Genetic Analysis of West Nile Virus Isolates from an Outbreak in Idaho, United States, 2006–2007

Abstract
:1. Introduction

2. Experimental Section
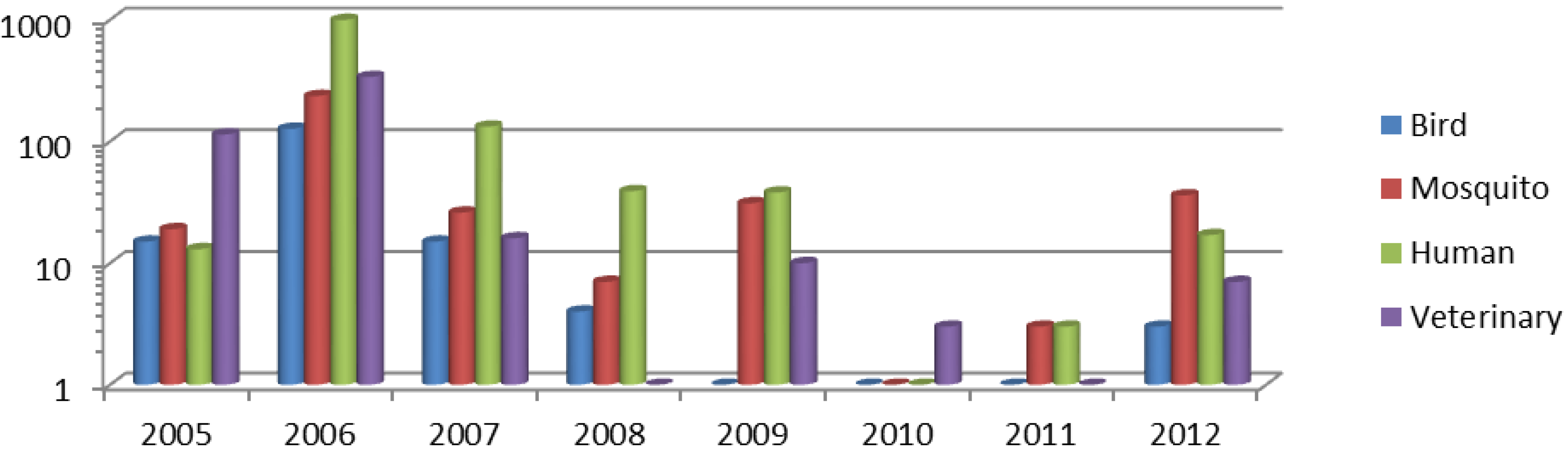
2.1. Study Sample

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Isolate | Host | Year | State | GenBank | ID-Δ13 |
|---|---|---|---|---|---|---|
| of isolation | accession no. | |||||
| 1 | ARC1-06 | Human | 2006 | ID | N/A | N |
| 2 | ARC10-06 | Human | 2006 | ID | JF957161 | Y |
| 3 | ARC104-06 | Human | 2006 | ID | N/A | Y |
| 4 | ARC105-06 | Human | 2006 | ID | N/A | Y |
| 5 | ARC106-06 | Human | 2006 | ID | N/A | Y |
| 6 | ARC108-06 | Human | 2006 | ID | N/A | Y |
| 7 | ARC112-06 | Human | 2006 | ID | N/A | Y |
| 8 | ARC13-06 | Human | 2006 | ID | JF957162 | Y |
| 9 | ARC17-06 | Human | 2006 | ID | JF957163 | Y |
| 10 | ARC19-06 | Human | 2006 | ID | N/A | N |
| 11 | ARC22-06 | Human | 2006 | ID | N/A | N |
| 12 | ARC23-06 | Human | 2006 | ID | JF957164 | N |
| 13 | ARC26-06 | Human | 2006 | ID | N/A | N |
| 14 | ARC27-06 | Human | 2006 | ID | JF957165 | Y |
| 15 | ARC28-06 | Human | 2006 | ID | N/A | N |
| 16 | ARC30-06 | Human | 2006 | ID | N/A | N |
| 17 | ARC31-06 | Human | 2006 | ID | N/A | N |
| 18 | ARC32-06 | Human | 2006 | ID | N/A | N |
| 19 | ARC41-06 | Human | 2006 | ID | N/A | N |
| 20 | ARC42-06 | Human | 2006 | ID | N/A | Y |
| 21 | ARC57-06 | Human | 2006 | ID | N/A | N |
| 22 | ARC60-06 | Human | 2006 | ID | N/A | N |
| 23 | ARC61-06 | Human | 2006 | ID | N/A | Y |
| 24 | ARC-Z-06 | Human | 2006 | ID | N/A | N |
| 25 | ARC140-07 | Human | 2007 | ID | JF957168 | Y |
| 26 | ID7mq-07 | Mosquito | 2007 | ID | N/A | Y |
| 27 | ID19bd-07 | Avian | 2007 | ID | N/A | Y |
| 28 | ID20bd-07 | Avian | 2007 | ID | N/A | Y |
| 29 | ID21bd-07 | Avian | 2007 | ID | JF957171 | Y |
| 30 | ID28bd-07 | Avian | 2007 | ID | JF957172 | Y |
| 31 | ID29bd-07 | Avian | 2007 | ID | N/A | Y |
| 32 | ARC135-06 | Human | 2006 | IL | N/A | N |
| 33 | ARC126-06 | Human | 2006 | KS | N/A | N |
