Effects of Tetrodotoxin on the Mammalian Cardiovascular System

Abstract
:1. Introduction
2. TTX Sensitive Na+ Channels in the Mammalian Heart—A Brief Summary
3. Systemic Effects of TTX on the Cardiovascular System—Lessons from Animal Experimentations
3.1. Hypotension
3.2. Bradycardia
3.3. Stoke volume reduction
3.4. Cardiac conduction disturbances
4. Human Tetrodotoxication
4.1. Epidemiology, symptoms, and prognosis
4.2. Correlation between blood TTX levels and intoxication symptoms
4.3. Cardiac excitation and performance in intoxicated patients
5. Conclusions
- Samples Availability: Available from the authors.
References and Notes
- Catterall, WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar]
- Catterall, WA; Goldin, AL; Waxman, SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 2005, 57, 397–409. [Google Scholar]
- Gellens, ME; George, AL; Chen, LQ; Chahine, M; Horn, R; Barchi, RL; Kallen, RG. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci USA 1992, 89, 554–558. [Google Scholar]
- Gaborit, N; Le Bouter, S; Szuts, V; Varro, A; Escande, D; Nattel, S; Demolombe, S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol 2007, 582, 675–693. [Google Scholar]
- Blechschmidt, S; Haufe, V; Benndorf, K; Zimmer, T. Voltage-gated Na+ channel transcript patterns in the mammalian heart are species-dependent. Prog Biophys Mol Biol 2008, 98, 309–318. [Google Scholar]
- Zimmer, T; Surber, R. SCN5A channelopathies - an update on mutations and mechanisms. Prog Biophys Mol Biol 2008, 98, 120–136. [Google Scholar]
- Lei, M; Huang, CL; Zhang, Y. Genetic Na+ channelopathies and sinus node dysfunction. Prog Biophys Mol Biol 2008, 98, 171–178. [Google Scholar]
- Goldin, AL. Resurgence of sodium channel research. Annu Rev Physiol 2001, 63, 871–894. [Google Scholar]
- Haufe, V; Chamberland, C; Dumaine, R. The promiscuous nature of the cardiac sodium current. J Mol Cell Cardiol 2007, 42, 469–477. [Google Scholar]
- Verkerk, AO; van Ginneken, AC; van Veen, TA; Tan, HL. Effects of heart failure on brain-type Na+ channels in rabbit ventricular myocytes. Europace 2007, 9, 571–577. [Google Scholar]
- Maier, SK; Westenbroek, RE; Schenkman, KA; Feigl, EO; Scheuer, T; Catterall, WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci USA 2002, 99, 4073–4078. [Google Scholar]
- Maier, SK; Westenbroek, RE; Yamanushi, TT; Dobrzynski, H; Boyett, MR; Catterall, WA; Scheuer, T. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci USA 2003, 100, 3507–3512. [Google Scholar]
- Baruscotti, M; DiFrancesco, D; Robinson, RB. A TTX-sensitive inward sodium current contributes to spontaneous activity in newborn rabbit sino-atrial node cells. J Physiol 1996, 492, 21–30. [Google Scholar]
- Baruscotti, M; Westenbroek, R; Catterall, WA; DiFrancesco, D; Robinson, RB. The newborn rabbit sino-atrial node expresses a neuronal type I-like Na+ channel. J Physiol 1997, 498, 641–648. [Google Scholar]
- Lei, M; Jones, SA; Liu, J; Lancaster, MK; Fung, SS; Dobrzynski, H; Camelliti, P; Maier, SK; Noble, D; Boyett, MR. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. J Physiol 2004, 559.3, 835–848. [Google Scholar]
- Maier, SKG; Westenbroek, RE; McCormick, KA; Curtis, R; Scheuer, T; Catterall, WA. Distinct subcellular localization of different sodium channel α and β subunits in single ventricular myocytes from mouse heart. Circulation 2004, 109, 1421–1427. [Google Scholar]
- Coraboeuf, E; Deroubaix, E; Coulombe, A. Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. Am J Physiol 1979, 236, H561–567. [Google Scholar]
- Haufe, V; Cordeiro, JM; Zimmer, T; Wu, YS; Schiccitano, S; Benndorf, K; Dumaine, R. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res 2005, 65, 117–127. [Google Scholar]
