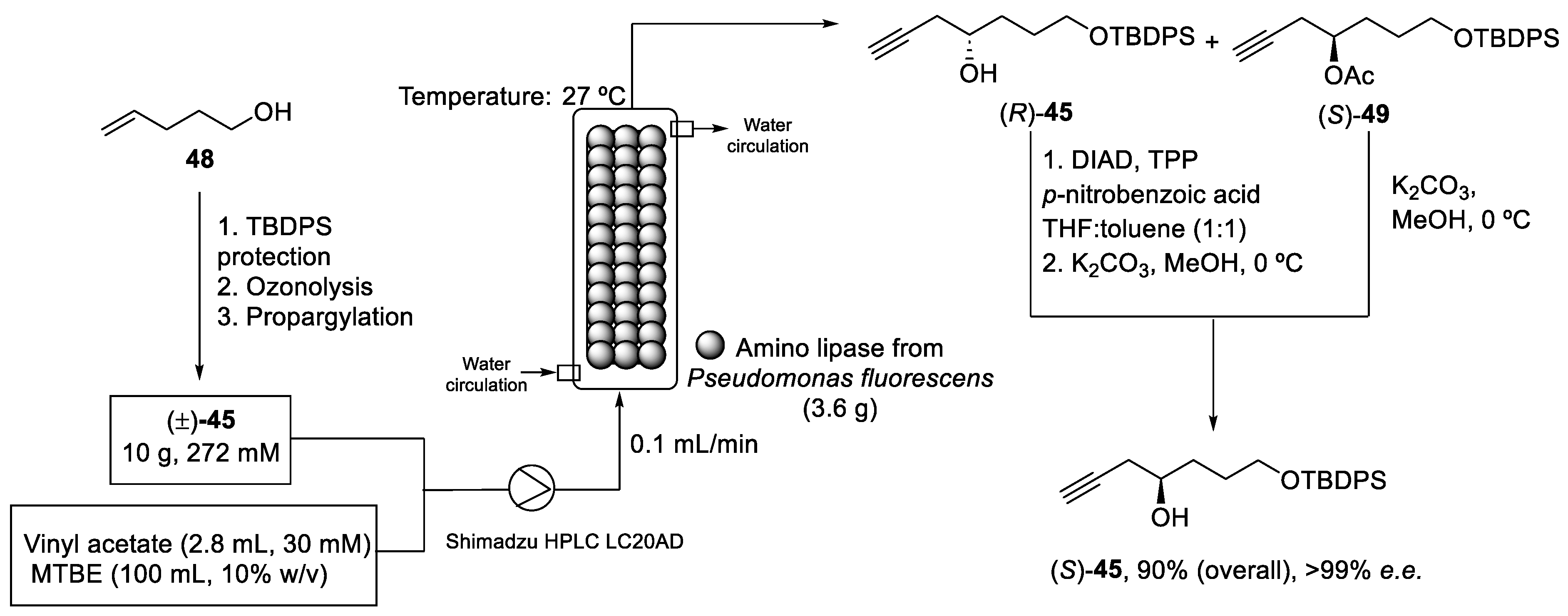
2.4. Synthesis of Eribulin
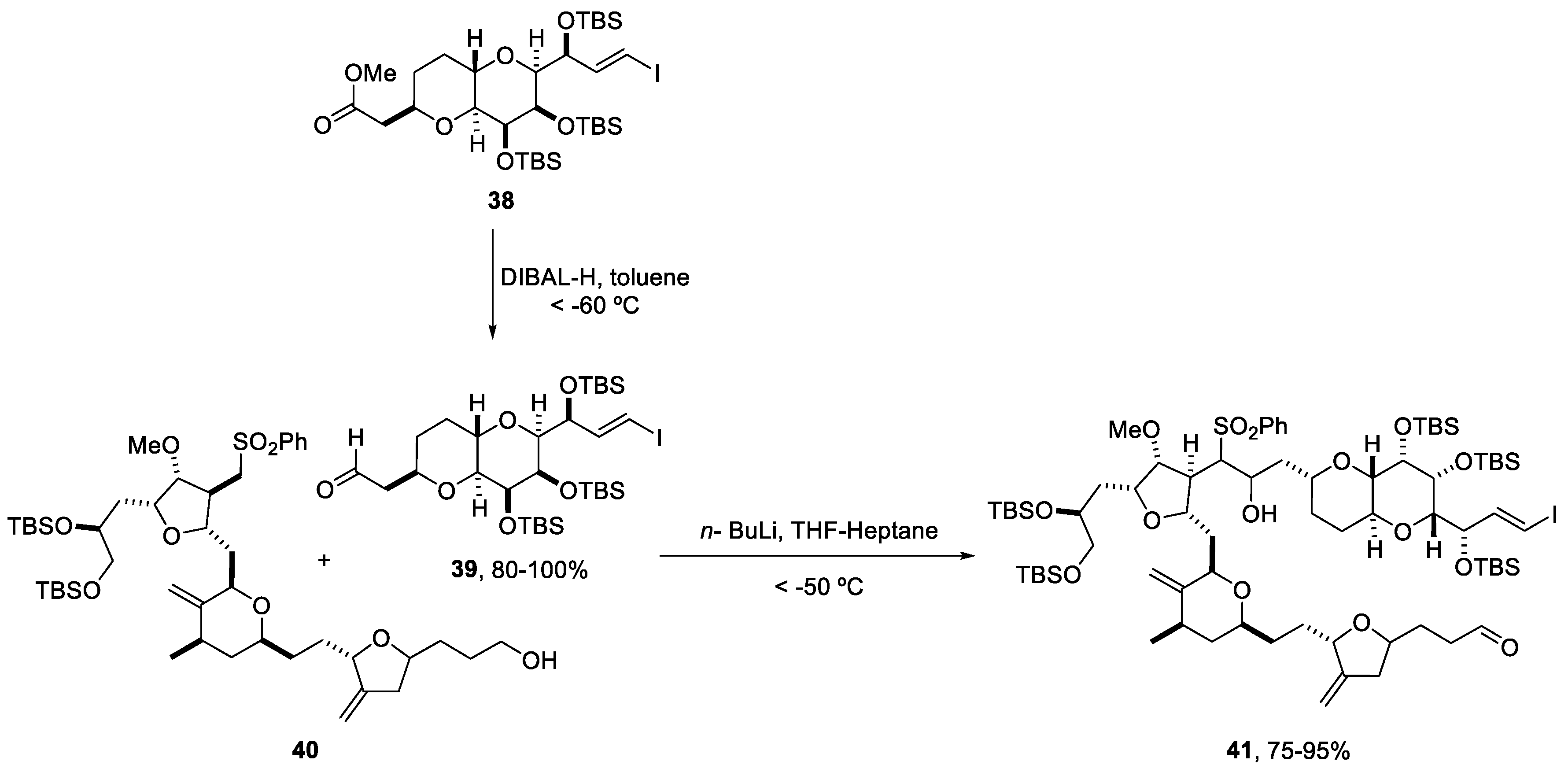
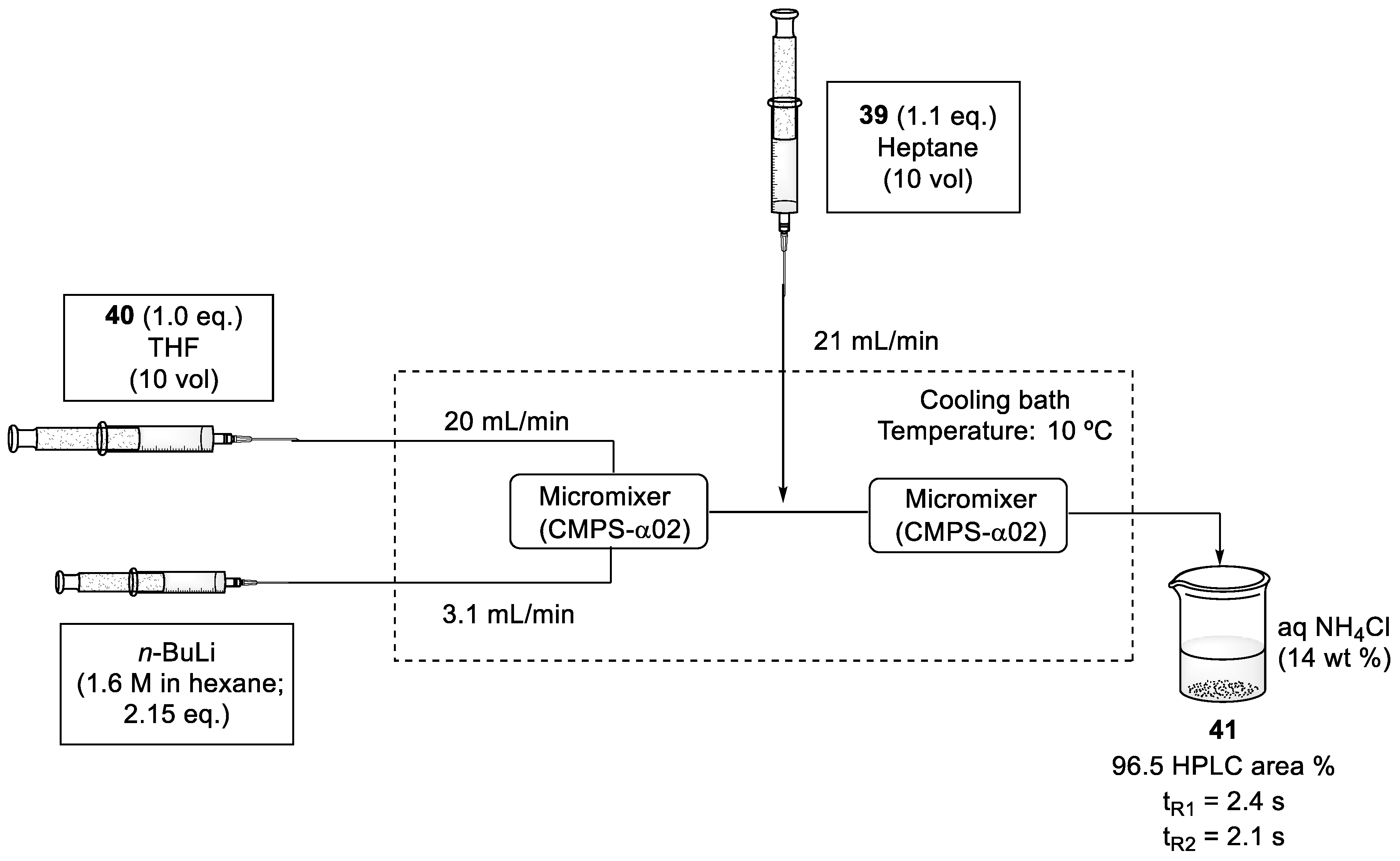
2.5. Synthesis of Yessotoxin
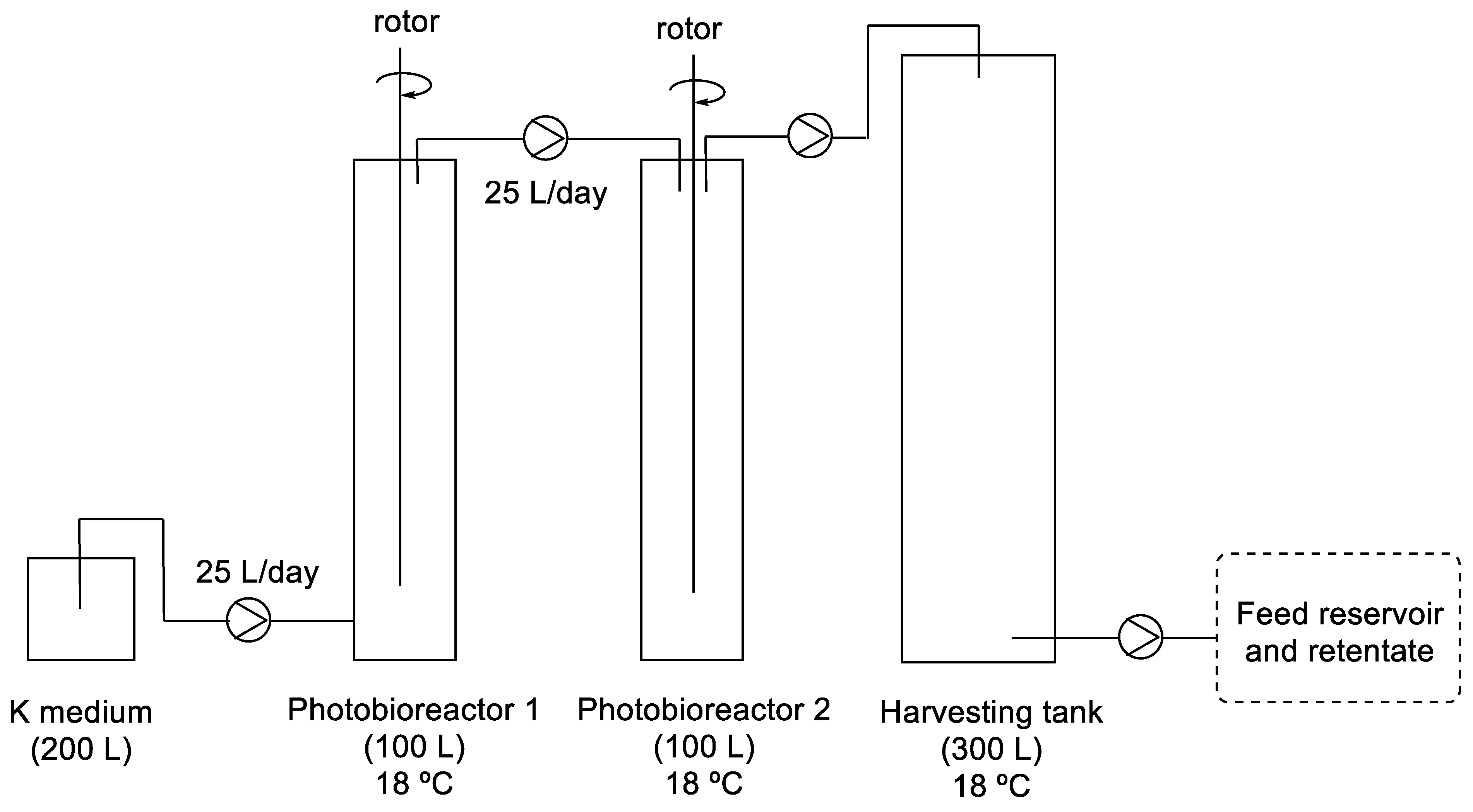
2.6. Synthesis of Azaspiracids through Flow Photochemistry
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef]
- Bana, P.; Örkényi, R.; Lövei, K.; Lakó, Á.; Túrós, G.I.; Éles, J.; Faigl, F.; Greiner, I. The route from problem to solution in multistep continuous flow synthesis of pharmaceutical compounds. Bioorg. Med. Chem. 2017, 25, 6180–6189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdan, A.R.; Dombrowski, A.W. Emerging Trends in Flow Chemistry and Applications to the Pharmaceutical Industry. J. Med. Chem. 2019, 62, 6422–6468. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Brown, D.G.; Young, R.J.; Keserü, G.M. Expanding the medicinal chemistry synthetic toolbox. Nat. Rev. Drug Discov. 2018, 17, 709–727. [Google Scholar] [CrossRef]
- Alcázar, J. Sustainable Flow Chemistry in Drug Discovery. In Sustainable Flow Chemistry; Wiley: Hoboken, NJ, USA, 2017; pp. 135–164. [Google Scholar]
- Bogdan, A.R.; Organ, M.G. Flow Chemistry as a Drug Discovery Tool: A Medicinal Chemistry Perspective. In Flow Chemistry for the Synthesis of Heterocycles; Sharma, U.K., Van der Eycken, E.V., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 319–341. [Google Scholar]
- Alcazar, J.; de la Hoz, A.; Díaz-Ortiz, A. Flow Chemistry in Drug Discovery. In Green Synthetic Processes and Procedures; Ballini, R., Ed.; The Royal Society of Chemistry: London, UK, 2019; pp. 53–78. [Google Scholar]
- Ürge, L.; Alcazar, J.; Huck, L.; Dormán, G. Chapter Four—Recent Advances of Microfluidics Technologies in the Field of Medicinal Chemistry. In Annual Reports in Medicinal Chemistry; Goodnow, R.A., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 50, pp. 87–147. [Google Scholar]
- Gioiello, A.; Piccinno, A.; Lozza, A.M.; Cerra, B. The Medicinal Chemistry in the Era of Machines and Automation: Recent Advances in Continuous Flow Technology. J. Med. Chem. 2020, 63, 6624–6647. [Google Scholar] [CrossRef]
- Gérardy, R.; Emmanuel, N.; Toupy, T.; Kassin, V.-E.; Tshibalonza, N.N.; Schmitz, M.; Monbaliu, J.-C.M. Continuous Flow Organic Chemistry: Successes and Pitfalls at the Interface with Current Societal Challenges. Eur. J. Org. Chem. 2018, 2018, 2301–2351. [Google Scholar] [CrossRef]
- Cole, K.P.; Johnson, M.D. Continuous flow technology vs. the batch-by-batch approach to produce pharmaceutical compounds. Expert Rev. Clin. Pharmacol. 2018, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, B.; Kappe, C.O. Forbidden chemistries—Paths to a sustainable future engaging continuous processing. J. Flow. Chem. 2017, 7, 65–71. [Google Scholar] [CrossRef]