| 34 | ARC134-06 | Human | 2006 | MD | N/A | N |
| 35 | ARC-W-06 | Human | 2006 | MN | N/A | N |
| 36 | ARC-Y-06 | Human | 2006 | MN | N/A | N |
| 37 | ARC127-06 | Human | 2006 | NE | N/A | N |
| 38 | ARC128-06 | Human | 2006 | NE | N/A | N |
| 39 | ARC131-06 | Human | 2006 | NE | N/A | N |
| 40 | ARC2-06 | Human | 2006 | NE | N/A | N |
| 41 | ARC3-06 | Human | 2006 | NE | N/A | N |
| 42 | ARC38-06 | Human | 2006 | NE | N/A | N |
| 43 | ARC125-06 | Human | 2006 | NY | N/A | N |
| 44 | ARC132-06 | Human | 2006 | SD | N/A | N |
| 45 | ARC133-06 | Human | 2006 | TX | N/A | N |
| 46 | ARC102-06 | Human | 2006 | UT | N/A | N |
| 47 | ARC11-06 | Human | 2006 | UT | N/A | N |
| 48 | ARC25-06 | Human | 2006 | UT | N/A | N |
| 49 | ARC33-06 | Human | 2006 | UT | JF957166 | N |
| 50 | BSL103-06 | Human | 2006 | SD | N/A | N |
| 51 | BSL106-06 | Human | 2006 | ND | JF957167 | Y |
| 52 | BSL107-06 | Human | 2006 | ND | N/A | N |
| 53 | BSL110-06 | Human | 2006 | SD | N/A | N |
| 54 | CO2-07 | Human | 2007 | CO | N/A | N |
| 55 | CO3-07 | Human | 2007 | CO | N/A | N |
| 56 | CO4-07 | Human | 2007 | CO | JF957169 | N |
| 57 | CO5-07 | Human | 2007 | CO | JF957170 | N |
2.2. Virus Isolation
2.3. RNA Extraction and Polymerase Chain Reaction (PCR)

2.4. Multiplex PCR
2.5. DNA Sequencing, Assembly and Analysis
2.6. Illumina NGS
2.7. NGS Data Analysis
3. Results and Discussion
3.1. Nucleotide Mutations and Amino Acid Substitutions Identified by Sanger Sequencing
| Gene | prM | E | NS1 | NS2A | NS2B | NS3 | NS4A | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nt # | 660 | 1320 | 1442 | 1974 | 2466 | 2661 | 3228 | 3399 | 3927 | 4146 | 4255 | 4803 | 6138 | 6238 | 6426 | 6721 | 6765 | ||||||||||
| NY99 | C | A | T | C | C | G | T | T | T | A | C | C | C | C | C | G | T | ||||||||||
| ARC10-06 | T | G | C | T | T | A | C | C | C | G | T | T | T | T | T | A | C | ||||||||||
| ARC13-06 | T | G | C | T | T | A | C | C | C | G | T | T | T | T | T | A | C | ||||||||||