- Qu, Y; Karnabi, E; Chahine, M; Vassalle, M; Boutjdir, M. Expression of skeletal muscle Na(V)1.4 Na channel isoform in canine cardiac Purkinje myocytes. Biochem Biophys Res Commun 2007, 355, 28–33. [Google Scholar]
- Sills, MN; Xu, XC; Baracchini, E; Goodman, RH; Cooperman, SS; Mandel, G; Chien, KR. Expression of diverse Na+ channel messenger RNAs in rat myocardium. Evidence for a cardiac-specific Na+ channel. J Clin Invest 1989, 84, 331–336. [Google Scholar]
- Malhotra, JD; Chen, C; Rivolta, I; Abriel, H; Malhotra, R; Mattei, LN; Brosius, FC; Kass, RS; Isom, LL. Characterization of sodium channel α- and β-subunits in rat and mouse cardiac myocytes. Circulation 2001, 103, 1303–1310. [Google Scholar]
- Huang, B; El-Sherif, T; Gidh-Jain, M; Qin, D; El-Sherif, N. Alterations of sodium channel kinetics and gene expression in the postinfarction remodeled myocardium. J Cardiovasc Electrophysiol 2001, 12, 218–225. [Google Scholar]
- Haufe, V; Camacho, JA; Dumaine, R; Gunther, B; Bollensdorff, C; von Banchet, GS; Benndorf, K; Zimmer, T. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J Physiol 2005, 564.3, 683–696. [Google Scholar]
- Nikmaram, MR; Liu, J; Abdelrahman, M; Dobrzynski, H; Boyett, MR; Lei, M. Characterization of the effects of ryanodine, TTX, E-4031 and 4-AP on the sinoatrial and atrioventricular nodes. Prog Biophys Mol Biol 2008, 96, 452–464. [Google Scholar]
- Remy, C. Sur les poissons toxiques du Japon. Comp Rend Soc de Biol 1883, 35, 1–28. [Google Scholar]
- Takahashi, D; Inoko, Y. Experimentelle Untersuchungen über das Fugugift. Ein Beitrag zur Kenntnis der Fischgifte. Arch Exp Pathol Pharmakol 1890, 26, 401–418. [Google Scholar]
- Ishihara, F. Über die physiologischen Wirkungen des Fugutoxins. Mittheil Med Fak Tokio Univ 1918, 20, 375–426. [Google Scholar]
- Tsuda, K; Ikuma, S; Kawamura, M; Tachikawa, R; Sakai, K; Tamura, C; Amakasu, D. Tetrodotoxin. VII. On the structures of tetrodotoxin and its derivatives. Chem Pharm Bull 1964, 12, 1357–1374. [Google Scholar]
- Saint, DA. The cardiac persistent sodium current: an appealing therapeutic target. Br J Pharmacol 2008, 153, 1133–1142. [Google Scholar]
- Böhle, T; Benndorf, K. Voltage-dependent properties of three different gating modes in single cardiac Na+ channels. Biophys J 1995, 69, 873–882. [Google Scholar]
- Undrovinas, A; Maltsev, VA. Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovasc Hematol Agents Med Chem 2008, 6, 348–359. [Google Scholar]
- Protas, L; Dun, W; Jia, Z; Lu, J; Bucchi, A; Kumari, S; Chen, M; Cohen, IS; Rosen, MR; Entcheva, E; Robinson, RB. Expression of skeletal but not cardiac Na+ channel isoform preserves normal conduction in a depolarized cardiac syncytium. Cardiovasc Res 2009, 81, 528–535. [Google Scholar]
- Rogart, RB; Cribbs, LL; Muglia, LK; Kephart, DD; Kaiser, MW. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc Natl Acad Sci USA 1989, 86, 8170–8174. [Google Scholar]
- Protas, L; Oren, RV; Clancy, CE; Robinson, RB. Age-dependent changes in Na+ current magnitude and TTX-sensitivity in the canine sinoatrial node. J Mol Cell Cardiol 2010, 48, 172–180. [Google Scholar]
- Murtha, EF; Stabile, DE; Wills, JH. Some pharmacological effects of puffer poison. J Pharmacol Exp Ther 1958, 112, 247–254. [Google Scholar]
- Murtha, EF. Pharmacological study of poisons from shellfish and puffer fish. Ann NY Acad Sci 1960, 90, 820–836. [Google Scholar]
- Feinstein, MB; Paimre, M. Mechanism of cardiovascular action of tetrodotoxin in the cat. Block of conduction in peripheral sympathetic fibers. Circ Res 1968, 23, 553–565. [Google Scholar]
- Bernstein, ME. Pharmacologic effects of tetrodotoxin: cardiovascular and antiarrhythmic activities. Toxicon 1969, 7, 287–302. [Google Scholar]
- Kao, CY; Nagasawa, J; Spiegelstein, MY; Cha, YN. Vasodilatory effects of tetrodotoxin in the cat. J Pharmacol Exp Ther 1971, 178, 110–121. [Google Scholar]
