The Tetrodotoxin Binding Site Is within the Outer Vestibule of the Sodium Channel

Abstract
:1. Introduction
- Guanidinium ions are (slightly) permeable, implying that they can interact within the pore as deep as the selectivity filter. TTX and STX, although somewhat larger than guanidinium ions, have guanidinium moietie(s), plausibly allowing them to reach the narrowest part of the pore and interact with the selectivity filter, but block because they are too large to pass through [7].
- Protons and the guanidinium toxins both have positive charges. Protons reduce both Na permeation and TTX block [5]. Their block is field-dependent; both appear to bind about one-third of the distance through the membrane electric field [7,8], presumably interacting with carboxylates involved in permeation.
2. The Role of Gating in TTX Block
3. Cloning of the Na Channel Family
4. Early Post-Cloning Insights
5. Mutations Affecting TTX Binding
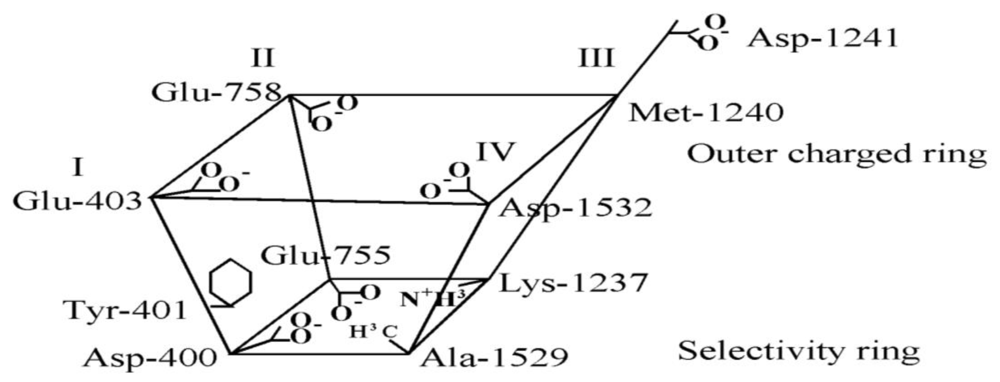
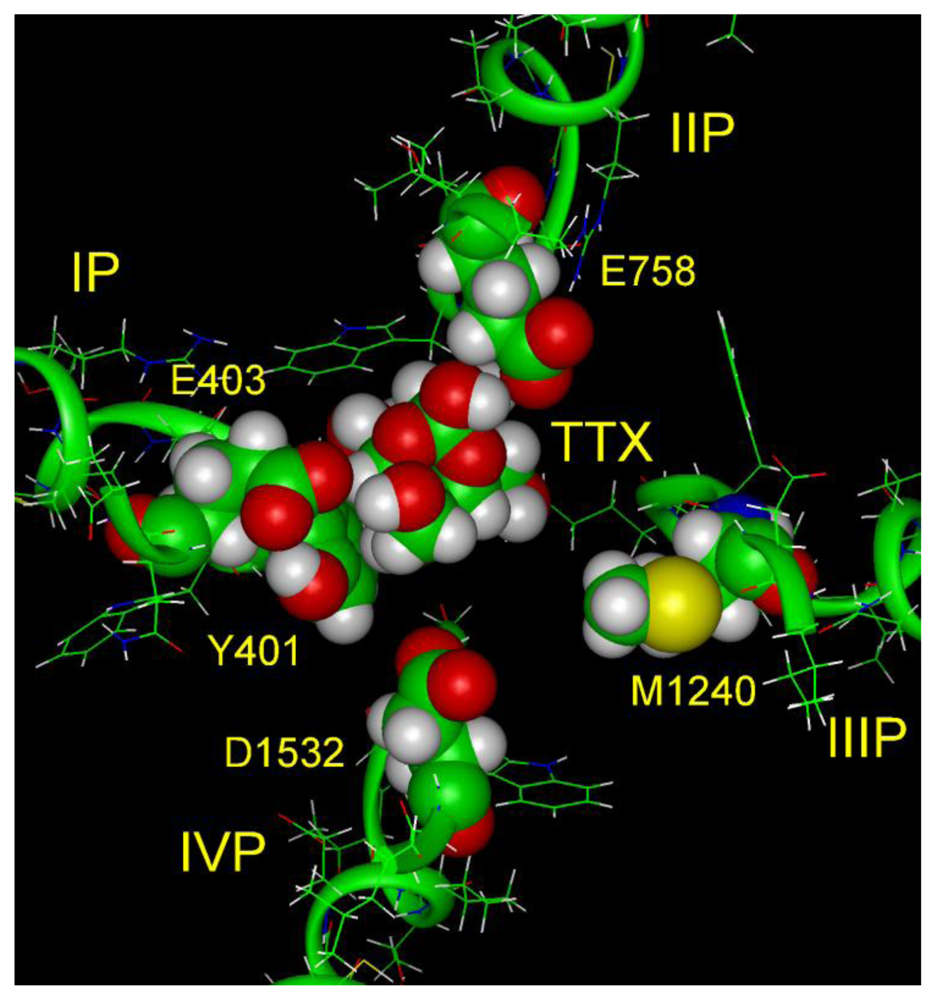
6. Modeling the TTX Binding Site
- The 1,2,3 guanidinium group of TTX and the 7,8,9 guanidinium group of STX are directed into the pore, where they interact most strongly with Glu-755 of domain II and Asp-400 of domain I.