- Capaldo, L.; Wen, Z.; Noël, T. A field guide to flow chemistry for synthetic organic chemists. Chem. Sci. 2023, 14, 4230–4247. [Google Scholar] [CrossRef]
- Tomarelli, E.; Cerra, B.; Mutti, F.G.; Gioiello, A. Merging Continuous Flow Technology, Photochemistry and Biocatalysis to Streamline Steroid Synthesis. Adv. Synth. Catal. 2023, 365, 1–26. [Google Scholar] [CrossRef]
- Frederick, M.O.; Pietz, M.A.; Kjell, D.P.; Richey, R.N.; Tharp, G.A.; Touge, T.; Yokoyama, N.; Kida, M.; Matsuo, T. Development of a Leuckart–Wallach Reaction in Flow for the Synthesis of Abemaciclib. Org. Process Res. Dev. 2017, 21, 1447–1451. [Google Scholar] [CrossRef] [Green Version]
- Verghese, J.; Kong, C.J.; Rivalti, D.; Yu, E.C.; Krack, R.; Alcázar, J.; Manley, J.B.; McQuade, D.T.; Ahmad, S.; Belecki, K.; et al. Increasing global access to the high-volume HIV drug nevirapine through process intensification. Green Chem. 2017, 19, 2986–2991. [Google Scholar] [CrossRef]
- Hughes, D.L. Applications of Flow Chemistry in Drug Development: Highlights of Recent Patent Literature. Org. Process Res. Dev. 2018, 22, 13–20. [Google Scholar] [CrossRef]
- Defrance, T.; Septavaux, J.; Nuel, D. Process for Preparing Brivaracetam. WO 2017076738 A1, 11 May 2017. [Google Scholar]
- Jain, S.; Ansari, K.; Maddala, S.; Meenakshisunderam, S. Process for the Preparation of Valacyclovir. WO 2017/149420, 9 September 2017. [Google Scholar]
- Hong, H.; Gage, J.; Lu, J.; Li, J.; Shen, L.S. Synthetic Method of Crizotinib Intermediate. CN 105906656, 31 August 2016. [Google Scholar]
- López, E.; Linares, M.L.; Alcázar, J. Flow chemistry as a tool to access novel chemical space for drug discovery. Future Med. Chem. 2020, 12, 1547–1563. [Google Scholar] [CrossRef] [PubMed]
- Linares, M.L.; López, E.; Palao, E.; Alcázar, J. Flow Chemistry Opportunities for Drug Discovery. In Flow and Microreactor Technology in Medicinal Chemistry; Methods and Principles in Medicinal Chemistry; John Wiley & Sons: New York, NY, USA, 2022; pp. 67–102. [Google Scholar]
- Malve, H. Exploring the ocean for new drug developments: Marine pharmacology. J. Pharm. Bioallied Sci. 2016, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [Green Version]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed Marine Natural Products in the Pharmaceutical and Cosmeceutical Industries: Tips for Success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [Green Version]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar] [CrossRef]
- Fernández-Peña, L.; Matos, M.J.; López, E. Recent Advances in Biologically Active Coumarins from Marine Sources: Synthesis and Evaluation. Mar. Drugs 2023, 21, 37. [Google Scholar] [CrossRef]