| ARC17-06 | T | G | C | T | T | A | C | C | C | G | T | T | T | T | T | A | C | ||||||||||
| ARC23-06 | T | G | C | T | T | C | G | T | T | T | A | C | |||||||||||||||
| ARC27-06 | T | G | C | T | T | A | C | C | C | G | T | T | T | T | T | A | C | ||||||||||
| ARC33-06 | T | G | C | T | T | C | G | T | T | T | T | A | C | ||||||||||||||
| BSL106-06 | T | G | C | T | T | A | C | C | C | G | T | T | T | T | T | A | C | ||||||||||
| ARC140-07 | T | G | C | T | T | A | C | C | C | G | T | T | T | T | T | A | C | ||||||||||
| ID21bd-07 | T | G | C | T | T | A | C | C | C | G | T | T | T | T | T | A | C | ||||||||||
| ID28bd-07 | T | G | C | T | T | A | C | C | C | G | T | T | T | T | T | A | C | ||||||||||
| CO4-07 | T | C | T | G | T | T | T | T | |||||||||||||||||||
| CO5-07 | T | G | C | T | T | C | G | T | T | T | T | A | C | ||||||||||||||
| Gene | NS4B | NS5 | 3′UTR | ||||||||||||||||||||||||
| nt # | 6936 | 6996 | 7015 | 7209 | 7245 | 7269 | 7938 | 8550 | 8621 | 8811 | 9264 | 9352 | 9660 | 10062 | Δ13 | 10851 | |||||||||||
| NY99 | T | C | T | A | T | T | T | C | A | T | T | C | C | T | N | A | |||||||||||
| ARC10-06 | C | T | C | T | C | C | C | T | G | C | C | T | T | C | Y | G | |||||||||||
| ARC13-06 | C | T | C | T | C | C | C | T | G | C | C | T | T | C | Y | G | |||||||||||
| ARC17-06 | C | T | C | T | C | C | C | T | G | C | C | T | T | C | Y | G | |||||||||||
| ARC23-06 | C | T | C | C | C | T | G | C | C | T | T | C | N | G | |||||||||||||
| ARC27-06 | C | T | C | T | C | C | C | T | G | C | C | T | T | C | Y | G | |||||||||||
| ARC33-06 | C | T | C | C | C | T | G | C | C | T | T | C | N | G | |||||||||||||
| BSL106-06 | C | T | C | T | C | C | C | T | G | C | C | T | T | C | Y | G | |||||||||||
| ARC140-07 | C | T | C | T | C | C | C | T | G | C | C | T | T | C | Y | G | |||||||||||
| ID21bd-07 | C | T | C | T | C | C | C | T | G | C | C | T | T | C | Y | ||||||||||||
| ID28bd-07 | C | T | C | T | C | C | C | T | G | C | C | T | T | C | Y | G | |||||||||||
