Rubella Virus Infection, the Congenital Rubella Syndrome, and the Link to Autism


Abstract
:1. Introduction
2. Maternal Rubella, Cataract, and the Congenital Rubella Syndrome
- bulging fontanelle and rash at birth;
- fetal growth restriction (FGR) and low birth weight;
- seizures;
- hearing and cardiovascular defects (most commonly patent ductus arteriosus);
- microcephaly;
- psychomotor retardation;
- behavioral and speech disorders;
- thrombocytopenic purpura;
- hepatitis;
- hepatosplenomegaly;
- bone lesions;
- pneumonitis;
- diabetes mellitus;
- thyroid disorders;
- progressive rubella panencephalitis
3. Rubella Linked to Autism
4. Congenital Rubella Syndrome—Case Study
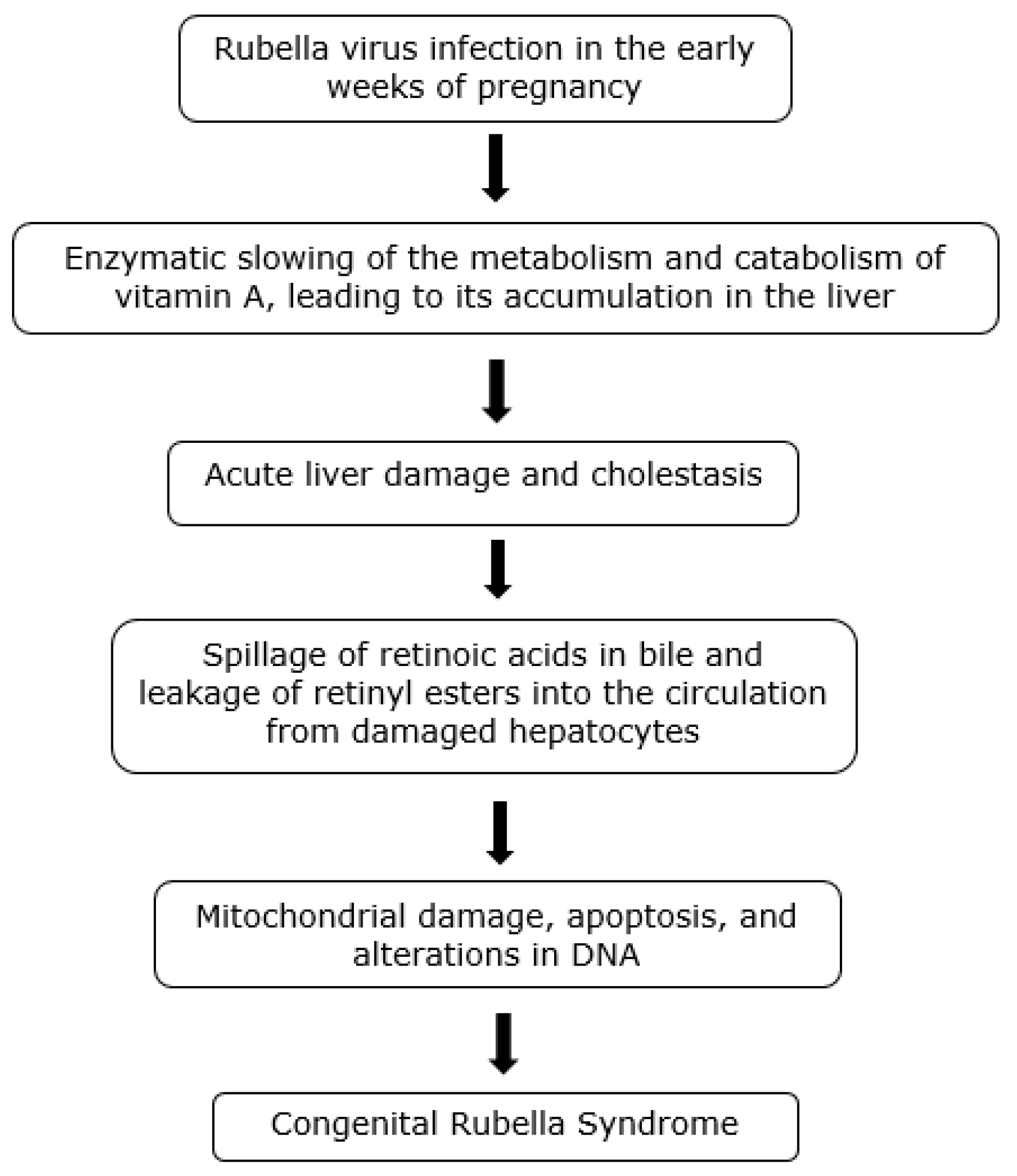
5. Rubella Infection and CRS—Introduction
Retinoid Toxicity Hypothesis of Rubella Infection
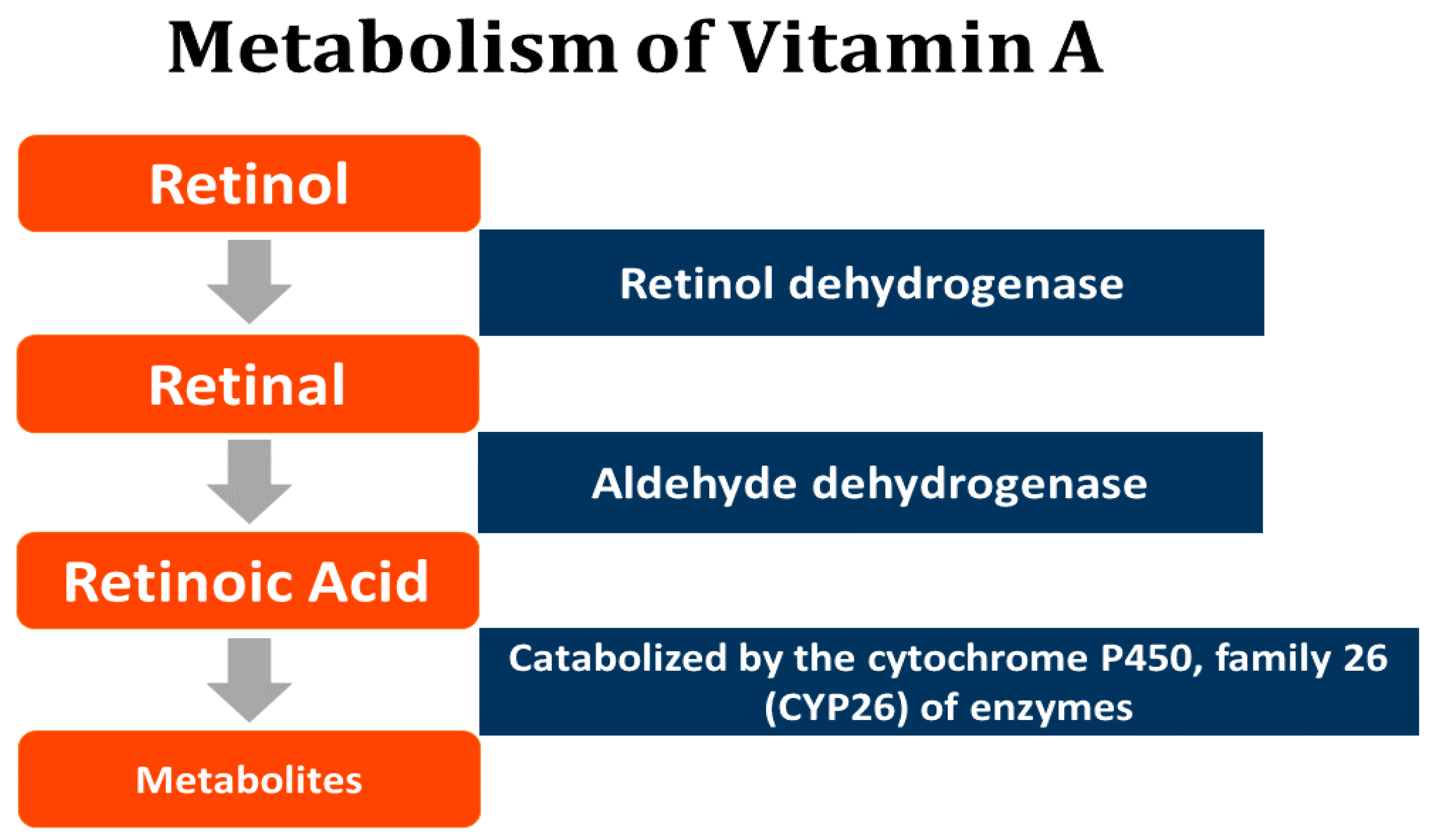
6. Retinoids—A Summary
7. Rubella Symptoms and Hypervitaminosis A
8. Congenital Rubella Syndrome (CRS)
9. Pathology of CRS
10. CRS and Hypervitaminosis A-Associated Embryopathy
11. Preventing CRS
- stopping all alcohol for a period of at least 4 weeks;
- stopping all liver-damaging drugs that are not essential to life (e.g., anabolic steroids, recreational drugs, antidepressants, anxiolytics and hypnotics);
- maintaining good hydration by drinking plain, non-fluoridated water (and not through drinking coffee or tea, both of which are dehydrating);
- eliminating vitamin A from the diet for one month (the main dietary source is liver, but vitamin A is also found in milk, cheese, egg yolks and fish oils);
- (optional) reducing circulating retinoids, under medical supervision, through phlebotomy, plasmapheresis or hirudotherapy.
12. Retinoid Hypothesis of Regressive Autism