- Brown, D.G.; Boström, J. Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem. 2016, 59, 4443–4458. [Google Scholar] [CrossRef] [Green Version]
- Xynas, R.; Capon, R.J. Two New Bromotyrosine-Derived Metabolites from an Australian Marine Sponge, Aplysina sp. Aust. J. Chem. 1989, 42, 1427–1433. [Google Scholar] [CrossRef]
- Jurek, J.; Yoshida, W.Y.; Scheuer, P.J.; Kelly-Borges, M. Three New Bromotyrosine-Derived Metabolites of the Sponge Psammaplysilla purpurea. J. Nat. Prod. 1993, 56, 1609–1612. [Google Scholar] [CrossRef]
- Tran, T.D.; Pham, N.B.; Fechner, G.; Hooper, J.N.A.; Quinn, R.J. Bromotyrosine Alkaloids from the Australian Marine Sponge Pseudoceratina verrucosa. J. Nat. Prod. 2013, 76, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, M.S.; Carroll, A.R.; Fechner, G.A.; Boyle, A.; Simpson, M.; Addepalli, R.; Avery, V.M.; Hooper, J.N.A.; Cheung, T.; Chen, H.; et al. Aplysamine 6, an Alkaloidal Inhibitor of Isoprenylcysteine Carboxyl Methyltransferase from the Sponge Pseudoceratina sp. J. Nat. Prod. 2008, 71, 1066–1067. [Google Scholar] [CrossRef]
- Ullah, N.; Arafeh, K.M. The first total synthesis of aplysamine 6, an inhibitor of isoprenylcysteine carboxy methyltransferase. Tetrahedron Lett. 2009, 50, 158–160. [Google Scholar] [CrossRef]
- Achanta, S.; Liautard, V.; Paugh, R.; Organ, M.G. The Development of a General Strategy for the Synthesis of Tyramine-Based Natural Products by Using Continuous Flow Techniques. Chem. Eur. J. 2010, 16, 12797–12800. [Google Scholar] [CrossRef]
- Ichiba, T.; Yoshida, W.Y.; Scheuer, P.J.; Higa, T.; Gravalos, D.G. Hennoxazoles, bioactive bisoxazoles from a marine sponge. J. Am. Chem. Soc. 1991, 113, 3173–3174. [Google Scholar] [CrossRef]
- Yokokawa, F.; Asano, T.; Shioiri, T. Total synthesis of (−)-hennoxazole A. Tetrahedron 2001, 57, 6311–6327. [Google Scholar] [CrossRef]
- Wipf, P.; Lim, S. Total Synthesis of the Enantiomer of the Antiviral Marine Natural Product Hennoxazole A. J. Am. Chem. Soc. 1995, 117, 558–559. [Google Scholar] [CrossRef]
- Fernández, A.; Levine, Z.; Baumann, M.; Sulzer-Mossé, S.; Sparr, C.; Schläger, S.; Metzger, A.; Baxendale, I.; Ley, S. Synthesis of (−)-Hennoxazole A: Integrating Batch and Flow Chemistry Methods. Synlett 2013, 24, 514–518. [Google Scholar]