| CO4-07 | T | C | C | C | T | N | G | ||||||||||||||||||||
| CO5-07 | C | T | C | C | C | T | G | C | C | T | T | C | N | G | |||||||||||||
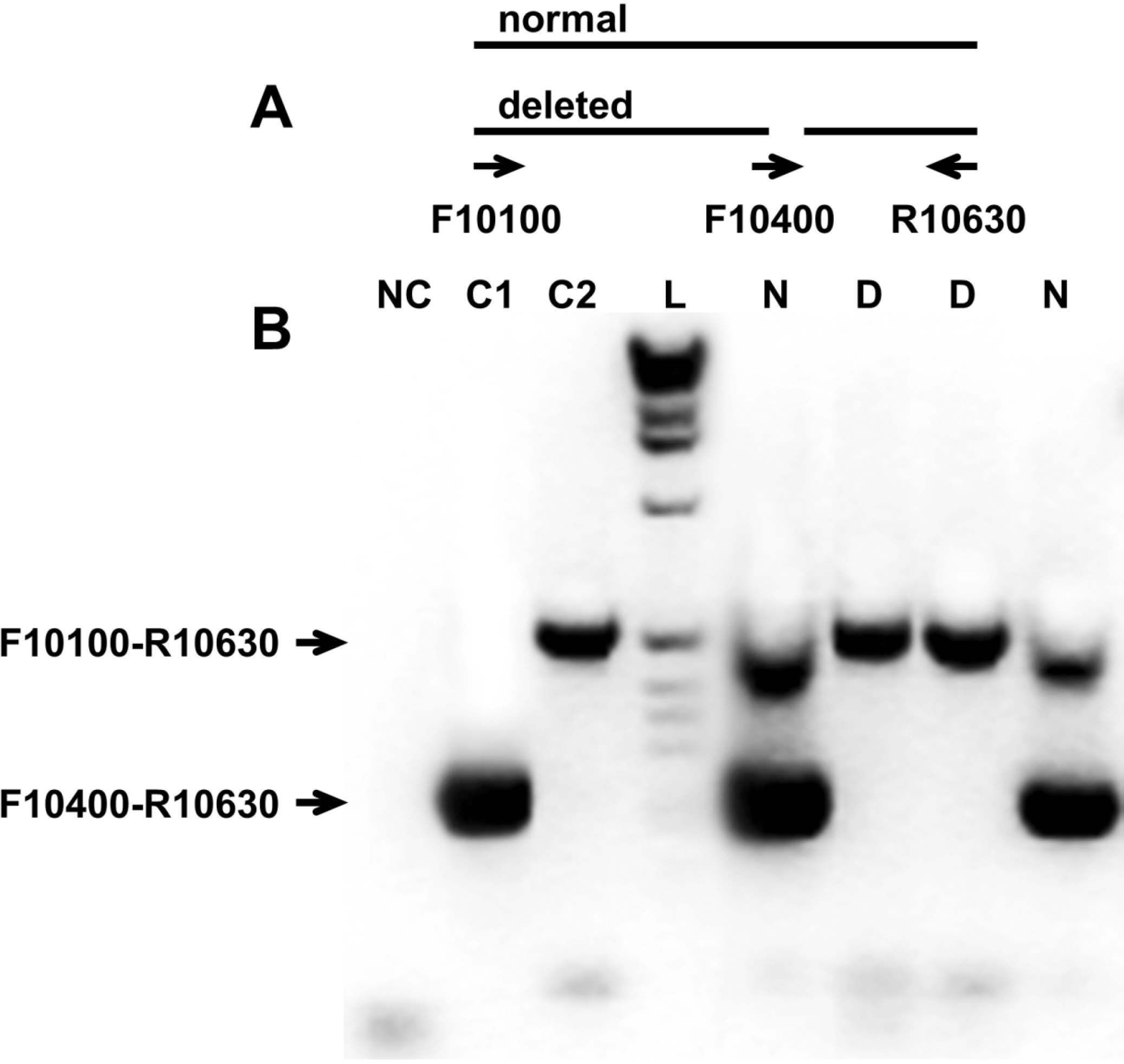
3.2. Analysis of the Variable Region of the 3′UTR

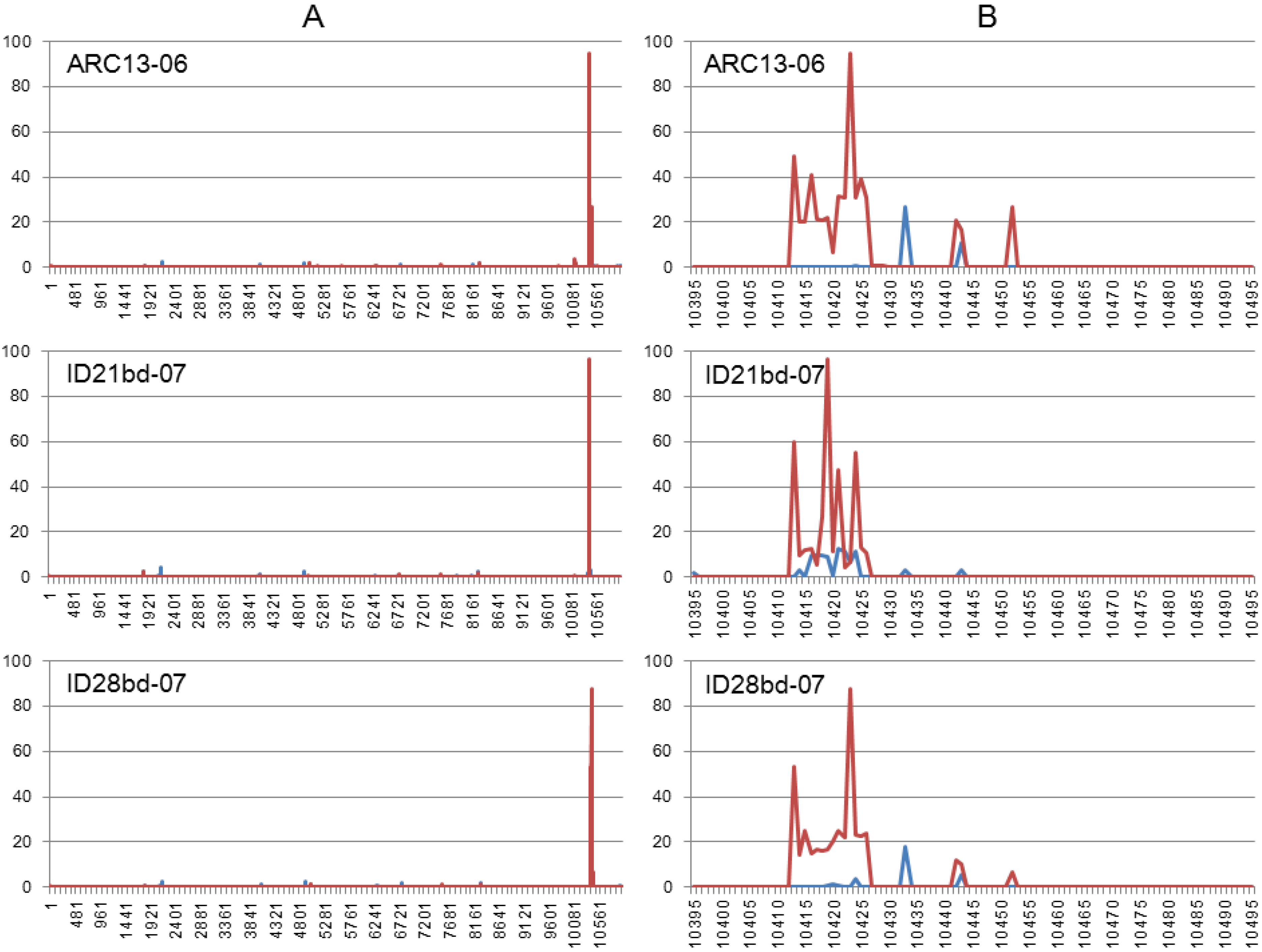
3.3. Illumina NGS Data Analysis
| Mutation frequencies | |||||
|---|---|---|---|---|---|
| Isolate | Read Count, ×106 | Total nucleotides read, ×109 | Substitutions, % | Insertions, % | deletions, % |
| ARC13-06 | 80.18 | 8.02 | 0.89 | 0.64 | 1.29 |
| ID21bd-07 | 91.23 | 9.13 | 0.86 | 0.78 | 1.09 |
| ID28bd-07 | 83.32 | 8.27 | 0.82 | 0.54 | 1.27 |

4. Discussion
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Añez, G.; Chancey, C.; Grinev, A.; Rios, M. Dengue virus and other arboviruses: A global view of risks. ISBT Sci. Ser. 2012, 7, 274–282. [Google Scholar] [CrossRef]
- Brinton, M.A. Molecular Biology of West Nile Virus. In West Nile Encephalitis Virus Infection: Viral Pathogenesis and the Host Immune Response; Diamond, M., Ed.; Springer: New York, NY, USA, 2009; pp. 97–136. [Google Scholar]
- Markoff, L. 5′- and 3′-noncoding regions in flavivirus RNA. Adv. Virus Res. 2003, 59, 177–228. [Google Scholar] [CrossRef]
- Gritsun, T.S.; Gould, E.A. Direct repeats in the flavivirus 3′ untranslated region; a strategy for survival in the environment? Virology 2007, 358, 258–265. [Google Scholar] [CrossRef]
- Murray, K.O.; Mertens, E.; Despres, P. West Nile virus and its emergence in the United States of America. Vet. Res. 2010, 41, 67–81. [Google Scholar] [CrossRef]
- Kauffman, E.B.; Franke, M.A.; Wong, S.J.; Kramer, L.D. Detection of West Nile virus. Methods Mol. Biol. 2011, 665, 383–413. [Google Scholar]
- Hayes, E.B.; Gubler, D.J. West Nile virus: Epidemiology and clinical features of an emerging epidemic in the United States. Annu. Rev. Med. 2006, 57, 181–194. [Google Scholar]
- Centers for Disease Control and Prevention. Available online: http://www.cdc.gov/westnile/statsMaps/ (accessed on 26 June 2013).