- The vaccination schedule has been expanded and accelerated during the same period of rapid increase in rates of autism and other chronic illnesses. The schedule now involves 69 doses of 16 vaccines between the day of birth and age 18, with 50 doses before age six [155], which is over three times the number of vaccinations recommended in 1983 [156]. The clinical outcomes of this expanded schedule on children’s health, where many vaccinations are administered at the same time or in close succession, are virtually unknown [157].
- Vaccines are known on occasion to cause severe adverse effects, including immune system disorders, brain damage and death [164]. These outcomes are considered so rare that vaccines are believed safe to administer to all healthy infants and children [165,166]. However, the extent of serious injury from vaccines remains uncertain. One study of infants who were hospitalized or had died after receiving vaccinations, based on 38,801 reports to the Vaccine Adverse Events Reporting System (VAERS), showed a linear relationship between the number of vaccine doses administered at one time and the rate of hospitalization and death; furthermore, the younger the infant at the time of vaccination, the higher was the rate of hospitalization and death. The rate of hospitalization for two vaccine doses was 11% and increased to 23.5% for eight doses (r2 = 0.91). The case fatality rate increased from 3.6% for 1–4 doses, increasing to 5.4% among those receiving 5–8 doses [167].
13. Could Other Risk Factors Be Involved?
14. Nutritional Implications of the Model
15. Environmental Factors—Herbicides
16. Discussion and Conclusions
- The signs and symptoms of rubella may be due to virus-induced alterations in vitamin A metabolism and its accumulation in the liver, leading to mild hepatic inflammation and dysfunction and to the spillage of stored vitamin A compounds into the circulation in correspondingly low concentrations and hence mild toxicity.