- Bergmann, W.; Feeney, R.J. Contributions to the Study of Marine Products, XXXII. The nucleosides of sponges. I.1. J. Org. Chem. 1951, 16, 981–987. [Google Scholar] [CrossRef]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Santi, M.; Sancineto, L.; Nascimento, V.; Braun Azeredo, J.; Orozco, E.V.M.; Andrade, L.H.; Gröger, H.; Santi, C. Flow Biocatalysis: A Challenging Alternative for the Synthesis of APIs and Natural Compounds. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Tamborini, L.; Previtali, C.; Annunziata, F.; Bavaro, T.; Terreni, M.; Calleri, E.; Rinaldi, F.; Pinto, A.; Speranza, G.; Ubiali, D.; et al. An Enzymatic Flow-Based Preparative Route to Vidarabine. Molecules 2020, 25, 1223. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, F.; Fernández-Lucas, J.; de la Fuente, D.; Zheng, C.; Bavaro, T.; Peters, B.; Massolini, G.; Annunziata, F.; Conti, P.; de la Mata, I.; et al. Immobilized enzyme reactors based on nucleoside phosphorylases and 2′-deoxyribosyltransferase for the in-flow synthesis of pharmaceutically relevant nucleoside analogues. Bioresour. Technol. 2020, 307, 123258. [Google Scholar] [CrossRef]
- Serra, I.; Daly, S.; Alcantara, A.R.; Bianchi, D.; Terreni, M.; Ubiali, D. Redesigning the synthesis of vidarabine via a multienzymatic reaction catalyzed by immobilized nucleoside phosphorylases. RSC Adv. 2015, 5, 23569–23577. [Google Scholar] [CrossRef]
- Glaudemans, C.P.J.; Fletcher, H.G., Jr. Syntheses with Partially Benzylated Sugars. III.1 A Simple Pathway to a “cis- Nucleoside”, 9-β-D-Arabinofuranosyladenine (Spongoadenosine). J. Org. Chem. 1963, 28, 3004–3006. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D. Halichondrins - antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Swami, U.; Shah, U.; Goel, S. Eribulin in Cancer Treatment. Mar. Drugs 2015, 13, 5016–5058. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.; O’Shaughnessy, J.; Loesch, D.; Blum, J.L.; Vahdat, L.T.; Petrakova, K.; Chollet, P.; Manikas, A.; Diéras, V.; Delozier, T.; et al. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): A phase 3 open-label randomised study. Lancet 2011, 377, 914–923. [Google Scholar] [CrossRef]
- Lin, N.U.; Burstein, H.J. EMBRACE, eribulin, and new realities of advanced breast cancer. Lancet 2011, 377, 878–880. [Google Scholar] [CrossRef]
- Osgood, C.L.; Chuk, M.K.; Theoret, M.R.; Huang, L.; He, K.; Her, L.; Keegan, P.; Pazdur, R. FDA Approval Summary: Eribulin for Patients with Unresectable or Metastatic Liposarcoma Who Have Received a Prior Anthracycline-Containing Regimen. Clin. Cancer Res. 2017, 23, 6384–6389. [Google Scholar] [CrossRef] [Green Version]
- Murthy, A.S.; Mahipal, B.; Chandrasekhar, S. Asymmetric Synthesis of the C14–C26 Building Block of Eribulin Mesylate. Eur. J. Org. Chem. 2012, 2012, 6959–6966. [Google Scholar] [CrossRef]
- Melvin, J.Y.; Zheng, W.; Seletsky, B.M. From micrograms to grams: Scale-up synthesis of eribulin mesylate. Nat. Prod. Rep. 2013, 30, 1158–1164. [Google Scholar] [CrossRef]