- U.S. Department of the Interior, U.S. Geological Survey. Available online: http://diseasemaps.usgs.gov/wnv_historical.html (accessed on 26 June 2013).
- Berthet, F.X.; Zeller, H.G.; Drouet, M.T.; Rauzier, J.; Digoutte, J.P.; Deubel, V. Extensive nucleotide changes and deletions within the envelope glycoprotein gene of Euro-African West Nile viruses. J. Gen. Virol. 1997, 78, 2293–2297. [Google Scholar]
- Lanciotti, R.S.; Ebel, G.D.; Deubel, V.; Kerst, A.J.; Murri, S.; Meyer, R.; Bowen, M.; McKinney, N.; Morrill, W.E.; Crabtree, M.B.; et al. Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology 2002, 298, 96–105. [Google Scholar] [CrossRef]
- Bakonyi, T.; Hubálek, Z.; Rudolf, I.; Nowotny, N. Novel flavivirus or new lineage of West Nile virus, Central Europe. Emerg. Infect. Dis. 2005, 11, 225–231. [Google Scholar] [CrossRef]
- Bondre, V.P.; Jadi, R.S.; Mishra, A.C.; Yergolkar, P.N.; Arankalle, V.A. West Nile virus isolates from India: Evidence for a distinct genetic lineage. J. Gen. Virol. 2007, 88, 875–884. [Google Scholar] [CrossRef]
- May, F.J.; Davis, C.T.; Tesh, R.B.; Barrett, A.D. Phylogeography of West Nile virus: From the cradle of evolution in Africa to Eurasia, Australia, and the Americas. J. Virol. 2011, 85, 2964–2974. [Google Scholar] [CrossRef]
- Davis, C.T.; Ebel, G.D.; Lanciotti, R.S.; Brault, A.C.; Guzman, H.; Siirin, M.; Lambert, A.; Parsons, R.E.; Beasley, D.W.; Novak, R.J.; et al. Phylogenetic analysis of North American West Nile virus isolates, 2001–2004: Evidence for the emergence of a dominant genotype. Virology 2005, 342, 252–265. [Google Scholar] [CrossRef]
- Snapinn, K.W.; Holmes, E.C.; Young, D.S.; Bernard, K.A.; Kramer, L.D.; Ebel, G.D. Declining growth rate of West Nile virus in North America. J. Virol. 2007, 81, 2531–2534. [Google Scholar] [CrossRef]
- Moudy, R.M.; Meola, M.A.; Morin, L.L.; Ebel, G.D.; Kramer, L.D. A newly emergent genotype of West Nile virus is transmitted earlier and more efficiently by Culex mosquitoes. Am. J. Trop. Med. Hyg. 2007, 77, 365–370. [Google Scholar]
- Vanlandingham, D.L.; McGee, C.E.; Klingler, K.A.; Galbraith, S.E.; Barrett, A.D.T.; Higgs, S. Comparison of oral infectious dose of West Nile virus isolates representing three distinct genotypes in Culex quinquefasciatus. Am. J. Trop. Med. Hyg. 2008, 79, 951–954. [Google Scholar]
- McMullen, A.R.; May, F.J.; Li, L.; Guzman, H.; Bueno, R., Jr.; Dennett, J.A.; Tesh, R.B.; Barrett, A.D. Evolution of new genotype of West Nile virus in North America. Emerg. Infect. Dis. 2011, 17, 785–793. [Google Scholar] [CrossRef]
- Añez, G.; Grinev, A.; Chancey, C.; Ball, C.; Akolkar, N.; Land, K.J.; Winkelman, V.; Stramer, S.L.; Kramer, L.D.; Rios, M. Evolutionary dynamics of West Nile virus in the United States, 1999–2011: Phylogeny, selection pressure and evolutionary time-scale analysis. PLoS Negl. Trop. Dis. 2013, 7. [Google Scholar] [CrossRef]
- Aaskov, J.; Buzacott, K.; Field, E.; Lowry, K.; Berlioz-Arthaud, A.; Holmes, E.C. Multiple recombinant dengue type 1 viruses in an isolate from a dengue patient. J. Gen. Virol. 2007, 88, 3334–3340. [Google Scholar] [CrossRef]
- Domingo, E.; Martin, V.; Perales, C.; Grande-Pérez, A.; García-Arriaza, J.; Arias, A. Viruses as quasispecies: Biological implications. Curr. Top. Microbiol. Immunol. 2006, 299, 51–82. [Google Scholar] [CrossRef]
- Ruiz-Jarabo, C.M.; Arias, A.; Baranowski, E.; Escarmís, C.; Domingo, E. Memory in viral quasispecies. J. Virol. 2000, 74, 3543–3547. [Google Scholar] [CrossRef]
- Rogers, Y.H.; Venter, J.C. Genomics: Massively parallel sequencing. Nature 2005, 437, 326–327. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-generation DNA sequencing methods. Annu. Rev. Genomics. Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Rozera, G.; Abbate, I.; Bruselles, A.; Vlassi, C.; D’Offizi, G.; Narciso, P.; Chillemi, G.; Prosperi, M.; Ippolito, G.; Capobianchi, M.R. Massively parallel pyrosequencing highlights minority variants in the HIV-1 env quasispecies deriving from lymphomonocyte sub-populations. Retrovirology 2009, 6. [Google Scholar] [CrossRef]
