Lipid Metabolism in Late-Onset Alzheimer’s Disease Differs from Patients Presenting with Other Dementia Phenotypes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. ApoE Genotyping
2.3. Quantification of CSF Total Protein, Tau, and Aβ42
2.4. Enzyme Activity Assays
2.5. Lipid Extraction
2.6. LC-MS/MS of Lipids
2.7. LC-MS/MS Data Analysis
2.8. Statistical Methods
3. Results
3.1. Demographic Data and CSF Biomarkers
3.2. Dementia Risk Factors and Medication Use
3.3. GPs in LOAD and OD
3.4. Sphingolipids in LOAD and OD
3.5. Enzyme Activities of LOAD and OD CSF Samples
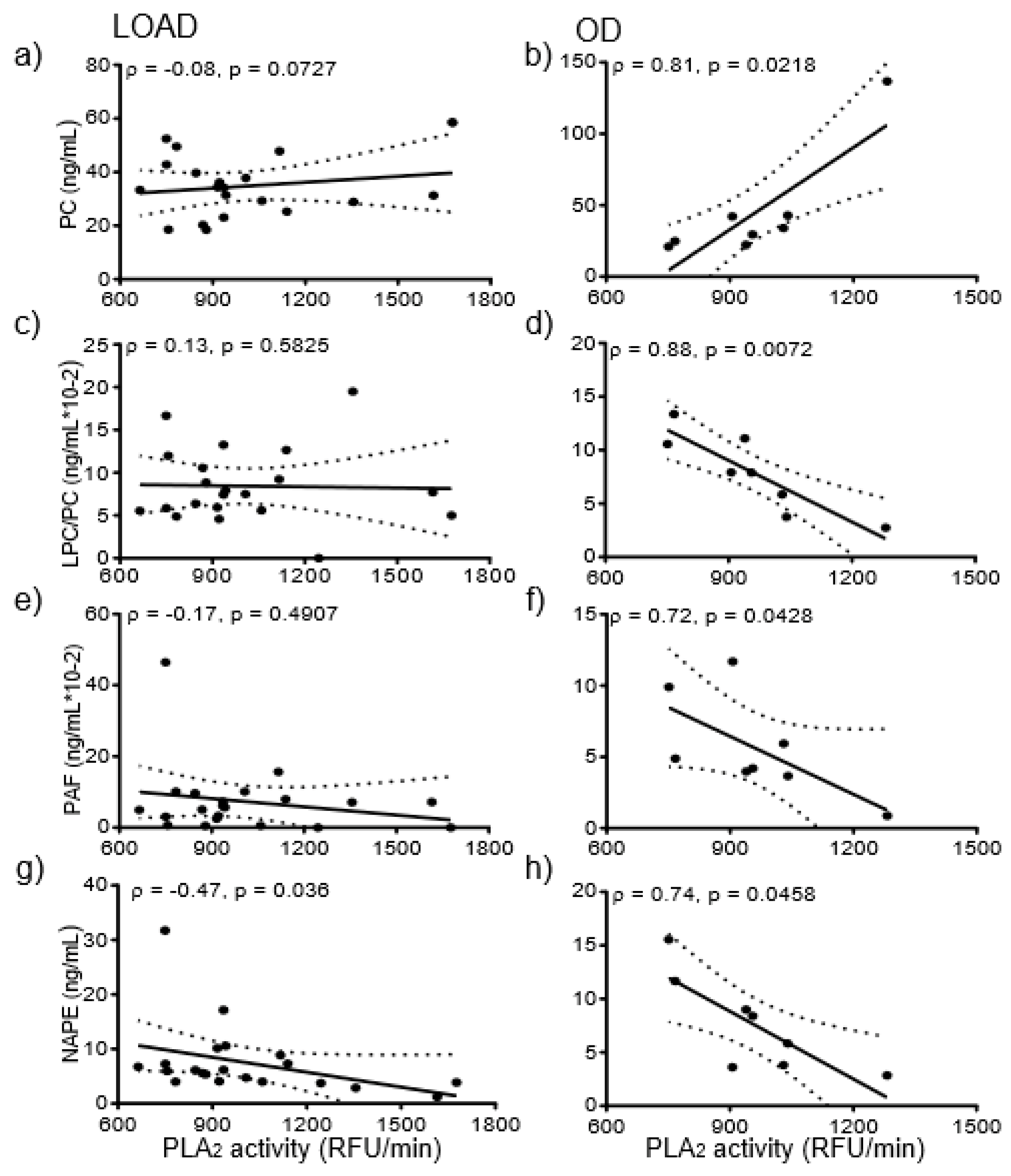
3.6. Correlation of Glycerophospholipids and Aβ42 or Tau
3.7. Correlation of Sphingolipids and Aβ42 or Tau
3.8. Correlation of Enzyme Activities with CSF Lipids
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CSF | cerebrospinal fluid |
| CM | ceramide |
| dhCM | dihydroceramide |
| LC | liquid chromatography |
| LOAD | late-onset Alzheimer’s Disease |
| LPC | lysophosphatidylcholine |
| PAF_LL | platelet-activating factor-like lipids |
| PE | phosphatidylethanolamine |
| PC | phosphatidylcholine |
| PS | phosphatidylserine |
| GP | glycerophospholipid |
| PLA2 | phospholipase A2 |
| NAPE | N-acyl phosphatidylethanolamine |
| NP | nanoparticles |
| SM | sphingomyelin |
| SMase | sphingomyelinase |
| SP | sphingolipids |
| SF | supernatant fluid |
| xPE | unknown lipid containing phosphoethanolamine |
References
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Hirtz, D.; Thurman, D.J.; Gwinn-Hardy, K.; Mohamed, M.; Chaudhuri, A.R.; Zalutsky, R. How common are the “common” neurologic disorders? Neurology 2007, 68, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Irie, F.; Fitzpatrick, A.L.; Lopez, O.L.; Kuller, L.H.; Peila, R.; Newman, A.B.; Launer, L.J. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE ε4: The cardiovascular health study cognition study. Arch. Neurol. 2008, 65, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-beta (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Andreasson, U.; Persson, S.; Arai, H.; Batish, S.D.; Bernardini, S.; Bocchio-Chiavetto, L.; Blankenstein, M.A.; Carrillo, M.C.; Chalbot, S.; et al. The Alzheimer’s association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement. 2011, 7, 386–395.e6. [Google Scholar] [CrossRef]
- Fagan, A.M.; Holtzman, D.M. Cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark. Med. 2010, 4, 51–63. [Google Scholar] [CrossRef]
- Blennow, K.; Vanmechelen, E.; Hampel, H. Csf total tau, Aβ42 and phosphorylated tau protein as biomarkers for Alzheimer’s disease. Mol. Neurobiol. 2001, 24, 87–97. [Google Scholar] [CrossRef]
- Rademakers, R.; Neumann, M.; Mackenzie, I.R. Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 2012, 8, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.K. Parkinson’s disease with dementia, lewy-body disorders and alpha-synuclein: Recent advances and a case report. Acta Neurol. Taiwan. 2011, 20, 4–14. [Google Scholar]