- Murthy, A.S.; Chandrasekhar, S. Practical and stereoselective synthesis of [6,6,5]-tricyclic core (C1–C13) of eribulin mesylate. Tetrahedron Lett. 2015, 56, 4280–4282. [Google Scholar] [CrossRef]
- Lavanya, N.; Kiranmai, N.; Mainkar, P.S.; Chandrasekhar, S. A practical synthesis of C14–C26 fragment of anticancer drug, eribulin mesylate. Tetrahedron Lett. 2015, 56, 4283–4285. [Google Scholar] [CrossRef]
- Austad, B.C.; Calkins, T.L.; Chase, C.E.; Fang, F.G.; Horstmann, T.E.; Hu, Y.; Lewis, B.M.; Niu, X.; Noland, T.A.; Orr, J.D.; et al. Commercial Manufacture of Halaven®: Chemoselective Transformations En Route to Structurally Complex Macrocyclic Ketones. Synlett 2013, 24, 333–337. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, T.; Chiba, H.; Kuroda, H.; Takigawa, T.; Kayano, A.; Tagami, K. Application of Continuous Flow for DIBAL-H Reduction and n-BuLi Mediated Coupling Reaction in the Synthesis of Eribulin Mesylate. Org. Process Res. Dev. 2016, 20, 503–509. [Google Scholar] [CrossRef]
- Krishna, A.S.; Basetty, S.; Nasam, R.; Ralte, S.L.; Reddy, C.R.; Sudhakar, G.; Pabbaraja, S.; Chandrasekhar, S.; Mainkar, P.S.; Kumaraguru, T.; et al. Chemoenzymatic Process for the Preparation of (S)-7-((tert-Butyldiphenylsilyl)oxy)hept-1-yn-4-ol in a Continuous Packed-Bed Reactor, a Key Intermediate for Eribulin Synthesis. Org. Process Res. Dev. 2020, 24, 2657–2664. [Google Scholar] [CrossRef]
- Liu, S.; Kim, J.T.; Dong, C.-G.; Kishi, Y. Catalytic Enantioselective Cr-Mediated Propargylation: Application to Halichondrin Synthesis. Org. Lett. 2009, 11, 4520–4523. [Google Scholar] [CrossRef] [Green Version]
- Murata, M.; Kumagai, M.; Lee, J.S.; Yasumoto, T. Isolation and structure of yessotoxin, a novel polyether compound implicated in diarrhetic shellfish poisoning. Tetrahedron Lett. 1987, 28, 5869–5872. [Google Scholar] [CrossRef]
- Satake, M.; MacKenzie, L.; Yasumoto, T. Identification of Protoceratium reticulatum as the biogenetic origin of yessotoxin. Nat. Toxins 1997, 5, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Satake, M.; Terasawa, K.; Kadowaki, Y.; Yasumoto, T. Relative configuration of yessotoxin and isolation of two new analogs from toxic scallops. Tetrahedron Lett. 1996, 37, 5955–5958. [Google Scholar] [CrossRef]
- Takahashi, H.; Kusumi, T.; Kan, Y.; Satake, M.; Yasumoto, T. Determination of the absolute configuration of yessotoxin, a polyether compound implicated in diarrhetic shellfish poisoning, by NMR spectroscopic method using a chiral anisotropic reagent, methoxy-(2-naphthyl)acetic acid. Tetrahedron Lett. 1996, 37, 7087–7090. [Google Scholar] [CrossRef]