- RS and associated autism due to rubella infection in the early weeks of pregnancy may similarly result from maternal liver dysfunction and exposure of the fetus to excess endogenous vitamin A, resulting in embryopathy and long-term metabolic and neurodevelopmental disorders.
- Increasing rates of regressive autism result primarily from post-natal factors to which almost all children are exposed, causing liver dysfunction and both acute and chronic forms of hypervitaminosis A.
- These post-natal factors may include: multiple vaccinations and their ingredients administered together or in close succession, especially when combined with the physiological impact of preterm birth; the intake of high vitamin A-containing and vitamin A-fortified foods such as dairy products, infant formula, and vitamin A supplements; and exposure to certain ubiquitous herbicides that may influence retinoid metabolism. Further studies are awaited on the overall safety and health outcomes of the current vaccination schedule, with a view to optimizing the beneficial impact of vaccines on children’s health.
- An early diagnosis of ASD and CRS-like features in infants may be due to maternal exposures of a liver-damaging nature in the early weeks of pregnancy.
- Young children with liver-related conditions that include preterm birth and metabolic disorders, as well as genetic factors associated with alterations in vitamin A metabolism, may be especially susceptible to autism and related neurodevelopmental disorders.
Author Contributions
Funding
Conflicts of Interest
References
- Hawker, J.; Begg, N.; Reintjes, R.; Ekdahl, K.; Edeghere, O.; van Steenbergen, J. Communicable Disease Control and Health Protection Handbook, 4th ed.; Wiley Blackwell: Oxford, UK, 2019; pp. 205–207. [Google Scholar]
- Gershon, A.A. Rubella virus (German Measles). In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; John, E., Bennett, R.D., Blaser, M.J., Eds.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Lambert, N.; Strebel, P.; Orenstein, W.; Icenogle, J.; Poland, G.A. Rubella. Lancet 2015, 385, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention. Epidemiology and Prevention of Vaccine-Preventable Diseases, 5th ed.; Atkinson, W., Humiston, S., Wolfe, C., Nelson, R., Eds.; Department of Health and Human Services: Washington, DC, USA, 1999. [Google Scholar]
- Miller, E.; Waight, P.A.; Gay, N.; Ramsay, M.; Vurdien, J.; Morgan-Capner, P.; Hesketh, L.; Brown, D.; Tookey, P.; Peckham, C. The epidemiology of rubella in England and Wales before and after the 1994 measles and rubella vaccination campaign: Fourth joint report from the PHLS and the National Congenital Rubella Surveillance Program. Commun. Dis. Rep. 1997, 7, R26–R32. [Google Scholar]
- Cutts, F.T.; Vynnycky, E. Modelling the incidence of congenital rubella syndrome in developing countries. Int. J. Epidemiol. 1999, 28, 1176–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banatvala, J.E.; Brown, D.W.G. Rubella. Lancet 2004, 363, 1127–1137. [Google Scholar] [CrossRef]
- Atreya, C.D.; Mohan, K.V.; KulKarni, B. Rubella virus and birth defects: Molecular insights into the viral teratogenesis at the cellular level. Birth Defects Res. A Clin. Mol. Teratol. 2004, 70, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC. Global Health: Measles & Rubella Move Fast Infographic. Available online: https://www.cdc.gov/globalhealth/immunization/infographic/measles.htm (accessed on 18 September 2019).
- Toizumi, M.; Vo, H.M.; Dang, D.A.; Moriuchi, H.; Yoshida, L.M. Clinical manifestations of congenital rubella syndrome: A review of our experience in Vietnam. Vaccine 2019, 37, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Webster, W.S. Teratogen update: Congenital rubella. Teratology 1998, 58, 13–23. [Google Scholar] [CrossRef]
- Gregg, N.M. Congenital cataract following German measles in the mother. Trans. Ophthalmol. Soc. Aust. 1941, 3, 35–46. [Google Scholar]
- Givens, K.T.; Lee, D.A.; Jones, T.; Ilstrup, D.M. Congenital rubella syndrome: Ophthalmic manifestations and associated systemic disorders. Br. J. Ophthalmol. 1993, 77, 358–363. [Google Scholar] [CrossRef]
- Vandenbosche, R.C.; Kirschner, J.T. Intrauterine growth retardation. Am. Fam. Physician 1998, 58, 1384–1390. [Google Scholar]
- Desmond, M.M.; Wilson, S.; Melnick, J.L.; Singer, D.B.; Zion, T.E.; Rudolph, A.J.; Pineda, R.G.; Ziai, M.H. Congenital rubella encephalitis. J. Pediatr. 1967, 71, 311–331. [Google Scholar] [CrossRef]