- Lebedev, A.V.; Beyer, M.K.; Fritze, F.; Westman, E.; Ballard, C.; Aarsland, D. Cortical changes associated with depression and antidepressant use in Alzheimer and lewy body dementia: An mri surface-based morphometric study. Am. J. Geriatr. Psychiatry 2014, 22, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Gomperts, S.N.; Locascio, J.J.; Marquie, M.; Santarlasci, A.L.; Rentz, D.M.; Maye, J.; Johnson, K.A.; Growdon, J.H. Brain amyloid and cognition in lewy body diseases. Mov. Disord. 2012, 27, 965–973. [Google Scholar] [CrossRef]
- Hampel, H.; Broich, K.; Hoessler, Y.; Pantel, J. Biological markers for early detection and pharmacological treatment of Alzheimer’s disease. Dialogues Clin. Neurosci. 2009, 11, 141–157. [Google Scholar] [PubMed]
- Piomelli, D.; Astarita, G.; Rapaka, R. A neuroscientist’s guide to lipidomics. Nat. Rev. Neurosci. 2007, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, F.N.; Allsop, D. Alpha-synuclein interacts with lipoproteins in plasma. J. Mol. Neurosci. 2017, 63, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, A.A.; Liss, L.; Horrocks, L.A. Neurochemical aspects of Alzheimer’s disease: Involvement of membrane phospholipids. Metab. Brain Dis. 1988, 3, 19–35. [Google Scholar] [CrossRef]
- Florent-Bechard, S.; Desbene, C.; Garcia, P.; Allouche, A.; Youssef, I.; Escanye, M.C.; Koziel, V.; Hanse, M.; Malaplate-Armand, C.; Stenger, C.; et al. The essential role of lipids in Alzheimer’s disease. Biochimie 2009, 91, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Han, X. Potential mechanisms contributing to sulfatide depletion at the earliest clinically recognizable stage of Alzheimer’s disease: A tale of shotgun lipidomics. J. Neurochem. 2007, 103, 171–179. [Google Scholar] [CrossRef]
- Kosicek, M.; Hecimovic, S. Phospholipids and Alzheimer’s disease: Alterations, mechanisms and potential biomarkers. Int. J. Mol. Sci. 2013, 14, 1310–1322. [Google Scholar] [CrossRef]
- van Echten-Deckert, G.; Walter, J. Sphingolipids: Critical players in Alzheimer’s disease. Prog. Lipid Res. 2012, 51, 378–393. [Google Scholar] [CrossRef]
- Penke, B.; Paragi, G.; Gera, J.; Berkecz, R.; Kovacs, Z.; Crul, T.; László, V. The role of lipids and membranes in the pathogenesis of Alzheimer’s disease: A comprehensive view. Curr. Alzheimer Res 2018, 15, 1191–1212. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J.A.; Bevan, D.R. Lipid composition influences the release of Alzheimer’s amyloid beta-peptide from membranes. Protein Sci. 2011, 20, 1530–1545. [Google Scholar] [CrossRef] [PubMed]