- de la Rosa, L.A.; Alfonso, A.; Vilariño, N.; Vieytes, M.R.; Botana, L.M. Modulation of cytosolic calcium levels of human lymphocytes by yessotoxin, a novel marine phycotoxin. Biochem. Pharmacol. 2001, 61, 827–833. [Google Scholar] [CrossRef]
- Alfonso, A.; de la Rosa, L.; Vieytes, M.R.; Yasumoto, T.; Botana, L.M. Yessotoxin, a novel phycotoxin, activates phosphodiesterase activity: Effect of yessotoxin on cAMP levels in human lymphocytes. Biochem. Pharmacol. 2003, 65, 193–208. [Google Scholar] [CrossRef]
- Korsnes, M.S.; Hetland, D.L.; Espenes, A.; Aune, T. Induction of apoptosis by YTX in myoblast cell lines via mitochondrial signalling transduction pathway. Toxicol. In Vitro 2006, 20, 1419–1426. [Google Scholar] [CrossRef]
- Halim, R.; Brimble, M.A.; Merten, J. Synthesis of the ABC Fragment of the Pectenotoxins. Org. Lett. 2005, 7, 2659–2662. [Google Scholar] [CrossRef]
- Oishi, T.; Suzuki, M.; Watanabe, K.; Murata, M. Synthesis of the ABC and IJ ring fragments of yessotoxin. Tetrahedron Lett. 2006, 47, 3975–3978. [Google Scholar] [CrossRef]
- Oishi, T.; Suzuki, M.; Watanabe, K.; Murata, M. Convergent synthesis of the CDEF ring fragment of yessotoxin via α-cyano ethers. Heterocycles 2006, 69, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Minato, H.; Murata, M.; Oishi, T. Synthesis of the JK ring fragments of yessotoxin and 42, 43, 44, 45, 46, 47, 55-heptanor-41-oxoyessotoxin. Heterocycles 2007, 72, 207–212. [Google Scholar] [CrossRef]
- Oishi, T.; Watanabe, K.; Murata, M. Convergent synthesis of trans-fused 6/n/6/6 (n=7, 8) tetracyclic ether system via α-cyano ethers. Tetrahedron Lett. 2003, 44, 7315–7319. [Google Scholar] [CrossRef]
- Tohru, O.; Tomoyoshi, I.; Michio, M. Reductive Etherification under Microfluidic Conditions: Application to Practical Synthesis of the FGHIJ-Ring System of Yessotoxin. Chem. Lett. 2010, 39, 108–109. [Google Scholar] [CrossRef]
- Oishi, T. Convergent method via α-cyano ethers: A powerful strategy for synthesizing ladder-shaped polyethers. J. Syn. Chem. Jpn. 2012, 70, 1170–1177. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Terrett, J.A.; Zbieg, J.R. Visible-Light Photocatalysis as an Enabling Technology for Drug Discovery: A Paradigm Shift for Chemical Reactivity. ACS Med. Chem. Lett. 2020, 11, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Politano, F.; Oksdath-Mansilla, G. Light on the Horizon: Current Research and Future Perspectives in Flow Photochemistry. Org. Process Res. Dev. 2018, 22, 1045–1062. [Google Scholar] [CrossRef] [Green Version]
- Beeler, A.B.; Corning, S.R. Photochemistry in flow. In Photochemistry: Volume 43; Fasani, E., Albini, A., Eds.; The Royal Society of Chemistry: London, UK, 2015; Volume 43, pp. 175–190. [Google Scholar]