- Margeridon-Thermet, S.; Shulman, N.S.; Ahmed, A.; Shahriar, R.; Liu, T.; Wang, C.; Holmes, S.P.; Babrzadeh, F.; Gharizadeh, B.; Hanczaruk, B.; et al. Ultra-deep pyrosequencing of hepatitis B virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)-treated patients and NRTI-naive patients. J. Infect. Dis. 2009, 199, 1275–1285. [Google Scholar] [CrossRef]
- Baillie, G.J.; Galiano, M.; Agapow, P.M.; Myers, R.; Chiam, R.; Gall, A.; Palser, A.L.; Watson, S.J.; Hedge, J.; Underwood, A.; et al. Evolutionary dynamics of local pandemic H1N1/09 influenza lineages revealed by whole genome analysis. J. Virol. 2011, 86, 11–18. [Google Scholar]
- Wright, C.F.; Morelli, M.J.; Thebaud, G.; Knowles, N.J.; Herzyk, P.; Paton, D.J.; Haydon, D.T.; King, D.P. Beyond the consensus: Dissecting within-host viral population diversity of foot-and-mouth disease virus using next-generation genome sequencing. J. Virol. 2010, 85, 2266–2275. [Google Scholar]
- Daly, G.M.; Be xfield, N.; Heaney, J.; Stubbs, S.; Mayer, A.P.; Palser, A.; Kellam, P.; Drou, N.; Caccamo, M.; Tiley, L.; et al. A viral discovery methodology for clinical biopsy samples utilising massively parallel next generation sequencing. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Grinev, A.; Daniel, S.; Stramer, S.; Rossmann, S.; Caglioti, S.; Rios, M. Genetic variability of West Nile virus in US blood donors, 2002–2005. Emerg. Infect. Dis. 2008, 14, 436–444. [Google Scholar] [CrossRef]
- HIVE: High-performance Integrated Virtual Environment Provides Solutions for Next-Generation Sequencing Data Storage and Analysis. Available online: https://hive.biochemistry.gwu.edu/HIVEWhitePaper.pdf (accessed on 26 June 2013).
- Ball, E.V.; Stenson, P.D.; Abeysinghe, S.S.; Krawczak, M.; Cooper, D.N.; Chuzhanova, N.A. Microdeletions and microinsertions causing human genetic disease: Common mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum. Mutat. 2005, 26, 205–213. [Google Scholar] [CrossRef]
- Chen, C.Y.; Shyu, A.B. AU-rich elements: Characterization and importance in mRNA degradation. Trends. Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Roehrig, J.T.; Deubel, V.; Smith, J.; Parker, M.; Steele, K.; Crise, B.; Volpe, K.E.; Crabtree, M.B.; Scherret, J.H.; et al. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 1999, 286, 2333–2337. [Google Scholar] [CrossRef]
- Beasley, D.W.; Davis, C.T.; Guzman, H.; Vanlandingham, D.L.; Travassos da Rosa, A.P.; Parsons, R.E.; Higgs, S.; Tesh, R.B.; Barrett, A.D. Limited evolution of West Nile virus has occurred during its southwesterly spread in the United States. Virology 2003, 309, 190–195. [Google Scholar] [CrossRef]
- Davis, C.T.; Beasley, D.W.; Guzman, H.; Raj, R.; D’Anton, M.; Novak, R.J.; Unnasch, T.R.; Tesh, R.B.; Barrett, A.D. Genetic variation among temporally and geographically distinct West Nile virus isolates, United States, 2001, 2002. Emerg. Infect. Dis. 2003, 9, 1423–1429. [Google Scholar] [CrossRef]
- Ebel, G.D.; Carricaburu, J.; Young, D.; Bernard, K.A.; Kramer, L.D. Genetic and phenotypic variation of West Nile virus in New York, 2000–2003. Am. J. Trop. Med. Hyg. 2004, 71, 493–500. [Google Scholar]
- Herring, B.L.; Bernardin, F.; Caglioti, S.; Stramer, S.; Tobler, L.; Andrews, W.; Cheng, L.; Rampersad, S.; Cameron, C.; Saldanha, J.; et al. Phylogenetic analysis of WNV in North American blood donors during the 2003–2004 epidemic seasons. Virology 2007, 363, 220–228. [Google Scholar] [CrossRef]
- Bertolotti, L.; Kitron, U.; Goldberg, T.L. Diversity and evolution of West Nile virus in Illinois and the United States, 2002–2005. Virology 2007, 360, 143–149. [Google Scholar] [CrossRef]
- Bertolotti, L.; Kitron, U.D.; Walker, E.D.; Ruiz, M.O.; Brawn, J.D.; Loss, S.R.; Hamer, G.L.; Goldberg, T.L. Fine-scale genetic variation and evolution of West Nile Virus in a transmission “hot spot” in suburban Chicago, USA. Virology 2008, 374, 381–389. [Google Scholar] [CrossRef]
- Gray, R.R.; Veras, N.M.; Santos, L.A.; Salemi, M. Evolutionary characterization of the West Nile Virus complete genome. Mol. Phylogenet. Evol. 2010, 56, 195–200. [Google Scholar] [CrossRef]