- Fonteh, A.N.; Chiang, J.; Cipolla, M.; Hale, J.; Diallo, F.; Chirino, A.; Arakaki, X.; Harrington, M.G. Alterations in cerebrospinal fluid glycerophospholipids and phospholipase A2 activity in Alzheimer’s disease. J. Lipid Res. 2013, 54, 2884–2897. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.K.; Tsai, Y.T.; Ariga, T. Functional roles of gangliosides in neurodevelopment: An overview of recent advances. Neurochem. Res. 2012, 37, 1230–1244. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.; Fabelo, N.; Ferrer, I.; Marin, R. “Lipid raft aging” in the human frontal cortex during nonpathological aging: Gender influences and potential implications in Alzheimer’s disease. Neurobiol. Aging 2018, 67, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef]
- Haughey, N.J.; Bandaru, V.V.; Bae, M.; Mattson, M.P. Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim. Biophys. Acta 2010, 1801, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Chi, E.Y.; Ege, C.; Winans, A.; Majewski, J.; Wu, G.; Kjaer, K.; Lee, K.Y. Lipid membrane templates the ordering and induces the fibrillogenesis of Alzheimer’s disease amyloid-beta peptide. Proteins 2008, 72, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Berkowitz, M.L. Interaction between amyloid-beta (1–42) peptide and phospholipid bilayers: A molecular dynamics study. Biophys. J. 2009, 96, 785–797. [Google Scholar] [CrossRef]
- Fan, J.; Donkin, J.; Wellington, C. Greasing the wheels of abeta clearance in Alzheimer’s disease: The role of lipids and apolipoprotein e. Biofactors 2009, 35, 239–248. [Google Scholar] [CrossRef]
- He, X.; Huang, Y.; Li, B.; Gong, C.X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.M.; Michikawa, M.; Kim, S.U.; Nagai, A. Lysophosphatidylcholine increases the neurotoxicity of Alzheimer’s amyloid beta1-42 peptide: Role of oligomer formation. Neuroscience 2015, 292, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Alessenko, A.V.; Bugrova, A.E.; Dudnik, L.B. Connection of lipid peroxide oxidation with the sphingomyelin pathway in the development of Alzheimer’s disease. Biochem. Soc. Trans. 2004, 32, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Malaplate-Armand, C.; Florent-Bechard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.L.; Oster, T.; Pillot, T. Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 2006, 23, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of gamma-secretase with lipid rafts in post-golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [PubMed]
- Hagen-Euteneuer, N.; Lutjohann, D.; Park, H.; Merrill, A.H., Jr.; van Echten-Deckert, G. Sphingosine 1-phosphate (S1P) lyase deficiency increases sphingolipid formation via recycling at the expense of de novo biosynthesis in neurons. J. Biol. Chem. 2012, 287, 9128–9136. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Wasser, C.; Shakirzyanova, A.; Giniatullin, A.; Goodman, K.; Munoz-Bravo, J.L.; Raingo, J.; Jorgacevski, J.; Kreft, M.; Zorec, R.; et al. Sphingosine facilitates snare complex assembly and activates synaptic vesicle exocytosis. Neuron 2009, 62, 683–694. [Google Scholar] [CrossRef]