- Bousquet, P; Feldman, J; Bloch, R; Schwartz, J. Medullary cardiovascular effects of tetrodotoxin in anaesthetized cats. Eur J Pharmacol 1980, 65, 293–296. [Google Scholar]
- Kao, CY. Tetrodotoxin, saxitoxin, and their significance in the study of excitation phenomena. Pharmacol Rev 1966, 18, 997–1049. [Google Scholar]
- Ogura, Y. Some recent problems on fugu-toxin, particularly on crystalline tetrodotoxin. Seitai No Kagaku 1958, 9, 281–287. [Google Scholar]
- Cheng, CPK; Cheng, KK; Wang, JCC. The action of tetrodotoxin on the heart. J Pathol 1970, 100, 121–126. [Google Scholar]
- Mackenzie, CF; Smalley, AJ; Barnas, GM; Park, SG. Tetrodotoxin infusion: nonventilatory effects and role in toxicity models. Acad Emerg Med 1996, 3, 1106–1112. [Google Scholar]
- Chang, FC; Benton, BJ; Salyer, JL; Foster, RE; Franz, DR. Respiratory and cardiovascular effects of tetrodotoxin in urethane-anesthetized guinea pigs. Brain Res 1990, 528, 259–268. [Google Scholar]
- Cheng, KK; Li, KM. The hypotensive action of puffer fish toxin. J Pathol Bacteriol 1966, 92, 471–476. [Google Scholar]
- Tomlinson, JC; James, TN. Pharmacologic actions of tetrodotoxin studied by direct perfusion of the sinus node. Circ Res 1968, 23, 501–506. [Google Scholar]
- Flachsenberger, WA. Respiratory failure and lethal hypotension due to blue-ringed octopus and tetrodotoxin envenomation observed and counteracted in animal models. J. Toxicol. Clin. Toxicol. 1986–1987, 24, 485–502. [Google Scholar]
- Kao, CY; Fuhrman, FA. Pharmacological studies on tarichatoxin, a potent neurotoxin. J Pharmacol Exp Ther 1963, 140, 31–40. [Google Scholar]
- Kreitner, D. Evidence for the existence of a rapid sodium channel in the membrane of rabbit sinoatrial cells. J Mol Cell Cardiol 1975, 7, 655–662. [Google Scholar]
- Sano, T; Yamagishi, S; Iida, Y. Several aspects on the spontaneous activity of the sinus node and its spread. Jpn Circulation J 1966, 30, 134–138. [Google Scholar]
- Verkerk, AO; Wilders, R; van Borren, MM; Tan, HL. Is sodium current present in human sinoatrial node cells. Int J Biol Sci 2009, 5, 201–204. [Google Scholar]
- Lei, M; Goddard, C; Liu, J; Léoni, AL; Royer, A; Fung, SS; Xiao, G; Ma, A; Zhang, H; Charpentier, F; Vandenberg, JI; Colledge, WH; Grace, AA; Huang, CL. Sinus node dysfunction following targeted disruption of the murine cardiac sodium channel gene Scn5a. J Physiol 2005, 567.2, 387–400. [Google Scholar]
- Baer, M; Best, PM; Reuter, H. Voltage-dependent action of tetrodotoxin in mammalian cardiac muscle. Nature 1976, 263, 344–345. [Google Scholar]
- Honerjäger, P; Reiter, M. The relation between the effects of veratridine on action potential and contraction in mammalian ventricular myocardium. Naunyn Schmiedebergs Arch Pharmacol 1975, 289, 1–28. [Google Scholar]
- Honerjäger, P; Loibl, E; Steidl, I; Schönsteiner, G; Ulm, K. Negative inotropic effects of tetrodotoxin and seven class 1 antiarrhythmic drugs in relation to sodium channel blockade. Naunyn Schmiedebergs Arch Pharmacol 1986, 332, 184–195. [Google Scholar]
- Tsukuda, O. Sur le mechanism de la bradycardie provoquée par la tetrodotoxine cristallisée. Ann Rep Inst Food Microbiol Chiba Univ 1957, 10, 78–83. [Google Scholar]
- Papadatos, GA; Wallerstein, PM; Head, CE; Ratcliff, R; Brady, PA; Benndorf, K; Saumarez, RC; Trezise, AE; Huang, CL; Vandenberg, JI; Colledge, WH; Grace, AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA 2002, 99, 6210–6215. [Google Scholar]
- Yoo, S; Dobrzynski, H; Fedorov, VV; Xu, SZ; Yamanushi, TT; Jones, SA; Yamamoto, M; Nikolski, VP; Efimov, IR; Boyett, MR. Localization of Na+ channel isoforms at the atrioventricular junction and atrioventricular node in the rat. Circulation 2006, 114, 1360–1371. [Google Scholar]
- Noguchi, T; Arakawa, O. Tetrodotoxin-distribution and accumulation in aquatic organisms, and cases of human intoxication. Mar Drugs 2008, 6, 220–242. [Google Scholar]