- Armstrong, P.M.; Vossbrinck, C.R.; Andreadis, T.G.; Anderson, J.F.; Pesko, K.N.; Newman, R.M.; Lennon, N.J.; Birren, B.W.; Ebel, G.D.; Henn, M.R. Molecular evolution of West Nile virus in a northern temperate region: Connecticut, USA 1999–2008. Virology 2011, 417, 203–210. [Google Scholar] [CrossRef]
- Chuzhanova, N.A.; Anassis, E.; Ball, E.; Krawczak, M.; Cooper, D.N. Meta-analysis of indels causing human genetic disease: Mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum. Mutat. 2003, 21, 28–44. [Google Scholar] [CrossRef]
- Kondrashov, A.S.; Rogozin, I.B. Context of deletions and insertions in human coding sequences. Hum. Mutat. 2004, 23, 177–185. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Sverdlov, A.V.; Babenko, V.N.; Koonin, E.V. Analysis of evolution of exon-intron structure of eukaryotic genes. Brief. Bioinform. 2005, 6, 118–134. [Google Scholar] [CrossRef]
- DeMaria, C.T.; Brewer, G. AUF1 binding affinity to A + U-rich elements correlates with rapid mRNA degradation. J. Biol. Chem. 1996, 271, 12179–12184. [Google Scholar] [CrossRef]
- Peng, S.S.; Chen, C.Y.; Xu, N.; Shyu, A.B. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 1998, 17, 3461–3470. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Kedersha, N.; Anderson, P.; Emara, M.; Swiderek, K.M.; Moreno, G.T.; Brinton, M.A. Cell proteins TIA-1 and TIAR interact with the 3′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J. Virol. 2002, 76, 11989–12000. [Google Scholar] [CrossRef]
- Nadar, M.; Chan, M.Y.; Huang, S.W.; Huang, C.C.; Tseng, J.T.; Tsai, C.H. HuR binding to AU-rich elements present in the 3′ untranslated region of Classical swine fever virus. Virol. J. 2011, 8. [Google Scholar] [CrossRef] [Green Version]
- Tilgner, M.; Deas, T.S.; Shi, P.Y. The flavivirus-conserved penta-nucleotide in the 3′ stem-loop of the West Nile virus genome requires a specific sequence and structure for RNA synthesis, but not for viral translation. Virology 2005, 331, 375–386. [Google Scholar] [CrossRef]
- Tajima, S.; Nukui, Y.; Ito, M.; Takasaki, T.; Kurane, I. Nineteen nucleotides in the variable region of 3′ non-translated region are dispensable for the replication of dengue type 1 virus in vitro. Virus Res. 2006, 116, 38–44. [Google Scholar] [CrossRef]
- Kato, F.; Kotaki, A.; Yamaguchi, Y.; Shiba, H.; Hosono, K.; Harada, S.; Saijo, M.; Kurane, I.; Takasaki, T.; Tajima, S. Identification and characterization of the short variable region of the Japanese encephalitis virus 3′NTR. Virus Genes 2012, 44, 191–197. [Google Scholar] [CrossRef]
- Men, R.; Bray, M.; Clark, D.; Chanock, R.M.; Lai, C.J. Dengue type 4 virus mutants containing deletions in the 3′ noncoding region of the RNA genome: Analysis of growth restriction in cell culture and altered viremia pattern and immunogenicity in rhesus monkeys. J. Virol. 1996, 70, 3930–3937. [Google Scholar]
- Yun, S.I.; Choi, Y.J.; Song, B.H.; Lee, Y.M. 3′ cis-acting elements that contribute to the competence and efficiency of Japanese encephalitis virus genome replication: Functional importance of sequence duplications, deletions, and substitutions. J. Virol. 2009, 83, 7909–7930. [Google Scholar] [CrossRef]
- Alvarez, D.E.; de Lella Ezcurra, A.L.; Fucito, S.; Gamarnik, A.V. Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 2005, 339, 200–212. [Google Scholar] [CrossRef]
- Tajima, S.; Nukui, Y.; Takasaki, T.; Kurane, I. Characterization of the variable region in the 3′ non-translated region of dengue type 1 virus. J. Gen. Virol. 2007, 88, 2214–2222. [Google Scholar] [CrossRef]
- Funk, A.; Truong, K.; Nagasaki, T.; Torres, S.; Floden, N.; Balmori Melian, E.; Edmonds, J.; Dong, H.; Shi, P.Y.; Khromykh, A.A. RNA structures required for production of subgenomic flavivirus RNA. J. Virol. 2010, 84, 11407–11417. [Google Scholar] [CrossRef]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef]
- Hughes, A.L.; Piontkivska, H.; Foppa, I. Rapid fixation of a distinctive sequence motif in the 3′ noncoding region of the clade of West Nile virus invading North America. Gene 2007, 399, 152–161. [Google Scholar] [CrossRef]
- Holmes, E.C. The RNA virus quasispecies: Fact or fiction? J. Mol. Biol. 2010, 400, 271–273. [Google Scholar] [CrossRef]