- Popp, J.; Lewczuk, P.; Kolsch, H.; Meichsner, S.; Maier, W.; Kornhuber, J.; Jessen, F.; Lutjohann, D. Cholesterol metabolism is associated with soluble amyloid precursor protein production in Alzheimer’s disease. J. Neurochem. 2012, 123, 310–316. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Thinakaran, G. Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim. Biophys. Acta 2010, 1801, 860–867. [Google Scholar] [CrossRef]
- Wood, W.G.; Schroeder, F.; Igbavboa, U.; Avdulov, N.A.; Chochina, S.V. Brain membrane cholesterol domains, aging and amyloid beta-peptides. Neurobiol. Aging 2002, 23, 685–694. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, S.; Salmon, D.; Mercaldo, N.; Ferris, S.; Graff-Radford, N.R.; Chui, H.; Cummings, J.; DeCarli, C.; Foster, N.L.; Galasko, D.; et al. The Alzheimer’s disease centers’ uniform data set (UDS): The neuropsychologic test battery. Alzheimer Dis. Assoc. Disord. 2009, 23, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Beekly, D.L.; Ramos, E.M.; Lee, W.W.; Deitrich, W.D.; Jacka, M.E.; Wu, J.; Hubbard, J.L.; Koepsell, T.D.; Morris, J.C.; Kukull, W.A.; et al. The national Alzheimer’s coordinating center (NACC) database: The uniform data set. Alzheimer Dis. Assoc. Disord. 2007, 21, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Crum, R.M.; Anthony, J.C.; Bassett, S.S.; Folstein, M.F. Population-based norms for the mini-mental state examination by age and educational level. JAMA 1993, 269, 2386–2391. [Google Scholar] [CrossRef] [PubMed]
- Freitas, S.; Simoes, M.R.; Alves, L.; Santana, I. Montreal cognitive assessment: Validation study for mild cognitive impairment and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2013, 27, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C. Clinical dementia rating: A reliable and valid diagnostic and staging measure for dementia of the alzheimer type. Int. Psychogeriatr. 1997, 9, 173–176; discussion 177–178. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with lewy bodies: Third report of the dlb consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- Chui, H.C.; Victoroff, J.I.; Margolin, D.; Jagust, W.; Shankle, R.; Katzman, R. Criteria for the diagnosis of ischemic vascular dementia proposed by the state of california alzheimer’s disease diagnostic and treatment centers. Neurology 1992, 42, 473–480. [Google Scholar] [CrossRef]
- Harrington, M.G.; Chiang, J.; Pogoda, J.M.; Gomez, M.; Thomas, K.; Marion, S.D.; Miller, K.J.; Siddarth, P.; Yi, X.; Zhou, F.; et al. Executive function changes before memory in preclinical Alzheimer’s pathology: A prospective, cross-sectional, case control study. PLoS ONE 2013, 8, e79378. [Google Scholar] [CrossRef] [PubMed]
- Calero, O.; Hortiguela, R.; Bullido, M.J.; Calero, M. Apolipoprotein e genotyping method by real time PCR, a fast and cost-effective alternative to the taqman and fret assays. J. Neurosci. Methods 2009, 183, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Fonteh, A.N.; Ormseth, C.; Chiang, J.; Cipolla, M.; Arakaki, X.; Harrington, M.G. Sphingolipid metabolism correlates with cerebrospinal fluid beta amyloid levels in Alzheimer’s disease. PLoS ONE 2015, 10, e0125597. [Google Scholar] [CrossRef] [PubMed]