- Hwang, DF; Noguchi, T. Tetrodotoxin poisoning. Adv Food Nutr Res 2007, 52, 142–236. [Google Scholar]
- Azizul Haque, M; Tarikul Islam, Q; Razzak, MA; Faiz, MA; Iqbal Bari, MI. Neurological manifestations of puffer fish poisoning and it’s outcome: Study of 83 cases. TAJ 2008, 21, 121–125. [Google Scholar]
- Ahasan, HA; Mamun, AA; Karim, SR; Bakar, MA; Gazi, EA; Bala, CS. Paralytic complications of puffer fish (tetrodotoxin) poisoning. Singapore Med J 2004, 45, 73–74. [Google Scholar]
- Chowdhury, FR; Nazmul Ahasan, HA; Mamunur Rashid, AK; Al Mamun, A; Khaliduzzaman, SM. Tetrodotoxin poisoning: a clinical analysis, role of neostigmine and short-term outcome of 53 cases. Singapore Med J 2007, 48, 830–833. [Google Scholar]
- Fernández-Ortega, JF; Santos, JM; Herrera-Gutiérrez, ME; Fernández-Sánchez, V; Loureo, PR; Rancano, AA; Téllez-Andrade, A. Seafood intoxication by tetrodotoxin: First case in Europe. J Emerg Med 2009. [Google Scholar] [CrossRef]
- Rodriguez, P; Alfonso, A; Vale, C; Alfonso, C; Vale, P; Tellez, A; Botana, LM. First toxicity report of tetrodotoxin and 5,6,11-trideoxyTTX in the trumpet shell Charonia lampas lampas in Europe. Anal Chem 2008, 80, 5622–5629. [Google Scholar]
- Forster, G. A Voyage Round the World; London, UK, 1777; Volume 2, pp. 403–424.
- Fukuda, A; Tani, A. Records of puffer poisonings. Report 3. Nippon Igaku Oyobi Kenko Hoken 1941, 3528, 7–13. [Google Scholar]
- Benzer, T. Tetrodotoxin toxicity. E-Medicine. Updated October 2, 2009. Available online: http://emedicine.medscape.com/article/818763-print accessed on 18 March 2010.
- Sims, JK; Ostman, DC. Pufferfish poisoning: Emergency diagnosis and management of mild human tetrodotoxin. Ann Emerg Med 1986, 15, 1094–1098. [Google Scholar]
- Richardson, BW. Fish poisoning and the disease “siguatera”. Asclepiad (London) 1893, 10, 38–42. [Google Scholar]
- Noguchi, T; Ebesu, JSM. Puffer poisoning: epidemiology and treatment. J Toxicol-Toxin Rev 2001, 20, 1–10. [Google Scholar]
- Halstead, BW. Poisonous and Venomous Marine Animals of the World, 2nd ed; Darwin Press Inc.: Princeton, NJ, USA, 1988. [Google Scholar]
- Field, J. Puffer fish poisoning. J Accid Emerg Med 1998, 15, 334–336. [Google Scholar]
- Kanchanapongkul, J. Tetrodotoxin poisoning following ingestion of the toxic eggs of the horseshoe crab Carcinoscorpius rotundicauda a case series from 1994 through 2006. Southeast Asian J. Trop. Med Public Health 2008, 39, 303–306. [Google Scholar]
- How, CK; Chern, CH; Huang, YC; Wang, LM; Lee, CH. Tetrodotoxin poisoning. Am J Emerg Med 2003, 21, 51–54. [Google Scholar]
- Isbister, GK; Son, J; Wang, F; Maclean, CJ; Lin, CS; Ujma, J; Balit, CR; Smith, B; Milder, DG; Kiernan, MC. Puffer fish poisoning: a potentially life-threatening condition. Med J Aust 2002, 177, 650–653. [Google Scholar]
- Oda, K; Araki, K; Totoki, T; Shibasaki, H. Nerve conduction study of human tetrodotoxication. Neurology 1989, 39, 743–745. [Google Scholar]
- Yamazaki, M; Shibuya, N. Motor nerve conduction velocity is useful for patients with tetrodotoxin. Anesth Analg 1995, 80, 857. [Google Scholar]
- O’Leary, MA; Schneider, JJ; Isbister, GK. Use of high performance liquid chromatography to measure tetrodotoxin in serum and urine of poisoned patients. Toxicon 2004, 44, 549–553. [Google Scholar]
- Tsai, YH; Hwang, DF; Cheng, CA; Hwang, CC; Deng, JF. Determination of tetrodotoxin in human urine and blood using C18 cartridge column, ultrafiltration and LC-MS. J Chromatogr B Analyt Technol Biomed Life Sci 2006, 832, 75–80. [Google Scholar]
- Centers for Disease Control and Prevention. Tetrodotoxin poisoning associated with eating puffer fish transported from Japan-California, 1996. MMWR Morb. Mortal Wkly Rep 1996, 45, 389–391.