- The 1,2,3 guanidinium group of STX, which is located at right angles to the plane of the other guanidinium group in the rigid STX structure, interacts with Asp-1532 of domain IV.
- In the plane of the 1,2,3 guanidinium group on the opposite side of STX is a C-12 gem-diol, postulated to interact with Glu-758 of domain II.
- There is a strong nonbonded interaction between the aromatic ring of Tyr-401 of domain I and the nonpolar surface of TTX.
7. Comparison of Model with Experimental Identification of Guanidinium Toxin Interactive Groups
8. Summary
References and Notes
- Kao, CY. Tetrodotoxin, saxitoxin and their significance in the study of excitation phenomenon. Pharm Rev 1966, 18, 997–1049. [Google Scholar]
- Kao, CY; Levinson, SR. Tetrodotoxin, saxitoxin, and the molecular biology of the sodium channel. Ann NY Acad Sci 1986, 479, 1–443. [Google Scholar]
- Narahashi, T; Deguchi, T; Urakawa, N; Ohkubo, Y. Stabilization and rectification of muscle fiber membrane by tetrodotoxin. Amer J Physiol 1960, 198, 934–938. [Google Scholar]
- Narahashi, T; Moore, JW; Scott, W. Tetrodotoxin blockage of sodium conductance increase in excitation. J Gen Physiol 1964, 47, 965–974. [Google Scholar]
- Hille, B. Pharmacological modifications of the sodium channels of frog nerve. J Gen Physiol 1968, 51, 199–219. [Google Scholar]
- Hille, B. Ion Channels of Excitable Membranes; Sinauer Associates, Inc: Sunderland, MA, USA, 2001; p. 534. [Google Scholar]
- Kao, CY; Nishiyama, A. Actions of saxitoxin on peripheral neuromuscular systems. J Physiol 1965, 180, 50–66. [Google Scholar]
- Ulbricht, W; Wagner, HH. The influence of pH on equilibrium effects of tetrodotoxin on myelinated nerve fibres of Rana esculenta. J Physiol 1975, 252, 159–184. [Google Scholar]
- Ulbricht, W; Wagner, HH. The influence of pH on the rate of tetrodotoxin action of myelinated nerve fibres. J Physiol 1975, 252, 185–202. [Google Scholar]
- Reed, JK; Raftery, MA. Properties of the tetrodotoxin binding component in plasma membranes isolated from Electrophorus electricus. Biochemistry 1976, 15, 944–953. [Google Scholar]
- Spalding, BC. Properties of toxin-resistant sodium channels produced by chemical modification in frog skeletal muscle. J Physiol 1980, 305, 485–500. [Google Scholar]
- Worley, JF, III; French, RJ; Krueger, BK. Trimethyloxonium modification of single batrachotoxin-activated sodium channels in planar bilayers. J Gen Physiol 1986, 87, 327–349. [Google Scholar]
- Henderson, R; Ritchie, JM; Strichartz, GR. Evidence that tetrodotoxin and saxitoxin act at a metal cation binding site in the sodium channels of nerve membrane. Proc Natl Acad Sci USA 1974, 71, 3936–3940. [Google Scholar]
- Doyle, DD; Guo, Y; Lustig, SL; Satin, J; Rogart, RB; Fozzard, HA. Divalent cation competition with [3H]saxitoxin binding to tetrodotoxin-resistant and -sensitive sodium channels. J Gen Physiol 1993, 101, 153–182. [Google Scholar]
- Aggarwal, R; Shorofsky, SR; Goldman, L; Balke, CW. Tetrodotoxin-blockable calcium currents in rat ventricular myocytes: a third type of cardiac cell sodium current. J Physiol 1997, 505, 353–369. [Google Scholar]