- Harrington, M.G.; Fonteh, A.N.; Oborina, E.; Liao, P.; Cowan, R.P.; McComb, G.; Chavez, J.N.; Rush, J.; Biringer, R.G.; Huhmer, A.F. The morphology and biochemistry of nanostructures provide evidence for synthesis and signaling functions in human cerebrospinal fluid. Cerebrospinal Fluid Res. 2009, 6, 10. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Weder, N.D.; Aziz, R.; Wilkins, K.; Tampi, R.R. Frontotemporal dementias: A review. Ann. Gen. Psychiatry 2007, 6, 15. [Google Scholar] [CrossRef]
- Vance, J.E. Phosphatidylserine and phosphatidylethanolamine in mammalian cells: Two metabolically related aminophospholipids. J. Lipid Res. 2008, 49, 1377–1387. [Google Scholar] [CrossRef]
- Kay, J.G.; Grinstein, S. Sensing phosphatidylserine in cellular membranes. Sensors 2011, 11, 1744–1755. [Google Scholar] [CrossRef]
- Jenzer, C.; Simionato, E.; Largeau, C.; Scarcelli, V.; Lefebvre, C.; Legouis, R. Autophagy mediates phosphatidylserine exposure and phagosome degradation during apoptosis through specific functions of GABARAP/LGG-1 and LC3/LGG-2. Autophagy 2018, 1–14. [Google Scholar] [CrossRef]
- Fadok, V.A.; de Cathelineau, A.; Daleke, D.L.; Henson, P.M.; Bratton, D.L. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem. 2001, 276, 1071–1077. [Google Scholar] [CrossRef]
- Morgado, I.; Garvey, M. Lipids in amyloid-beta processing, aggregation, and toxicity. Adv. Exp. Med. Biol. 2015, 855, 67–94. [Google Scholar] [PubMed]
- Lahdo, R.; Coillet-Matillon, S.; Chauvet, J.P.; de La Fourniere-Bessueille, L. The amyloid precursor protein interacts with neutral lipids. Eur. J. Biochem. 2002, 269, 2238–2246. [Google Scholar] [CrossRef] [PubMed]
- Urmoneit, B.; Turner, J.; Dyrks, T. Cationic lipids (lipofectamine) and disturbance of cellular cholesterol and sphingomyelin distribution modulates gamma-secretase activity within amyloid precursor protein in vitro. Prostaglandins Other Lipid Mediat. 1998, 55, 331–343. [Google Scholar] [CrossRef]
- Muramatsu, R.; Kuroda, M.; Matoba, K.; Lin, H.; Takahashi, C.; Koyama, Y.; Yamashita, T. Prostacyclin prevents pericyte loss and demyelination induced by lysophosphatidylcholine in the central nervous system. J. Biol. Chem. 2015, 290, 11515–11525. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Lemere, C.A.; Selkoe, D.J.; Clemens, J.A. Cytosolic phospholipase A2 (cPLA2) immunoreactivity is elevated in alzheimer’s disease brain. Neurobiol. Dis. 1996, 3, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, A.A.; Horrocks, L.A. Plasmalogen-selective phospholipase A2 and its involvement in Alzheimer’s disease. Biochem. Soc. Trans. 1998, 26, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejia, R.O.; Mucke, L. Phospholipase A2 and arachidonic acid in Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Kosicek, M.; Zetterberg, H.; Andreasen, N.; Peter-Katalinic, J.; Hecimovic, S. Elevated cerebrospinal fluid sphingomyelin levels in prodromal Alzheimer’s disease. Neurosci. Lett. 2012, 516, 302–305. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Interactions between neural membrane glycerophospholipid and sphingolipid mediators: A recipe for neural cell survival or suicide. J. Neurosci. Res. 2007, 85, 1834–1850. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Lyketsos, C.G. Alterations of the sphingolipid pathway in Alzheimer’s disease: New biomarkers and treatment targets? Neuromol. Med. 2010, 12, 331–340. [Google Scholar] [CrossRef]
- Ng, C.Y.; Kannan, S.; Chen, Y.J.; Tan, F.C.K.; Ong, W.Y.; Go, M.L.; Verma, C.S.; Low, C.M.; Lam, Y. A new generation of arachidonic acid analogues as potential neurological agent targeting cytosolic phospholipase A2. Sci. Rep. 2017, 7, 13683. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Rapoport, S.I.; Kim, H.W. Altered neuroinflammatory, arachidonic acid cascade and synaptic markers in postmortem Alzheimer’s disease brain. Transl. Psychiatry 2017, 7, e1127. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Altered lipid metabolism in brain injury and disorders. Subcell. Biochem. 2008, 49, 241–268. [Google Scholar] [PubMed]
- Bazan, N.G.; Colangelo, V.; Lukiw, W.J. Prostaglandins and other lipid mediators in Alzheimer’s disease. Prostaglandins Other Lipid Mediat. 2002, 68–69, 197–210. [Google Scholar] [CrossRef]
- Combs, C.K.; Karlo, J.C.; Kao, S.C.; Landreth, G.E. Beta-amyloid stimulation of microglia and monocytes results in tnfalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Bazan, N.G. Neuroinflammatory signaling upregulation in Alzheimer’s disease. Neurochem. Res. 2000, 25, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.T.; Mello, A.P.; Damasceno, N.R. Antioxidant and inflammatory aspects of lipoprotein-associated phospholipase A(2) (LP-PLA(2)): A review. Lipids Health Dis. 2011, 10, 170. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Schafer-Elinder, L.; Wu, R.; Claesson, H.E.; Frostegard, J. Lysophosphatidylcholine (LPC) induces proinflammatory cytokines by a platelet-activating factor (PAF) receptor-dependent mechanism. Clin. Exp. Immunol. 1999, 116, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Grin’kina, N.M.; Abdel-Baki, S.G.; Bergold, P.J. Reversible behavioral deficits in rats during a cycle of demyelination-remyelination of the fimbria. PLoS ONE 2013, 8, e53775. [Google Scholar] [CrossRef] [PubMed]
- Kilaru, A.; Isaac, G.; Tamura, P.; Baxter, D.; Duncan, S.R.; Venables, B.J.; Welti, R.; Koulen, P.; Chapman, K.D. Lipid profiling reveals tissue-specific differences for ethanolamide lipids in mice lacking fatty acid amide hydrolase. Lipids 2010, 45, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Wellner, N.; Diep, T.A.; Janfelt, C.; Hansen, H.S. N-acylation of phosphatidylethanolamine and its biological functions in mammals. Biochim. Biophys. Acta 2013, 