- Desmond, M.M.; Montgomery, J.R.; Melnick, J.L.; Cochran, G.G.; Verniaud, W. Congenital rubella encephalitis. Effects on growth and early development. Am. J. Dis. Child. 1969, 118, 30–31. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, E.G. Autistic children exhibit undetectable hemagglutination-inhibition antibody titers despite previous rubella vaccination. J. Autism Child. Schizophr. 1976, 6, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Chess, S. Autism in children with congenital rubella. J. Autism Child. Schizophr. 1971, 1, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Kanner, L. Follow-up study of eleven autistic children originally reported in 1943. Autism Child. Schizophr. 1971, 1, 119–145. [Google Scholar] [CrossRef]
- Treffert, D.A. Epidemiology of infantile autism. Arch. Gen. Psychiatry 1970, 22, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Lotter, V. Epidemiology of autistic conditions in young children. I: Prevalence. Soc. Psychiatry 1966, 1, 124–137. [Google Scholar] [CrossRef]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Rosenberg, C.R.; White, T.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef]
- Chess, S. Follow-up report on autism in congenital rubella. J. Autism Child. Schizophr. 1977, 7, 69–81. [Google Scholar] [CrossRef]
- Libbey, J.E.; Sweeten, T.L.; McMahon, W.M.; Fujinami, R.S. Autistic disorder and viral infections. J. Neurol. Virol. 2005, 11, 1–10. [Google Scholar] [CrossRef]
- Karim, S.; Mirza, Z.; Kamal, M.A.; Abuzenadah, A.M.; Azhar, E.I.; Al-Qahtani, M.H.; Damanhouri, G.A.; Ahmad, F.; Gan, S.H.; Sohrab, S.S. The role of viruses in neurodegenerative and neurobehavioral diseases. CNS Neurol. Disord. Drug Targets 2014, 13, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Abib, R.T.; Gaman, A.; Dargél, A.A.; Tamouza, R.; Kapczinski, F.; Gottfried, C.; Leboyer, M. Intracellular Pathogen Infections and Immune Response in Autism. Neuroimmunomodulation 2018, 25, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Chess, S.; Fernandez, P.; Korn, S. Behavioral consequences of congenital rubella. J. Pediatr. 1978, 93, 699–703. [Google Scholar] [CrossRef]
- Sheridan, E.; Aitken, C.; Jeffries, D. Congenital rubella syndrome: A risk in immigrant populations. Lancet 2002, 359, 674–675. [Google Scholar] [CrossRef]
- Frey, T.K. Neurological aspects of rubella virus infection. Intervirology 1997, 40, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y. Rubella arthritis. An outbreak in Kyoto. J. Rheumatol. 1994, 21, 874–876. [Google Scholar] [PubMed]
- Menser, M.A.; Forrest, J.M.; Bransby, R.D. Rubella infection and diabetes mellitus. Lancet 1978, 311, 57–60. [Google Scholar] [CrossRef]
- Schattner, A.; Rager-Zisman, B. Virus-induced autoimmunity. Rev. Infect. Dis. 1990, 12, 204–222. [Google Scholar] [CrossRef]
- McIntosh, E.D.G.; Menser, M.A. A fifty-year follow-up of congenital defects. Lancet 1992, 340, 414–415. [Google Scholar] [CrossRef]
- Ovsyannikova, I.G.; Haralambieva, I.H.; Dhiman, N.; O’Byrne, M.M.; Pankratz, V.S.; Jacobson, R.M.; Poland, G.A. Polymorphisms in the vitamin A receptor and innate immunity genes influence the antibody response to rubella vaccination. J. Infect. Dis. 2010, 201, 207–213. [Google Scholar] [CrossRef]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wongsiriroj, N.; Blaner, W.S. The multifaceted nature of retinoid transport and metabolism. Hepatobiliary Surg. Nutr. 2014, 3, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Tanumihardjo, S.A.; Russell, R.M.; Stephensen, C.B.; Gannon, B.M.; Craft, N.E.; Haskell, M.J.; Lietz, G.; Schulze, K.; Raiten, D.J. Biomarkers of Nutrition for Development (BOND)-Vitamin A Review. J. Nutr. 2016, 146, S1816–S1848. [Google Scholar] [CrossRef] [PubMed]
- WHO. Serum Retinol Concentrations for Determining the Prevalence of Vitamin a Deficiency in Populations: Vitamin and Mineral Nutrition Information System; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Theodosiou, M.; Laudet, V.; Schubert, M. From carrot to clinic: An overview of the retinoic acid signaling pathway. Cell. Mol. Life Sci. 2010, 67, 1423–1445. [Google Scholar] [CrossRef]
- Blaner, W.S. Vitamin a signaling and homeostasis in obesity, diabetes, and metabolic disorders. Pharmacol. Ther. 2019, 197, 153–178. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef]
- Penniston, K.L.; Tanumihardjo, S.A. The acute and chronic toxic effects of vitamin A. Am. J. Clin. Nutr. 2006, 83, 191–201. [Google Scholar] [CrossRef]
- De Oliveira, M.R.; Da Rocha, R.F.; Pasquali, M.A.; Moreira, J.C. The effects of vitamin A supplementation for 3 months on adult rat nigrostriatal axis: Increased monoamine oxidase enzyme activity, mitochondrial redox dysfunction, increased β-amyloid (1–40) peptide and TNF-α contents, and susceptibility of mitochondria to an in vitro H2O2 challenge. Brain Res. Bull. 2012, 87, 432–444. [Google Scholar]