- Fishman, S.L.; Branch, A.D. The quasispecies nature and biological implications of the hepatitis C virus. Infect. Genet. Evol. 2009, 9, 1158–1167. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Kirkegaard, K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 2005, 1. [Google Scholar] [CrossRef]
- Martell, M.; Esteban, J.I.; Quer, J.; Genescà, J.; Weiner, A.; Esteban, R.; Guardia, J.; Gómez, J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: Quasispecies nature of HCV genome distribution. J. Virol. 1992, 66, 3225–3229. [Google Scholar]
- Plyusnin, A.; Cheng, Y.; Lehväslaiho, H.; Vaheri, A. Quasispecies in wild-type Tula hantavirus populations. J. Virol. 1996, 70, 9060–9063. [Google Scholar]
- Quiñones-Mateu, M.E.; Albright, J.L.; Mas, A.; Soriano, V.; Arts, E.J. Analysis of pol gene heterogeneity, viral quasispecies, and drug resistance in individuals infected with group O strains of human immunodeficiency virus type 1. J. Virol. 1998, 72, 9002–9015. [Google Scholar]
- Cottam, E.M.; King, D.P.; Wilson, A.; Paton, D.J.; Haydon, D.T. Analysis of foot-and-mouth disease virus nucleotide sequence variation within naturally infected epithelium. Virus Res. 2009, 140, 199–204. [Google Scholar] [CrossRef]
- Jerzak, G.; Bernard, K.A.; Kramer, L.D.; Ebel, G.D. Genetic variation in West Nile virus from naturally infected mosquitoes and birds suggests quasispecies structure and strong purifying selection. J. Gen. Virol. 2005, 86, 2175–2183. [Google Scholar] [CrossRef]
- Ciota, A.T.; Ngo, K.A.; Lovelace, A.O.; Payne, A.F.; Zhou, Y.; Shi, P.Y.; Kramer, L.D. Role of the mutant spectrum in adaptation and replication of West Nile virus. J. Gen. Virol. 2007, 88, 865–874. [Google Scholar] [CrossRef]
- Jerzak, G.V.; Bernard, K.; Kramer, L.D.; Shi, P.Y.; Ebel, G.D. The West Nile virus mutant spectrum is host-dependant and a determinant of mortality in mice. Virology 2007, 360, 469–476. [Google Scholar] [CrossRef]
- Ciota, A.T.; Lovelace, A.O.; Jia, Y.; Davis, L.J.; Young, D.S.; Kramer, L.D. Characterization of mosquito-adapted West Nile virus. J. Gen. Virol. 2008, 89, 1633–1642. [Google Scholar] [CrossRef]
- Liang, B.; Luo, M.; Scott-Herridge, J.; Semeniuk, C.; Mendoza, M.; Capina, R.; Sheardown, B.; Ji, H.; Kimani, J.; Ball, B.T.; et al. A comparison of parallel pyrosequencing and Sanger clone-based sequencing and its impact on the characterization of the genetic diversity of HIV-1. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Chin-inmanu, K.; Suttitheptumrong, A.; Sangsrakru, D.; Tangphatsornruang, S.; Tragoonrung, S.; Malasit, P.; Tungpradabkul, S.; Suriyaphol, P. Feasibility of using 454 pyrosequencing for studying quasispecies of the whole dengue viral genome. BMC Genomics 2012, 13. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Grinev, A.; Chancey, C.; Añez, G.; Ball, C.; Winkelman, V.; Williamson, P.; Foster, G.A.; Stramer, S.L.; Rios, M. Genetic Analysis of West Nile Virus Isolates from an Outbreak in Idaho, United States, 2006–2007. Int. J. Environ. Res. Public Health 2013, 10, 4486-4506. https://doi.org/10.3390/ijerph10094486
Grinev A, Chancey C, Añez G, Ball C, Winkelman V, Williamson P, Foster GA, Stramer SL, Rios M. Genetic Analysis of West Nile Virus Isolates from an Outbreak in Idaho, United States, 2006–2007. International Journal of Environmental Research and Public Health. 2013; 10(9):4486-4506. https://doi.org/10.3390/ijerph10094486
Chicago/Turabian StyleGrinev, Andriyan, Caren Chancey, Germán Añez, Christopher Ball, Valerie Winkelman, Phillip Williamson, Gregory A. Foster, Susan L. Stramer, and Maria Rios. 2013. "Genetic Analysis of West Nile Virus Isolates from an Outbreak in Idaho, United States, 2006–2007" International Journal of Environmental Research and Public Health 10, no. 9: 4486-4506. https://doi.org/10.3390/ijerph10094486
APA StyleGrinev, A., Chancey, C., Añez, G., Ball, C., Winkelman, V., Williamson, P., Foster, G. A., Stramer, S. L., & Rios, M. (2013). Genetic Analysis of West Nile Virus Isolates from an Outbreak in Idaho, United States, 2006–2007. International Journal of Environmental Research and Public Health, 10(9), 4486-4506. https://doi.org/10.3390/ijerph10094486