1831, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Kornhuber, J.; Muehlbacher, M.; Trapp, S.; Pechmann, S.; Friedl, A.; Reichel, M.; Muhle, C.; Terfloth, L.; Groemer, T.W.; Spitzer, G.M.; et al. Identification of novel functional inhibitors of acid sphingomyelinase. PLoS ONE 2011, 6, e23852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, B.; Jenkins, G.M.; Hannun, Y.A.; Obeid, L.M. Expression of neutral sphingomyelinase identifies a distinct pool of sphingomyelin involved in apoptosis. J. Biol. Chem. 1997, 272, 9609–9612. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dinkins, M.; He, Q.; Zhu, G.; Poirier, C.; Campbell, A.; Mayer-Proschel, M.; Bieberich, E. Astrocytes secrete exosomes enriched with proapoptotic ceramide and prostate apoptosis response 4 (PAR-4): Potential mechanism of apoptosis induction in Alzheimer disease (AD). J. Biol. Chem. 2012, 287, 21384–21395. [Google Scholar] [CrossRef] [PubMed]
- Apraiz, A.; Idkowiak-Baldys, J.; Nieto-Rementeria, N.; Boyano, M.D.; Hannun, Y.A.; Asumendi, A. Dihydroceramide accumulation and reactive oxygen species are distinct and nonessential events in 4-HPR-mediated leukemia cell death. Biochem. Cell Biol. 2012, 90, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.M.; Wang, Y.; Duan, X.; Wenk, M.R.; Kalaria, R.N.; Chen, C.P.; Lai, M.K.; Shui, G. Brain lipidomes of subcortical ischemic vascular dementia and mixed dementia. Neurobiol. Aging 2014, 35, 2369–2381. [Google Scholar] [CrossRef] [PubMed]
- Canals, D.; Perry, D.M.; Jenkins, R.W.; Hannun, Y.A. Drug targeting of sphingolipid metabolism: Sphingomyelinases and ceramidases. Br. J. Pharmacol. 2011, 163, 694–712. [Google Scholar] [CrossRef] [PubMed]
- Mohaibes, R.J.; Fiol-deRoque, M.A.; Torres, M.; Ordinas, M.; Lopez, D.J.; Castro, J.A.; Escriba, P.V.; Busquets, X. The hydroxylated form of docosahexaenoic acid (DHA-H) modifies the brain lipid composition in a model of Alzheimer’s disease, improving behavioral motor function and survival. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1596–1603. [Google Scholar] [CrossRef]


{kind=link}
| Categories | LOAD | OD | 1 Difference (% LOAD) |
|---|---|---|---|
| Number of Subjects | 29 | 10 | |
| Female (%) | 53% | 40% | |
| Mean ± SEM | |||
| Age (years) | 77.4 ± 1.8 | 77.3 ± 4.0 | −0.1 |
| Education (years) | 14.5 ± 2.8 | 14.0 ± 3.1 | −3.5 |
| ApoE Genotype | 3.6 ± 0.2 | 4 ± 0 | 11.4 |
| BMI | 1.6 ± 0.2 | 1.3 ± 0.3 | −19.4 |
| 2 Neuropsych Classification | 9.1 ± 0.2 | 8.8 ± 0.3 | −3.72 |
| MMSE | 15 ± 1.5 | 16.0 ± 2.7 | 7.5 |
| CSF Tau (pg/mL) | 472.6 ± 41.1 | 387.2 ± 100.6 | −18.1 |
| CSF Aβ42 (pg/mL) | 506.2 ± 43.1 | 432.3 ± 59.4 | −13.8 |
| Aβ42/Tau | 1.4 ± 0.2 | 1.2 ± 0.3 | −12.1 |
| Dementia duration (years) | 5.7 ± 0.9 | 3.5 ± 0.6 | −39.0 |
| Risk Factors | LOAD n (Frequency), % | OD n (Frequency), % | p Value 1 |
|---|---|---|---|
| Obesity # | 26 (3), 11.5 | 9 (0), 0 | 0.55 |
| Hyperlipidemia | 28 (9), 32.1 | 10 (4), 40 | 0.71 |
| Hypertension | 27 (15), 55.6 | 10 (4), 40 | 0.48 |