- Yang, CC; Han, KC; Lin, TJ; Tsai, WJ; Deng, JF. An outbreak of tetrodotoxin poisoning following gastropod mollusc consumption. Hum Exp Toxicol 1995, 14, 446–450. [Google Scholar]
- Ellis, RM; Jelinek, GA. Never eat an ugly fish: three cases of tetrodotoxin poisoning from Western Australia. Emerg Med 1997, 9, 136–142. [Google Scholar]
- Lau, FL; Wong, CK; Yip, SH. Puffer fish poisoning. J Accid Emerg Med 1995, 12, 214–215. [Google Scholar]
- Yin, HL; Lin, HS; Huang, CC; Hwang, DF; Liu, JS; Chen, WH. Tetrodotoxication with Nassauris glans: a possibility of tetrodotoxin spreading in marine products near Pratas Island. Am J Trop Med Hyg 2005, 73, 985–990. [Google Scholar]
- Trevett, AJ; Mavo, B; Warrell, DA. Tetrodotoxic poisoning from ingestion of a porcupine fish (Diodon hystrix) in Papua New Guinea: nerve conduction studies. Am J Trop Med Hyg 1997, 56, 30–32. [Google Scholar]
- Chew, SK; Goh, CH; Wang, KW; Mah, PK; Tan, BY. Puffer fish (tetrodotoxin) poisoning: clinical report and role of anti-cholinesterase drugs in therapy. Singapore Med J 1983, 24, 168–171. [Google Scholar]
- Tambyah, PA; Hui, KP; Gopalakrishnakone, P; Chin, NK; Chan, TB. Central-nervous-system effects of tetrodotoxin poisoning. Lancet 1994, 343, 538–539. [Google Scholar]
- Park, CW; Ryoo, E; Yang, HJ; Lee, K. Clinical analysis of puffer fish poisoning. Ann Emerg Med 1999, 34, S57. [Google Scholar]
- Cohen, SA. Immunocytochemical localization of rH1 sodium channel in adult rat heart atria and ventricle. Presence in terminal intercalated disks. Circulation 1996, 94, 3083–3086. [Google Scholar]
- Dudel, J; Peper, K; Rüdel, R; Trautwein, W. The effect of tetrodotoxin on the membrane current in cardiac muscle (Purkinje fibers). Pflugers Arch 1967, 295, 213–226. [Google Scholar]
- Turner, LA; Marijic, J; Kampine, JP; Bosnjak, ZJ. A comparison of the effects of halothane and tetrodotoxin on the regional repolarization characteristics of canine Purkinje fibers. Anesthesiology 1990, 73, 1158–1168. [Google Scholar]
- Brette, F; Orchard, CH. No apparent requirement for neuronal sodium channels in excitation-contraction coupling in rat ventricular myocytes. Circ Res 2006, 98, 667–674. [Google Scholar]


{kind=link}
| Study | Results and suggested function of TTXs Na+ channels | Species |
|---|---|---|
| Sinus node automaticity and control of heart rate | ||
| [13,14] | Nav1.1 transcripts and TTXs currents (IC50 ~ 26 nM) in newborn (but not adult) sinus node cells, suggesting that, depending on age, TTXs Na+ channels contribute to slow diastolic depolarization. | Rabbit |
| [12] | Reduction in spontaneous heart rate by blocking Nav1.1/Nav1.3 at 100 nM TTX; important contribution of TTXs Na+ channels to sinus node automaticity and rhythm, suggesting a possible contribution to SSS in man. | Mouse |
| [15] | Slowing of pacemaking in intact sinus node preparations and isolated cells at 10 and 100 nM TTX, slowing of both pacemaking and sinus node conduction at 1–30 μM TTX. | Mouse |