- Buglioni, L.; Raymenants, F.; Slattery, A.; Zondag, S.D.A.; Noël, T. Technological Innovations in Photochemistry for Organic Synthesis: Flow Chemistry, High-Throughput Experimentation, Scale-up, and Photoelectrochemistry. Chem. Rev. 2022, 122, 2752–2906. [Google Scholar] [CrossRef]
- Satake, M.; Ofuji, K.; Naoki, H.; James, K.J.; Furey, A.; McMahon, T.; Silke, J.; Yasumoto, T. Azaspiracid, a New Marine Toxin Having Unique Spiro Ring Assemblies, Isolated from Irish Mussels, Mytilus edulis. J. Am. Chem. Soc. 1998, 120, 9967–9968. [Google Scholar] [CrossRef]
- Ofuji, K.; Satake, M.; McMahon, T.; Silke, J.; James, K.J.; Naoki, H.; Oshima, Y.; Yasumoto, T. Two analogs of azaspiracid isolated from mussels, Mytilus edulis, involved in human intoxication in Ireland. Nat. Toxins 1999, 7, 99–102. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Li, Y.; Uesaka, N.; Koftis, T.V.; Vyskocil, S.; Ling, T.; Govindasamy, M.; Qian, W.; Bernal, F.; Chen, D.Y.K. Total Synthesis of the Proposed Azaspiracid-1 Structure, Part 1: Construction of the Enantiomerically Pure C1–C20, C21–C27, and C28–C40 Fragments. Angew. Chem. Int. Ed. 2003, 42, 3643–3648. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Chen, D.Y.K.; Li, Y.; Qian, W.; Ling, T.; Vyskocil, S.; Koftis, T.V.; Govindasamy, M.; Uesaka, N. Total Synthesis of the Proposed Azaspiracid-1 Structure, Part 2: Coupling of the C1–C20, C21–C27, and C28–C40 Fragments and Completion of the Synthesis. Angew. Chem. Int. Ed. 2003, 42, 3649–3653. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Vyskocil, S.; Koftis, T.V.; Yamada, Y.M.A.; Ling, T.; Chen, D.Y.K.; Tang, W.; Petrovic, G.; Frederick, M.O.; Li, Y.; et al. Structural Revision and Total Synthesis of Azaspiracid-1, Part 1: Intelligence Gathering and Tentative Proposal. Angew. Chem. Int. Ed. 2004, 43, 4312–4318. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Koftis, T.V.; Vyskocil, S.; Petrovic, G.; Ling, T.; Yamada, Y.M.A.; Tang, W.; Frederick, M.O. Structural Revision and Total Synthesis of Azaspiracid-1, Part 2: Definition of the ABCD Domain and Total Synthesis. Angew. Chem. Int. Ed. 2004, 43, 4318–4324. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Frederick, M.O.; Petrovic, G.; Cole, K.P.; Loizidou, E.Z. Total Synthesis and Confirmation of the Revised Structures of Azaspiracid-2 and Azaspiracid-3. Angew. Chem. Int. Ed. 2006, 45, 2609–2615. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Kværnø, L.; Mulder, J.A.; Raymer, B.; Dunn, T.B.; Beauchemin, A.; Olhava, E.J.; Juhl, M.; Kagechika, K. Total Synthesis of (+)-Azaspiracid-1. Part I: Synthesis of the Fully Elaborated ABCD Aldehyde. Angew. Chem. Int. Ed. 2007, 46, 4693–4697. [Google Scholar] [CrossRef]