| Heart disease | 28 (11), 39.3 | 10 (4), 40 | 0.99 |
| Diabetes | 29 (4), 13.9 | 10 (1) 10 | 0.99 |
| Family history of dementia | 29 (14), 48.3 | 10 (2), 20 | 0.15 |
| GPs in SF (ng/mL) | LOAD | OD | 1 Difference (% LOAD) |
| LPC | 15.6 ± 1.1 | 16.0 ± 2.1 | 2.8 |
| PC | 8169.1 ± 915.1 | 7064.3 ± 1238.3 | −13.5 |
| LPC/PC | 0.0022 ± 0.0002 | 0.0023 ± 0.0002 | 4.6 |
| PS | 6.2 ± 1.0 | 16.01 ± 6.6 | 149.4 * |
| NAPE | 10.6 ± 2.3 | 15.09 ± 3.1 | 42.6 |
| PE | 27.8 ± 3.2 | 16.4 ± 2.5 | −41.2 |
| xPE | 1.3 ± 0.1 | 1.40± 0.2 | 11.2 |
| GPs in NP (ng/mL) | LOAD | OD | % LOAD |
| LPC | 3.05 ± 0.3 | 2.7 ± 0.2 | −11.2 |
| PC | 40.05 ± 4.8 | 61.9 ± 23.4 | 54.5 |
| LPC/PC | 0.09 ± 0.01 | 0.08 ± 0.01 | −10.1 |
| PAF | 0.08 ± 0.02 | 0.06 ± 0.01 | −28.3 |
| NAPE | 6.8 ± 1.1 | 6.9 ± 1.3 | 0.8 |
| PE | 7.9 ± 0.6 | 9.5 ± 1.3 | 20.5 |
| xPE | 1.8 ± 0.5 | 1.7 ± 0.2 | −7.3 |
| SP Classes in SF | LOAD (ng/mL) | OD (ng/mL) | 1 Difference (% LOAD) |
| SM | 1888.2 ± 138.2 | 1892.1 ± 400.1 | 2.1 |
| CM | 60.0 ± 6.9 | 73.2 ± 20.9 | 22.2 |
| dhCM | 179.2 ± 18.9 | 186.3 ± 63.7 | 4.0 |
| CM/SM | 0.0309 ± 0.0022 | 0.0369 ± 0.0041 | 19.4 |
| SP Classes in NP | LOAD (ng/mL) | OD (ng/mL) | % LOAD |
| SM | 95.2 ± 8.1 | 110.7 ± 20.6 | 16.3 |
| CM | 5.9 ± 0.6 | 6.5 ± 0.4 | 9.4 * |
| dhCM | 53.9 ± 3.0 | 55.6 ± 5.7 | 3.2 |
| CM/SM | 0.07 ± 0.01 | 0.08 ± 0.01 | 2.7 |
| Enzymes | LOAD (RFU/min) | OD (RFU/min) | 1 Difference (% LOAD) |
|---|---|---|---|
| PLA2 | 1008.1 ± 59.2 | 959.2 ± 60.1 | −4.9 |
| nSMase | 24.3 ± 2.2 | 20.4 ± 1.9 | −18.9 |
| aSMase | 15.0 ± 1.5 | 20.7 ± 1.7 | 37.9 * |
| aSMase/nSMase | 0.8 ± 0.7 | 1.1 ± 0.1 | 39.3 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarrafpour, S.; Ormseth, C.; Chiang, A.; Arakaki, X.; Harrington, M.; Fonteh, A. Lipid Metabolism in Late-Onset Alzheimer’s Disease Differs from Patients Presenting with Other Dementia Phenotypes. Int. J. Environ. Res. Public Health 2019, 16, 1995. https://doi.org/10.3390/ijerph16111995
Sarrafpour S, Ormseth C, Chiang A, Arakaki X, Harrington M, Fonteh A. Lipid Metabolism in Late-Onset Alzheimer’s Disease Differs from Patients Presenting with Other Dementia Phenotypes. International Journal of Environmental Research and Public Health. 2019; 16(11):1995. https://doi.org/10.3390/ijerph16111995
Chicago/Turabian StyleSarrafpour, Syena, Cora Ormseth, Abby Chiang, Xianghong Arakaki, Michael Harrington, and Alfred Fonteh. 2019. "Lipid Metabolism in Late-Onset Alzheimer’s Disease Differs from Patients Presenting with Other Dementia Phenotypes" International Journal of Environmental Research and Public Health 16, no. 11: 1995. https://doi.org/10.3390/ijerph16111995
APA StyleSarrafpour, S., Ormseth, C., Chiang, A., Arakaki, X., Harrington, M., & Fonteh, A. (2019). Lipid Metabolism in Late-Onset Alzheimer’s Disease Differs from Patients Presenting with Other Dementia Phenotypes. International Journal of Environmental Research and Public Health, 16(11), 1995. https://doi.org/10.3390/ijerph16111995