| Efficient EC coupling and increased cardiac contractility | ||
| [11] | Reduction of left ventricular function at 100 and 200 nM TTX, suggesting an unexpected role of brain-type Na+ channels in excitation-contraction coupling. | Mouse, guinea pig |
| [16] | Localization of brain-type Na+ channels and two β subunits in transverse tubules of myocytes, suggesting AP propagation from the cell surface into the interior by defined α/β-channel complexes. | Mouse |
| Purkinje fibers: Efficient cardiac conduction and AP prolongation | ||
| [17] | Shortening of AP duration, but not of the maximum rate of rise, at low TTX (≥33 nM). | Dog |
| [18] | Higher transcript levels and TTXs currents in Purkinje fibers (35 and 22%), when compared to ventricular myocytes (<20 and 10%, respectively). | Dog |
| [19] | Expression of Nav1.4 in cardiac Purkinje myocytes (PCR, immunofluorescence). | Dog |
| Other reports | ||
| [20] | Detection of Nav1.1 transcripts in the heart. | Rat |
| [21] | Cardiac Na+ channels are composed of either Nav1.1 or Nav1.5, and both associate with β1 and β2. | Mouse, rat |
| [22] | Up-regulation of Nav1.1 and increased TTXs Na+ current in the postinfarction remodeled myocardium. | Rat |
| [5,23] | Large transcript pool in whole hearts (30–40%), smaller TTXs Na+ currents in ventricular myocytes (8%) of mice (not observed in pigs and humans). | Mouse |
| [10] | Middle region of ventricular myocytes contains only TTXs Na+ channels, that can be blocked by 50 nM TTX. | Rabbit |
| [24] | Prolongation of the cycle length of the spontaneous pacemaker activity at 100 nM TTX by 22% and 53% in sinoatrial and atrioventricular node preparations, respectively. | Mouse |
| Study | Species | TTX app-lication | TTX dose | Cardiovascular effects |
|---|---|---|---|---|
| [26] | dog, cat, rabbit, rat | s.c. | Tetrodon hard roe extracts | mild intoxication: ataxia and paresis at normal heart function and blood pressure severe intoxication: cyanosis, areflexia, respiratory arrest, hypotension, bradycardia, AV block |
| [27] | rabbit bufo | i.v. s.c. | ≥0.7 MLD | hypotension, SA and AV block, but no direct chronotropic or inotropic effects |
| [47] | dog | into sinus node artery | up to 310 nM 3.1 μM | unchanged heart rate bradycardia (immediate slowing by 26 beats/min) |
| [38] | dog cat | i.v. i.v. | 5 μg/kg 7 μg/kg | bradycardia of sinoatrial origin, decrease in conduction, hypotension respiratory arrest, no change or decrease in heart rate, hypotension |
| [35] | cat cat, dog | i.p. i.v. | 1 μg/kg daily 5 μg/kg | no pathological change rapid fall in blood pressure, cessation of respiration, bradycardia, no significant ECG abnormalities, reduced contractile force |
| [36] | dog cat rabbit | i.v. i.v. isolated heart | 5 μg/kg 5 μg/kg ~100 nM | no significant ECG abnormalities, reduced contractile force, hypotension respiratory arrest, bradycardia at otherwise unchanged ECG, hypotension no significant effect |