- Sha, Q; Robinson, SW; McCulle, SL; Shorofsky, SR; Welling, PA; Goldman, L; Balke, CW. An antisense oligonucleotide against H1 inhibits the classical sodium current but not ICa(TTX) in rat ventricular cells. J Physiol 2003, 547, 435–440. [Google Scholar]
- Sun, H; Varela, D; Chartier, D; Ruben, PC; Nattel, S; Zamponi, GW; LeBlanc, N. Differential interactions of Na channel toxins with T-type Ca channels. J Gen Physiol 2008, 132, 101–113. [Google Scholar]
- Boccaccio, A; Moran, O; Imoto, K; Conti, F. Tonic and phasic tetrodotoxin block of sodium channels with point mutations in the outer pore region. Biophysical J 1999, 77, 229–240. [Google Scholar]
- Sheets, MF; Hanck, DA. Molecular action of lidocaine on the voltage sensors of sodium channels. J Gen Physiol 2003, 121, 163–175. [Google Scholar]
- Bezanilla, F. The voltage sensor in voltage-dependent ion channels. Physiol Rev 2000, 80, 555–592. [Google Scholar]
- Armstrong, CM; Bezanilla, F. Charge movement associated with the opening and closing of the activation gates of the Na channel. J Gen Physiol 1974, 63, 533–552. [Google Scholar]
- Heggeness, ST; Starkus, JG. Saxitoxin and tetrodotoxin. Electrostatic effects on sodium channel gating current in crayfish axons. Biophys J 1986, 49, 629–643. [Google Scholar]
- Satin, J; Limbertis, JT; Kyle, JW; Rogart, RB; Fozzard, HA. The saxitoxin-tetrodotoxin binding site on cloned rat brain Iia Na channels is in the transmembrane electrical field. Biophys J 1994, 67, 1007–1014. [Google Scholar]
- Moczydlowski, E; Hall, S; Garber, SS; Strichartz, GS; Miller, C. Voltage-dependent blockade of muscle Na channels by guanidinium toxins. J Gen Physiol 1984, 84, 687–704. [Google Scholar]
- French, RJ; Worley, JF, III; Krueger, BK. Voltage-dependent block by saxitoxin of sodium channels incorporated, to planar lipid bilayers. Biophys J 1984, 45, 301–310. [Google Scholar]
- Khan, A; Romantseva, L; Lam, A; Lipkind, GM; Fozzard, HA. Role of the outer ring carboxylates of the rat skeletal muscle sodium channel pore in proton block. J Physiol 2002, 543, 71–85. [Google Scholar]
- Ritchie, JM; Rogart, RB. The binding of saxitoxin and tetrodotoxin to excitable tissue. Rev Physiol Biochem Pharmacol 1977, 79, 1–50. [Google Scholar]
- Agnew, WS; Levinson, SR; Brabson, JS; Raftery, MA. Purification of the tetrodotoxin-binding component associated with the voltage-sensitive sodium channel from Electrophorus electricus electroplax membranes. Proc Natl Acad Sci USA 1978, 75, 2606–2610. [Google Scholar]
- Noda, M; Shimizu, S; Tanabe, T; Takai, T; Kayano, T; Ikada, H; Takahashi, H; Nakayama, H; Kanaoka, Y; Minamino, N; Kangawa, K; Matsuo, H; Raftery, MA; Hirose, T; Furutani, Y; Inayama, S; Hayashida, H; Miyata, T; Numa, S. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature (Lond) 1984, 312, 121–127. [Google Scholar]
- Trimmer, JS; Cooperman, SS; Tomiko, SA; Zhou, J; Crean, SM; Boyle, MB; Kallen, RG; Sheng, Z; Barchi, RL; Sigworth, FJ; Goodman, RH; Agnew, WS; Mandel, G. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron 1989, 3, 33–49. [Google Scholar]