- Leo, M.A.; Lieber, C.S. Hypervitaminosis A: A liver lover’s lament. Hepatology 1988, 8, 412–417. [Google Scholar] [CrossRef]
- Mitra, A.K.; Alvarez, M.A.; Wahed, M.A.; Fuchs, G.J.; Stephenson, C.B. Predictors of serum retinol in children with shigellosis. Am. J. Clin. Nutr. 1998, 68, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Schweigert, F.J. Inflammation–induced changes in the nutritional biomarkers serum retinol and carotenoids. Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Gieng, S.H.; Green, M.H.; Green, J.B.; Rosales, F.J. Model-based compartmental analysis indicates a reduced mobilization of hepatic vitamin A during inflammation in rats. J. Lipid Res. 2007, 48, 904–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeldis, J.B.; Miller, J.G.; Dienstag, J.L. Hepatitis in an adult with rubella. Am. J. Med. 1985, 79, 515–516. [Google Scholar] [CrossRef]
- Tameda, Y.; Kosaka, Y.; Shiraki, K.; Ohashi, Y.; Hamada, M.; Miyazaki, M.; Ito, N.; Takase, K.; Nakano, T. Hepatitis in an adult with rubella. Intern. Med. 1993, 32, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.A.; de Oliveira, M.I.; Tarandachi, P.R.; de Carvalho, W.B.; Kanamura, C.T.; Scatena Rdos, S. Fatal acute liver failure in a child due to acquired rubella infection. J. Clin. Virol. 2014, 61, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Kantartzis, K.; Häring, H.U. Causes and metabolic consequences of fatty liver. Endocr. Rev. 2008, 29, 939–960. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Wada, N.; Maruyama, K.; Nomiyama, T.; Tanaka, S.; Okazaki, I. Acute hepatitis in an adult with acquired rubella infection. J. Gastroenterol. 1995, 30, 539–542. [Google Scholar] [CrossRef]
- Kalvenes, M.B.; Flo, R.; Kalland, K.H.; Haukenes, G. Elevated rubella antibodies in patients with chronic liver disease. J. Med. Virol. 1994, 44, 30–36. [Google Scholar] [CrossRef]
- Takenaka, H.; Kishimoto, S.; Ichikawa, R.; Shibagaki, R.; Kubota, Y.; Yamagata, N.; Gotoh, H.; Fujita, N.; Yasuno, H. Virus-associated haemophagocytic syndrome caused by rubella in an adult. Br. J. Dermatol. 1998, 139, 877–880. [Google Scholar] [CrossRef]
- Abdullah, A.M.; Al Fadel Saleh, M.; Al Madan, M.; El Mouzan, M.; Olasope, B. Infantile cholestasis in the Central-Eastern Province Saudi Arabia. J. Trop. Pediatr. 1997, 43, 138–142. [Google Scholar] [CrossRef]
- Moore, T. Vitamin A; Elsevier North Holland, Inc.: New York, NY, USA, 1957. [Google Scholar]
- Olson, J.A. Vitamin A—Functions, dietary requirements and safety in humans. In Present Knowledge in Nutrition, 7th ed.; Ziegler, E.E., Filer, L.J., Jr., Eds.; International Life Sciences Institute Press: Washington, DC, USA, 2001; pp. 109–119. [Google Scholar]
- Safran, A.B.; Halioua, B.; Roth, A.; Saurat, J.-A. Ocular side effects of oral treatment with retinoids. In Retinoids: 10 Years On; Saurat, J.-A., Ed.; Karger: Basel, Switzerland, 1991; pp. 315–326. [Google Scholar]
- DiGiovanna, J.J.; Sollitto, R.B.; Abangan, D.L.; Steinberg, S.M.; Reynolds, J.C. Osteoporosis is a toxic effect of long-term etretinate therapy. Arch. Dermatol. 1995, 131, 1263–1267. [Google Scholar] [CrossRef]
- De Luca, L.M.; Creek, K.E. Vitamin A and the liver. Prog. Liver Dis. 1986, 8, 81–98. [Google Scholar]
- Miksad, R.; Ledinghen, V.; McDougall, C.; Isabele, F.; Howard, R. Hepatic hydrothorax associated with vitamin A toxicity. J. Clin. Gastroenterol. 2002, 34, 275–279. [Google Scholar] [CrossRef]
- De Genaro, S.P.; Martini, L.A. Vitamin A supplementation and risk of skeletal fracture. Nutr. Rev. 2004, 62, 65–67. [Google Scholar]
- Pritchard, J. Guillain Barré in 13-cis-retinoic acid. Br. Med. J. 2004, 328, 1537. [Google Scholar] [CrossRef]
- Bhalla, K.; Ennis, D.M.; Ennis, E.D. Hypercalcemia caused by iatrogenic hypervitaminosis A. J. Am. Diet. Assoc. 2005, 105, 119–121. [Google Scholar] [CrossRef]
- Patatanian, E.; Thompson, D.F. Retinoic acid syndrome: A review. J. Clin. Pharm. Ther. 2008, 33, 331–338. [Google Scholar] [CrossRef]
- Johnson-Davis, K.L.; Moore, S.J.; Owen, W.E.; Cutler, J.M.; Frank, E.L. A rapid HPLC method used to establish pediatric reference intervals for vitamins A and E. Clin. Chim. Acta 2009, 405, 35–38. [Google Scholar] [CrossRef]
- Franssila, R.; Hedman, K. Infection and musculoskeletal conditions: Viral causes of arthritis. Best Pract. Res. Clin. Rheumatol. 2006, 20, 1139–1157. [Google Scholar] [CrossRef]