| [46] | rat | i.v. | 5 μg/kg | sharp fall of blood pressure at initially unchanged heart rate and stroke volume |
| [43] | rat | i.v. | 2.5 μg/kg 10–40 μg/kg 80 μg/kg | unchanged blood flow in the ascending aorta, hypotension and bradycardia at otherwise unchanged ECG dose-related reduction of blood flow in the ascending aorta, bradycardia appeared unrelated to the size of the TTX dose, first degree AV block, bundle brunch block, ventricular flutter/fibrillation ventricular asystole |
| [43] | pithed rat | i.v. | 10–20 μg/kg | reduction of blood flow in the ascending aorta, hypotension, transient mild bradycardia, transient first degree AV block, bundle brunch block |
| [43] | rat | isolated heart | 1.0 to 4.0 μg (3 μM solution) | dissociation/cessation of ventricular contractions depending on dose |
| [37] | cat | i.v. | 1 μg/kg 2.5–10 μg/kg | unchanged heart rate, hypotension bradycardia, hypotension, slight PR prolongation, unchanged QS interval, reduced left ventricular force and reduced stroke volume |
| [39] | cat | i.v. | 1 μg/kg | unchanged heart rate, hypotension due to a direct relaxing effect on vascular smooth muscles |
| [49] | cat | i.v. | 1.4–3 μg/kg | prompt fall of blood pressure at unchanged heart rate and pulse pressure; initially no striking ECG alterations, increased amplitude of QRS and T wave after the development of hypotension |
| [45] | guinea pig | i.p. | 15 μg/kg | response before respiratory arrest (≤10.3 min from injection time point): decline in blood pressure, but no change in heart rate and ECG waveform cardiac response shortly after respiratory arrest: paroxysmal ventricular tachycardia, sinus bradycardia, AV block |
| [48] | rat | i.a. | 20 μg/kg | rapid and severe hypotension, bradycardia, heart rate increased shortly after artificial respiration was commenced |
| [44] | dog | i.v. (slowly) | 9.3 μg/kg/hr | at apnoe: bradycardia at unchanged stroke volume, hypotension, decreased total peripheral resistance, increased pulmonary vascular resistance and increased pulmonary arterial pressure; at higher TTX concentrations (12–20 μg/kg/hr), dogs died before or shortly after apnoe, which was due to fatal hypotension |
| Degree | Symptoms |
|---|---|
| First | Oral numbness and paraesthesia, sometimes accompanied by gastrointestinal symptoms (nauseaa)) |
| Second | Numbness of face and other areas, advanced paraesthesia, motor paralysis of extremities, incoordination, slurred speech, but still normal reflexes |
| Third | Gross muscular incoordination, aphonia, dysphagia, dyspnoea, cyanosis, drop in blood pressure, fixed/dilated pupils, precordial pain, but victims are still conscious |
| Fourth | Severe respiratory failure and hypoxia, severe hypotension, bradycardia, cardiac arrhythmia, heart continues to pulsate for a short period |