- George, AL, Jr; Ledbetter, DH; Kallen, RG; Barchi, RL. Assignment of a human skeletal muscle sodium channel alpha-subunit gene (SCN4aA) to 17q23.–25.3. Genomics 1991, 9, 555–556. [Google Scholar]
- Rogart, RB; Cribbs, LL; Muglia, LK; Kephart, DD; Kaiser, MW. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na channel isoform. Proc Natl Acad Sci USA 1989, 86, 8170–8174. [Google Scholar]
- Gellens, ME; George, AL, Jr; Chen, L; Chahine, M; Horn, R; Barchi, RL; Kallen, RG. Primary structure and functional expression of the human cardiac, voltage-dependent sodium channel. Proc Natl Acad Sci USA 1992, 89, 554–558. [Google Scholar]
- Goldin, AL. Evolution of voltage-gated Na channels. J Exp Biol 2002, 205, 575–584. [Google Scholar]
- Creighton, TE. Proteins; WH Freeman and Company: New York, NY, USA, 1984. [Google Scholar]
- Guy, HR; Conti, F. Pursuing the structure and function of voltage-gated channels. Trends Neurosci 1990, 13, 201–206. [Google Scholar]
- Guy, HR; Seetharamulu, P. Molecular model of the action potential sodium channel. Proc Natl Acad Sci USA 1986, 83, 508–512. [Google Scholar]
- Hille, B. The permeability of the sodium channel to metal ions in myelinated nerve. J Gen Physiol 1971, 58, 599–619. [Google Scholar]
- Sigworth, FJ; Spalding, BC. Chemical modification reduces the conductance of sodium channels in nerve. Nature 1980, 283, 293–295. [Google Scholar]
- Guy, HR. A model relating the sodium channel’s structure to its function. In Molecular Biology of Ionic Channels; Agnew, WS, Claudio, T, Sigworth, FJ, Eds.; Academic Press: San Diego, CA, USA, 1988; Volume 33, pp. 289–308. [Google Scholar]
- Pusch, M; Noda, M; Stühmer, W; Numa, S; Conti, F. Single point mutations of the sodium channel drastically reduce the pore permeability without preventing its gating. Eur Biophys J 1991, 20, 127–133. [Google Scholar]
- Noda, M; Suzuki, H; Numa, S; Stühmer, W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett 1989, 259, 213–216. [Google Scholar]
- Terlau, H; Heinemann, SH; Stühmer, W; Pusch, M; Conti, F; Imoto, K; Numa, S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Letts 1991, 293, 93–96. [Google Scholar]
- Schleif, T; Schonherr, R; Imoto, K; Heinemann, SH. Pore properties of rat brain II sodium channels mutated in the selectivity filter domain. Eur Biophys J 1996, 25, 75–91. [Google Scholar]
- Chiamvimonvat, N; Perez-Garcia, MT; Tomaselli, GF; Marban, E. Control of ion flux and selectivity by negatively charged residues in the outer mouth of rat sodium channels. J Physiol 1996, 491, 51–59. [Google Scholar]
- Favre, I; Moczydlowski, E; Schild, L. On the structural basis for ionic selectivity among Na, K, and Ca in the voltage-gated sodium channel. Biophys J 1996, 71, 3110–3125. [Google Scholar]
- Satin, J; Kyle, JW; Chen, M; Bell, P; Cribbs, LL; Fozzard, HA; Rogart, RB. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science 1992, 256, 1202–1205. [Google Scholar]
- Favre, I; Moczydlowski, E; Schild, L. Specificity for block by saxitoxin and divalent cations at a residue which determines sensitivity of sodium channel subtypes to guanidinium toxins. J Gen Physiol 1995, 106, 203–229. [Google Scholar]