- Perl, A. Mechanisms of viral pathogenesis in rheumatic disease. Ann. Rheum. Dis. 1999, 58, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Frenkel, L.M.; Nielsen, K.; Garakian, A.; Jin, R.; Wolinsky, J.S.; Cherry, J.D. A search for persistent rubella virus infection in persons with chronic symptoms after rubella and rubella immunization and in patients with juvenile rheumatoid arthritis. Clin. Infect. Dis. 1996, 22, 287–294. [Google Scholar] [CrossRef]
- Guler, E.; Davutoglu, M.; Guler, S.; Citirik, D.; Karabiber, H. Encephalitis in a child during atypical course of rubella. Infection 2009, 37, 65–66. [Google Scholar] [CrossRef]
- Gülen, F.; Cagliyan, E.; Aydinok, Y.; Ozen, S.; Yildiz, B. A patient with rubella encephalitis and status epilepticus. Minerva Pediatr. 2008, 60, 141–144. [Google Scholar]
- Laroche, M.L.; Macian-Montoro, F.; Merle, L.; Vallat, J.M. Cerebral ischemia probably related to isotretinoin. Ann. Pharmacother. 2007, 41, 1073–1076. [Google Scholar] [CrossRef]
- Plotkin, S.A. The history of rubella and rubella vaccination leading to elimination. Clin. Infect. Dis. 2006, 43, S164–S168. [Google Scholar] [CrossRef]
- Forrest, J.M.; Turnbull, F.M.; Sholler, G.F.; Hawker, R.E.; Martin, F.J.; Doran, T.T.; Burgess, M.A. Gregg’s congenital rubella patients 60 years later. Med. J. Aust. 2002, 177, 664–667. [Google Scholar]
- Töndury, G.; Smith, D.W. Fetal rubella pathology. J. Pediatr. 1966, 68, 867–879. [Google Scholar] [CrossRef]
- Sever, J.L.; South, M.A.; Shaver, K.A. Delayed manifestations of congenital rubella. Rev. Infect. Dis. 1985, 7, S164–S169. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Pham, V.H.; Abe, K. Pathogenesis of congenital rubella virus infection in human fetuses: Viral infection in the ciliary body could play an important role in cataractogenesis. EBioMedicine 2014, 2, 59–63. [Google Scholar] [CrossRef]
- Lazar, M.; Perelygina, L.; Martines, R.; Greer, P.; Paddock, C.D.; Peltecu, G.; Lupulescu, E.; Icenogle, J.; Zaki, S.R. Immunolocalization and distribution of rubella antigen in fatal congenital rubella syndrome. EBioMedicine 2015, 3, 86–92. [Google Scholar] [CrossRef]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, e002. [Google Scholar] [CrossRef] [Green Version]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoid nuclear receptors: Lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 451–480. [Google Scholar] [CrossRef]
- De Oliveira, M.R.; Silvestrin, R.B.; Mello e Souza, T.; Moreira, J.C. Therapeutic vitamin A doses increase the levels of markers of oxidative insult in substantia nigra and decrease locomotor and exploratory activity in rats after acute and chronic supplementation. Neurochem. Res. 2008, 33, 378–383. [Google Scholar] [CrossRef]
- De Oliveira, M.R. Vitamin A and retinoids as mitochondrial toxicants. Oxid. Med. Cell. Longev. 2015, 2015, 140267. [Google Scholar] [CrossRef]
- Melnik, B.C. Apoptosis may explain the pharmacological mode of action and adverse effects of isotretinoin, including teratogenicity. Acta Derm. Venereol. 2017, 97, 173–181. [Google Scholar] [CrossRef]
- Soprano, D.R.; Soprano, K.J. Retinoids as teratogens. Annu. Rev. Nutr. 1995, 15, 111–132. [Google Scholar] [CrossRef]
- Monga, M. Vitamin A and its congeners. Semin. Perinatol. 1997, 21, 135–142. [Google Scholar] [CrossRef]
- Collins, M.D.; Mao, G.E. Teratology of retinoids. Annu. Rev. Pharm. Toxicol. 1999, 39, 399–430. [Google Scholar] [CrossRef]
- Lammer, E.J.; Chen, D.T.; Hoar, R.M.; Agnish, N.D.; Benke, P.J.; Braun, J.T.; Curry, C.J.; Fernhoff, P.M.; Grix, A.W., Jr.; Lott, I.T.; et al. Retinoic acid embryopathy. N. Engl. J. Med. 1985, 313, 837–841. [Google Scholar] [CrossRef]
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. American Heart Association Council on Cardiovascular Disease in the Young. Noninherited risk factors and congenital cardiovascular defects: Current knowledge: A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 2995–3014. [Google Scholar]
- Siu, B.L.; Alonzo, M.R.; Vargo, T.A.; Fenrich, A.L. Transient dilated cardiomyopathy in a newborn exposed to idarubicin and all-trans-retinoic acid (ATRA) early in the second trimester of pregnancy. Int. J. Gynecol. Cancer 2002, 12, 399–402. [Google Scholar] [CrossRef]
- Ganguly, J. The Biochemistry of Vitamin A; CRC Press: Boca Raton, FL, USA, 1989. [Google Scholar]
- Hathcock, J.N.; Hattan, D.G.; Jenkins, M.V. Evaluation of vitamin A toxicity. Am. J. Clin. Nutr. 1990, 52, 183–202. [Google Scholar] [CrossRef]
- Graham, E.M.; Petersen, S.M.; Christo, D.K. Intrapartum electronic fetal heart rate monitoring and the prevention of perinatal brain injury. Obstet. Gynecol. 2006, 108, 656–666. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Golczak, M.; Chou, S.; Desai, A.; Hoppel, C.L.; Matsuyama, S.; Palczewski, K. Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J. Biol. Chem. 2009, 284, 15173–15183. [Google Scholar] [CrossRef]