- Evans, D.A.; Dunn, T.B.; Kværnø, L.; Beauchemin, A.; Raymer, B.; Olhava, E.J.; Mulder, J.A.; Juhl, M.; Kagechika, K.; Favor, D.A. Total Synthesis of (+)-Azaspiracid-1. Part II: Synthesis of the EFGHI Sulfone and Completion of the Synthesis. Angew. Chem. Int. Ed. 2007, 46, 4698–4703. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Chen, D.Y.K.; Li, Y.; Uesaka, N.; Petrovic, G.; Koftis, T.V.; Bernal, F.; Frederick, M.O.; Govindasamy, M.; Ling, T.; et al. Total Synthesis and Structural Elucidation of Azaspiracid-1. Synthesis-Based Analysis of Originally Proposed Structures and Indication of Their Non-Identity to the Natural Product. J. Am. Chem. Soc. 2006, 128, 2258–2267. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Pihko, P.M.; Bernal, F.; Frederick, M.O.; Qian, W.; Uesaka, N.; Diedrichs, N.; Hinrichs, J.; Koftis, T.V.; Loizidou, E.; et al. Total Synthesis and Structural Elucidation of Azaspiracid-1. Construction of Key Building Blocks for Originally Proposed Structure. J. Am. Chem. Soc. 2006, 128, 2244–2257. [Google Scholar] [CrossRef]
- Tillmann, U.; Elbrächter, M.; Krock, B.; John, U.; Cembella, A. Azadinium spinosum gen. et sp. nov. (Dinophyceae) identified as a primary producer of azaspiracid toxins. Eur. J. Phycol. 2009, 44, 63–79. [Google Scholar] [CrossRef] [Green Version]
- Jauffrais, T.; Kilcoyne, J.; Séchet, V.; Herrenknecht, C.; Truquet, P.; Hervé, F.; Bérard, J.B.; Nulty, C.; Taylor, S.; Tillmann, U.; et al. Production and Isolation of Azaspiracid-1 and -2 from Azadinium spinosum Culture in Pilot Scale Photobioreactors. Mar. Drugs 2012, 10, 1360–1382. [Google Scholar] [CrossRef] [Green Version]

























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña, L.F.; González-Andrés, P.; Parte, L.G.; Escribano, R.; Guerra, J.; Barbero, A.; López, E. Continuous Flow Chemistry: A Novel Technology for the Synthesis of Marine Drugs. Mar. Drugs 2023, 21, 402. https://doi.org/10.3390/md21070402
Peña LF, González-Andrés P, Parte LG, Escribano R, Guerra J, Barbero A, López E. Continuous Flow Chemistry: A Novel Technology for the Synthesis of Marine Drugs. Marine Drugs. 2023; 21(7):402. https://doi.org/10.3390/md21070402
Chicago/Turabian StylePeña, Laura F., Paula González-Andrés, Lucía G. Parte, Raúl Escribano, Javier Guerra, Asunción Barbero, and Enol López. 2023. "Continuous Flow Chemistry: A Novel Technology for the Synthesis of Marine Drugs" Marine Drugs 21, no. 7: 402. https://doi.org/10.3390/md21070402
APA StylePeña, L. F., González-Andrés, P., Parte, L. G., Escribano, R., Guerra, J., Barbero, A., & López, E. (2023). Continuous Flow Chemistry: A Novel Technology for the Synthesis of Marine Drugs. Marine Drugs, 21(7), 402. https://doi.org/10.3390/md21070402