- Backx, PH; Yue, DT; Lawrence, JH; Marban, E; Tomaselli, GF. Molecular localization of an ion-binding site within the pore of mammalian sodium channels. Science 1992, 257, 248–251. [Google Scholar]
- Heinemann, SH; Terlau, H; Imoto, K. Molecular basis for pharmacological differences between brain and cardiac channels. Eur J Physiol 1992, 422, 90–92. [Google Scholar]
- Penzotti, JL; Fozzard, HA; Lipkind, GM; Dudley, SC, Jr. Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na channel outer vestibule. Biophys J 1998, 75, 2647–2657. [Google Scholar]
- Lipkind, GM; Fozzard, HA. A structural model of the tetrodotoxin and saxitoxin binding site of the Na channel. Biophys J 1994, 66, 1–13. [Google Scholar]
- Dudley, SC, Jr; Chang, N; Hall, J; Lipkind, GM; Fozzard, HA; French, RJ. μ-Conotoxin GmA interactions with the voltage-gated Na channel predict a clockwise arrangement of the domains. J Gen Physiol 2000, 116, 679–690. [Google Scholar]
- Li, RA; Ennis, IL; French, RJ; Dudley, SC, Jr; Tomaselli, GF; Marban, E. Clockwise domain arrangement of the sodium channel revealed byμ-conotoxin (GmA) docking orientation. J Biol Chem 2001, 276, 11072–11077. [Google Scholar]
- Doyle, DA; Cabral, JM; Pfuetzner, RA; Kuo, A; Gulbis, JM; Cohen, SL; Chait, BT; MacKinnon, R. The structure of the potassium channel: molecular basis of K conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar]
- Lipkind, GM; Fozzard, HA. KcsA crystal structure as framework for a molecular model of the Na channel pore. Biochemistry 2000, 40, 6786–6794. [Google Scholar]
- Yamagishi, T; Janecki, M; Marban, E; Tomaselli, GF. Topology of the P segments in the sodium channel revealed by cysteine mutagenesis. Biophys J 1997, 73, 195–204. [Google Scholar]
- Kao, CY; Walker, SE. Active groups of saxitoxin and tetrodotoxin as deduced from actions of saxitoxin analogues on frog muscle and squid axon. J Physiol 1982, 323, 619–637. [Google Scholar]
- Narahashi, T; Moore, JW; Posten, RN. Tetrodotoxin derivatives: Chemical structure and blockage of nerve membrane conductance. Science 1967, 156, 976–979. [Google Scholar]
- Kao, CY; Yasumoto, T. Actions of 4-epitetrodotoxin and anhydrotetrodotoxin. Toxicon 1985, 23, 725–729. [Google Scholar]
- Rosker, C; Lohberger, B; Hofer, D; Steinecker, B; Quasthoff, S; Schreibmayer, W. The TTX metabolite 4,9-anhydro-TTX is a highly specific blocker of the Nav1.6 voltage-dependent sodium channel. Amer J Physiol Cell Physiol 2007, 293, C783–789. [Google Scholar]
- Yang, L; Kao, CY; Yasumoto, T. Actions of 6-epi tetrodotoxin and 1-deoxytetrodotoxin on the frog skeletal muscle fiber. Toxicon 1992, 30, 635–643. [Google Scholar]
- Yotsu-Yamashita, M; Sugimoto, A; Takai, A; Yasumoto, T. Effects of specific modifications of several hydroxyls of tetrodotoxin on its affinity to rat brain membrane. J Pharmacol Exp Ther 1999, 289, 1688–1696. [Google Scholar]
- Chen, SF; Hartmann, HA; Kirsch, GE. Cysteine mapping in the ion selectivity and toxin binding region of the cardiac Na channel pore. J Membr Biol 1997, 155, 11–25. [Google Scholar]