- Lee, S.A.; Belyaeva, O.V.; Popov, I.K.; Kedishvili, N.Y. Overproduction of bioactive retinoic acid in cells expressing disease-associated mutants of retinol dehydrogenase 12. J. Biol. Chem. 2007, 282, 35621–35628. [Google Scholar] [CrossRef]
- Adams, J.; Lammar, E.J. Neurobehavioral teratology of isotretinoin. Reprod. Toxicol. 1993, 7, 175–177. [Google Scholar] [CrossRef]
- Steele, C.E.; Trasler, D.G.; New, D.A.T. An in vivo/in vitro evaluation of the teratogenic action of excess vitamin A. Teratology 1983, 28, 209–214. [Google Scholar] [CrossRef]
- Kurtz, P.J.; Emmerling, D.C.; Donofrio, D.J. Subchronic toxicity of all-trans-retinoic acid and retinylidene in Sprague-Dawley rats. Toxicology 1984, 30, 115–124. [Google Scholar] [CrossRef]
- Macapinlac, M.P.; Olson, J.A. A lethal hypervitaminosis A syndrome in young monkeys (Macacus fascicularis) following a single intramuscular dose of a water-miscible preparation containing vitamins A, D2 and E. Int. J. Vitam. Nutr. Res. 1981, 51, 331–341. [Google Scholar]
- Schurr, D.; Herbert, J.; Habibi, E.; Abrahamov, A. Unusual presentation of vitamin A intoxication. J. Pediatr. Gastroenterol. Nutr. 1983, 2, 705–707. [Google Scholar] [CrossRef]
- Marroni, M.; Bellomo, G.; Bucaneve, G.; Stagni, G.; Baldelli, F. Isotretinoin: Possible cause of acute seizure and confusion. Ann. Pharmacother. 1993, 27, 793–794. [Google Scholar] [CrossRef]
- Sayyah, M.; Rezaie, M.; Haghighi, S.; Amanzadeh, A. Intra-amygdala all-trans retinoic acid inhibits amygdala-kindled seizures in rats. Epilepsy Res. 2007, 75, 97–103. [Google Scholar] [CrossRef]
- Barua, A.R.; Duitsman, P.K.; Kostic, D.; Barua, M.; Olson, J.A. Reduction of serum retinol levels following a single oral dose of all-trans retinoic acid in humans. Int. J. Vit. Nutr. Res. 1997, 67, 423–426. [Google Scholar]
- Vahlquist, A. Long-term safety of retinoid therapy. J. Am. Acad. Dermatol. 1992, 27, S29–S33. [Google Scholar] [CrossRef]
- Raz, Y.; Kelley, M.W. Retinoic acid signaling is necessary for the development of the organ of Corti. Dev. Biol. 1999, 213, 180–193. [Google Scholar] [CrossRef]
- Barbero, P.; Lotersztein, V.; Bronberg, R.; Perez, M.; Alba, L. Acitretin embryopathy: A case report. Birth Defects Res. A Clin. Mol. Teratol. 2004, 70, 831–833. [Google Scholar] [CrossRef]
- Rosa, F.W.; Wilk, A.L.; Kelsey, F.O. Teratogen update: Vitamin A congeners. Teratology 1986, 33, 355–364. [Google Scholar] [CrossRef]
- Teratology Society. Recommendations for vitamin A use during pregnancy. Teratology 1987, 35, 269–275. [Google Scholar] [CrossRef]
- Rothman, K.J.; Moore, L.L.; Singer, M.R.; Nguyen, U.S.; Mannino, S.; Milunsky, A. Teratogenicity of high vitamin A intake. N. Engl. J. Med. 1995, 333, 1369–1373. [Google Scholar] [CrossRef]
- Botto, L.D.; Loffredo, C.; Scanlon, K.S.; Ferencz, C.; Khoury, M.J.; David Wilson, P.; Correa, A. Vitamin A and cardiac outflow tract defects. Epidemiology 2001, 12, 491–496. [Google Scholar] [CrossRef]
- Stånge, L.; Carlström, K.; Eriksson, M. Hypervitaminosis A in early human pregnancy and malformations of the central nervous system. Acta Obstet. Gynecol. Scand. 1978, 57, 289–291. [Google Scholar] [CrossRef]
- Irving, D.W.; Willhite, C.C.; Burk, D.T. Morphogenesis of isotretinoin-induced microcephaly and micrognathia studied by scanning electron microscopy. Teratology 1986, 34, 141–153. [Google Scholar] [CrossRef]
- Benke, P.J. The isotretinoin teratogen syndrome. JAMA 1984, 251, 3267–3269. [Google Scholar] [CrossRef]
- Mawson, A.R. Pathogenesis of zika virus-associated embryopathy. BioRes. Open Access 2016, 5, 171–176. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, H.N.; Pareek, V.; Raza, K.; Dantham, S.; Kumar, P.; Mochan, S.; Faiq, M.A. A possible mechanism of zika virus associated microcephaly: Imperative role of retinoic acid response element (RARE) consensus sequence repeats in the viral genome. Front. Hum. Neurosci. 2016, 10, 403. [Google Scholar] [CrossRef]
- Faiq, M.A.; Kumar, A.; Singh, H.N.; Pareek, V.; Kumar, P. Commentary: A Possible Mechanism of Zika Virus Associated Microcephaly: Imperative Role of Retinoic Acid Response Element (RARE) Consensus Sequence Repeats in the Viral Genome. Front. Microbiol. 2018, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Pick, E.; Charon, J.; Mizel, D. Toxicological aspects of vitamin A and other retinol derivatives. Clin. Haematol. 1981, 11, 117–124. [Google Scholar]