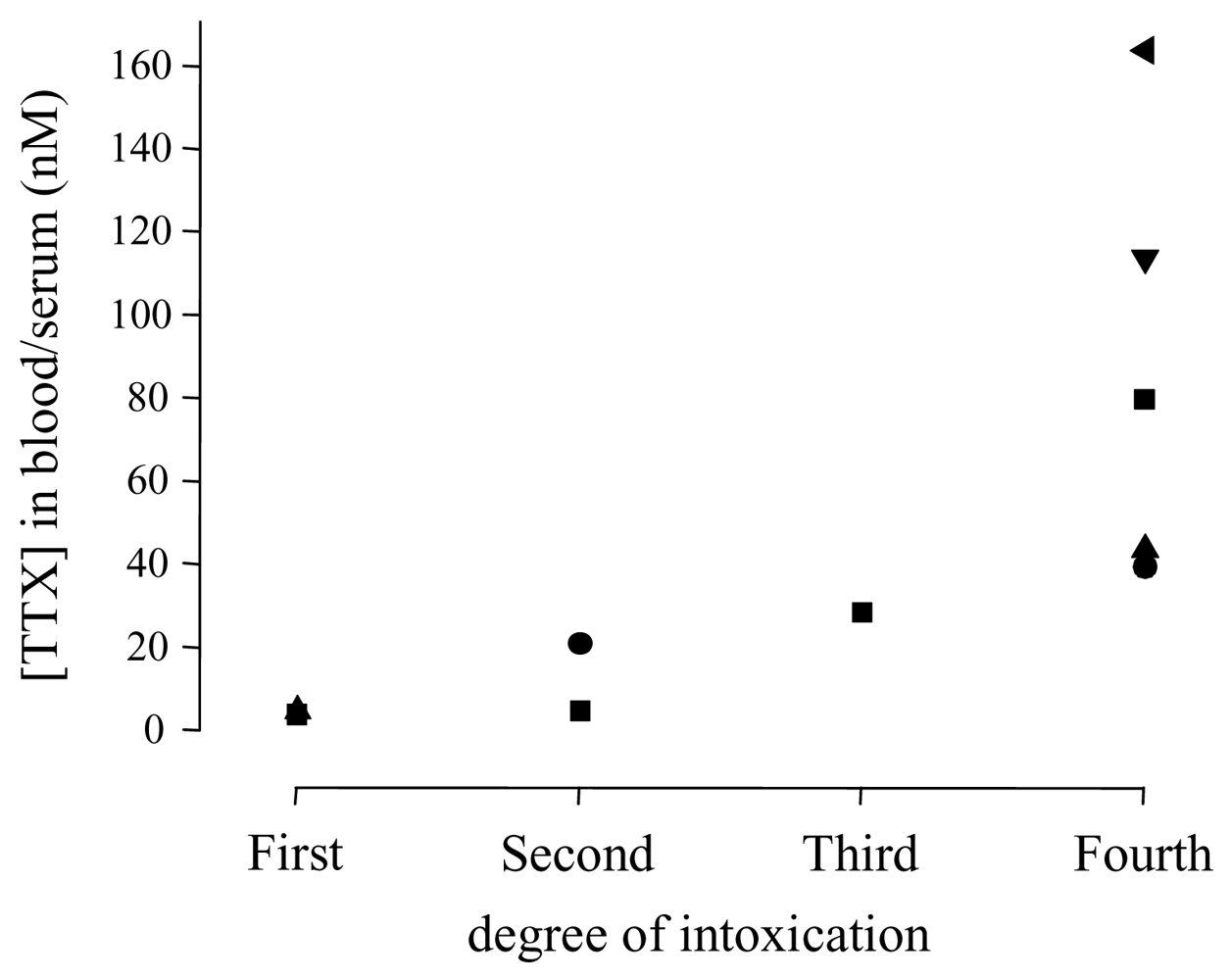
| Study | Cases | Grade | TTX (nM)a | Hypotension | Sinus bradycardia | ECG | Artificial respiration | Comments |
|---|---|---|---|---|---|---|---|---|
| [82] | 3 | 1–2 | no | no | no | oxygen saturation 96–99% | ||
| [76,81] | 4 | 1–2 | 4.5–21.1 | no or mild | no | normal | no | mild hypercapnia |
| [83] | 16 | 1–2 | no | eight patients had hypertension | ||||
| [84] | 3 | 1–2 | no | no | ||||
| [75] | 177 | 1–3 | no | no | all recovered completely | |||
| [85] | 6 | 1–3 | no | |||||
| [70] | 1 | 2–3 | yes | yes | normalb | no | hypoxemia, diabetes mellitusc | |
| [74] | 1 | 2–3 | no | no | no | normal arterial pO2 | ||
| [86] | 4 | 2–3 | no | intermittentd | normal | no | normal blood pressure, no hypoxia | |
| [77,80] | 11 | 2–3 | <5–5 (grade 2) | no | no | yes (grade 3) | no cardiovascular effects, no fatalities | |
| [87] | 1 | 3 | no | yes | normal | yes | reduced sensory and motor conduction velocities, decrease in evoked amplitudes | |
| [88] | 1 | 3 | no | no | normal | yes | decreased arterial pO2 | |
| [76,81] | 1 | 3 | 28.6 | yes | no | normal | yes | |
| [83] | 1 | 3–4 | hypertensionc | no | Yes (cyanosis) | diabetes mellitusc, renal failure and death (intoxication by molluscs) | ||
| [89] | 1 | 4 | yes | stable cardiovascular status after cardiopulmonary resuscitation; TTX-induced cranial diabetes insipidus | ||||
| [65,66] | 1 | 4 | 77/83 | no | no | normal | yes | non-excitability of sensory and motor nerves |
| [85] | 1 | 4 | yes | no | yes | cardiac arrest before admission to the ED, resuscitation to sinus rhythm, patient died | ||
| [76,81] | 1 | 4 | 40.6 | yes | yes | AV block | yes | diabetes mellitusc, full cardiac arrest, spontaneous circulation after resuscitation, but patient died due to multi-organ failure |
| [78] | 1 | 4 | 114 | yes | yes | normal | yes | hypothermia, blood gases were unremarkable |
| [79] | 1 | 4 | 164 | yes | yes | complete block of motor nerve conduction | ||
| [75] | 68 | 4 | Yes (14/68) | Yes (all) | five fatalities, one patient with brain damage | |||
| [62] | 83 | 1–4 | 5–43 e | seven fatal cases (respiratory arrest) | ||||
| [63] | 37 | 1–4 | eight fatalities | |||||
| [64] | 53 | 1–4 | eight fatalities | |||||
| [90] | 40 | 1–4 | no fatalities |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zimmer, T. Effects of Tetrodotoxin on the Mammalian Cardiovascular System. Mar. Drugs 2010, 8, 741-762. https://doi.org/10.3390/md8030741
Zimmer T. Effects of Tetrodotoxin on the Mammalian Cardiovascular System. Marine Drugs. 2010; 8(3):741-762. https://doi.org/10.3390/md8030741
Chicago/Turabian StyleZimmer, Thomas. 2010. "Effects of Tetrodotoxin on the Mammalian Cardiovascular System" Marine Drugs 8, no. 3: 741-762. https://doi.org/10.3390/md8030741
APA StyleZimmer, T. (2010). Effects of Tetrodotoxin on the Mammalian Cardiovascular System. Marine Drugs, 8(3), 741-762. https://doi.org/10.3390/md8030741