- Choudhary, G; Shang, L; Li, X; Dudley, SC, Jr. Energetic localization of saxitoxin in its channel binding site. Biophys J 2002, 83, 912–919. [Google Scholar]
- Santarelli, VP; Eastwood, AL; Dougherty, DA; Horn, R; Ahern, CA. A cation-π interactiion discriminates among sodium channels that are either sensitive or resistant to tetrodotoxin block. J Biol Chem 2007, 282, 8044–8051. [Google Scholar]
- Tikhonov, DB; Zhorov, BS. Modeling P-loops domain of sodium channel: Homology with potassium channels and interaction with ligands. Biophys J 2005, 88, 184–197. [Google Scholar]
- Guo, Z; Uehara, A; Ravindran, A; Bryant, SH; Hall, S; Moczydlowski, E. Kinetic basis for insensitivity to tetrodotoxin and saxitoxin in sodium channels of canine heart and denervated skeletal muscle. Biochemistry 1987, 26, 7346–7356. [Google Scholar]
- McNulty, MM; Edgerton, GB; Shah, RD; Hanck, DA; Fozzard, HA; Lipkind, GM. Charge at the lidocaine binding site residue Phe-1759 affects permeation in human cardiac voltage-gated sodium channels. J Physiol 2007, 581, 741–755. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Equilibrium IC50(μM) | kon (M−1s−1) | koff (s−1) | IC50 ratio |
|---|---|---|---|---|
| Native Nav1.4 | 0.036 ± 0.006 | 3.53 × 105 | 1.02 × 10−2. | 1 |
| D400A | 6.0 ± 0.7 | 2.93 × 102 | 1.26 × 10−2 | 168 |
| E403Q | 161 ± 14 | 2.48 × 102 | 1.57 × 10−2 | 4521 |
| E755A | 229 ± 21 | 2.90 × 102 | 2.69 × 10−2 | 6412 |
| E758Q | 6.2 ± 0.3 | 4.86 × 103 | 1.47 × 10−2 | 174 |
| K1237A | 23 ± 2 | 1.62 × 102 | 1.49 × 10−2 | 641 |
| M1240E | 87 ± 5 | 4.15 × 102 | 1.98 × 10−2 | 2433 |
| M1240K | 238 ± 17 | 1.02 × 102 | 1.58 × 10−2 | 6678 |
| D1532N | 1.5 ± 0.2 | 6.14 × 102 | 1.45 × 10−2 | 43 |
| Mutation | ΔΔGSTX ± SE | ΔΔGTTX ± SE | ΔΔGSTX/ΔΔGTTX ± SE |
|---|---|---|---|
| D400A | 3.8 ± 0.1 | 3.3 ± 0.3 | 1.2 ± 0.1 |
| Y401D | 2.4 ± 0.1 | 5.2 ± 0.3 | 0.5 ± 0.2 |
| Y401C | 2.7 ± 0.1 | 4.8 ± 0.3 | 0.6 ± 0.1 |
| E403Q | 6.2 ± 0.1 | 5.2 ± 0.3 | 1.2 ± 0.05 |
| E755A | 5.7 ± 0.1 | 5.4 ± 0.3 | 1.1 ± 0.1 |
| E758Q | 6.0 ± 0.1 | 3.3 ± 0.3 | 1.8 ± 0.05 |
| K1237A | 3.9 ± 0.2 | 4.1 ± 0.3 | 1.0 ± 0.1 |
| M1240E | 3.9 ± 0.1 | 4.8 ± 0.3 | 0.8 ± 0.1 |
| M1240K | 6.1 ± 0.2 | 5.4 ± 0.3 | 1.1 ± 0.1 |
| D1532N | 6.2 ± 0.2 | 2.4 ± 0.3 | 2.6 ± 0.1 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fozzard, H.A.; Lipkind, G.M. The Tetrodotoxin Binding Site Is within the Outer Vestibule of the Sodium Channel. Mar. Drugs 2010, 8, 219-234. https://doi.org/10.3390/md8020219
Fozzard HA, Lipkind GM. The Tetrodotoxin Binding Site Is within the Outer Vestibule of the Sodium Channel. Marine Drugs. 2010; 8(2):219-234. https://doi.org/10.3390/md8020219
Chicago/Turabian StyleFozzard, Harry A., and Gregory M. Lipkind. 2010. "The Tetrodotoxin Binding Site Is within the Outer Vestibule of the Sodium Channel" Marine Drugs 8, no. 2: 219-234. https://doi.org/10.3390/md8020219
APA StyleFozzard, H. A., & Lipkind, G. M. (2010). The Tetrodotoxin Binding Site Is within the Outer Vestibule of the Sodium Channel. Marine Drugs, 8(2), 219-234. https://doi.org/10.3390/md8020219