- O’Reilly, K.; Bailey, S.J.; Lane, M.A. Retinoid-mediated regulation of mood: Possible cellular mechanisms. Exp. Biol. Med. 2008, 233, 251–258. [Google Scholar] [CrossRef]
- Bremner, J.D.; McCaffrey, P. The neurobiology of retinoic acid in affective disorders. Prog. Neuropsychopharmacol. Biol. Psychiatr. 2008, 32, 315–331. [Google Scholar] [CrossRef] [Green Version]
- Crandall, J.; Sakai, Y.; Zhang, J.; Koul, O.; Mineur, Y.; Crusio, W.E.; McCaffery, P. 13-cis-retinoic acid suppresses hippocampal cell division and hippocampal-dependent learning in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 5111–5116. [Google Scholar] [CrossRef]
- Kempermann, G.; Jessberger, S.; Steiner, B.; Kronenberg, G. Milestones of neuronal development in the adult hippocampus. Trend Neurosci. 2004, 27, 447–452. [Google Scholar] [CrossRef]
- McCaffery, P.; Zhang, J.; Crandall, J.E. Retinoic acid signaling and function in the adult hippocampus. J. Neurobiol. 2006, 66, 780–791. [Google Scholar] [CrossRef]
- Dopheide, M.M.; Morgan, R.E. Isotretinoin (13-cis-retinoic acid) alters learning and memory, but not anxiety-like behavior, in the adult rat. Pharmacol. Biochem. Behav. 2008, 91, 243–251. [Google Scholar] [CrossRef]
- Ferguson, S.A.; Berry, K.J. Oral Accutane (13-cis-retinoic acid) has no effects on spatial learning and memory in male and female Sprague-Dawley rats. Toxicol. Teratol. 2007, 29, 219–227. [Google Scholar] [CrossRef]
- Geubel, A.; De Galocsy, C.; Alves, N.; Rahier, J.; Dive, C. Liver damage caused by therapeutic vitamin A administration: Estimate of dose-related toxicity in 41 cases. Gastroenterology 1991, 100, 1701–1709. [Google Scholar] [CrossRef]
- Dan, Z.; Popov, Y.; Patsenker, E.; Preimel, D.; Liu, C.; Wang, X.-D.; Seitz, H.K.; Schuppan, D.; Stickel, F. Hepatotoxicity of alcohol-induced polar retinol metabolites involves apoptosis via loss of mitochondrial membrane potential. FASEB J. 2005, 19, 845–847. [Google Scholar] [CrossRef]
- Russell, R.M.; Boyer, J.L.; Bagheri, S.A.; Hruban, Z. Hepatic injury from chronic hypervitaminosis A resulting in portal hypertension and ascites. N. Engl. J. Med. 1974, 291, 435–440. [Google Scholar] [CrossRef]
- Lieber, C.S.; Leo, M.A. Interaction of alcohol and nutritional factors with hepatic fibrosis. Prog. Liver Dis. 1986, 8, 253–272. [Google Scholar]
- Saltzman, M.D.; King, E.C. Central physeal arrests as a manifestation of hypervitaminosis A. J. Pediatr. Orthop. 2007, 27, 351–353. [Google Scholar] [CrossRef]
- Binkley, N.; Brueger, D. Hypervitaminosis A and bone. Nutr. Rev. 2000, 58, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Cione, E.; Caroleo, M.C.; Cannataro, R.; Perri, M.; Pingitore, A.; Genchi, G. Vitamin A and diabesity: New insight for drug discovery. Mini Rev. Med. Chem. 2016, 16, 738–742. [Google Scholar] [CrossRef]
- Ziring, P.R.; Fedun, B.A.; Cooper, L.Z. Thyrotoxicosis in congenital rubella. J. Pediatr. 1975, 87, 1002. [Google Scholar] [CrossRef]
- Ziring, P.R.; Gallo, G.; Finegold, M.; Buimovici-Klein, E.; Ogra, P. Chronic lymphocytic thyroiditis: Identification of rubella virus antigen in the thyroid of a child with congenital rubella. J. Pediatr. 1977, 90, 419–420. [Google Scholar] [CrossRef]
- Desailloud, R.; Hober, D. Viruses and thyroiditis: An update. Virol. J. 2009, 6, 5. [Google Scholar] [CrossRef]
- Croft, A.M.; Mawson, A.R. Evidence for neurotoxicity from quinoline antimalaria drugs: Four personal accounts. Open J. Anim. Sci. 2017, 7, 45–55. [Google Scholar] [CrossRef]
- Morley, J.E.; Melmed, S.; Reed, A.; Kasson, B.G.; Levin, S.R.; Pekary, A.E.; Hershman, J.E. Effect of vitamin A on the hypothalamo-pituitary-thyroid axis. Am. J. Physiol. Endocrinol. Metab. 1980, 238, E174–E179. [Google Scholar] [CrossRef]
- Townsend, J.J.; Baringer, J.R.; Wolinsky, J.S.; Malamud, N.; Mednick, J.P.; Panitch, H.S.; Scott, R.A.; Oshiro, L.; Cremer, N.E. Progressive rubella panencephalitis. Late onset after congenital rubella. N. Engl. J. Med. 1975, 292, 990–993. [Google Scholar] [CrossRef]
- Cheruvattath, R.; Orrego, M.; Gautam, M.; Byrne, T.; Alam, S.; Voltchenok, M.; Edwin, M.; Wilkens, J.; Williams, J.W.; Vargas, H.E. Vitamin A toxicity: When one a day doesn’t keep the doctor away. Liver Transpl. 2006, 12, 1888–1891. [Google Scholar] [CrossRef]
- Restak, R.M. Pseudotumor cerebri, psychosis, and hypervitaminosis A. J. Nerv. Ment. Dis. 1972, 155, 72–75. [Google Scholar] [CrossRef]
- Mikkelsen, B.; Ehlers, N.; Thomsen, H.G. Vitamin-A intoxication causing papilledema and simulating acute encephalitis. Acta Neurol. Scand. 1974, 50, 642–650. [Google Scholar] [CrossRef]
- Gungor, S.; Olmez, A.; Pinar Arikan Firat Haliloğlu, G.; Anlar, B. Serum retinol and beta-carotene levels in subacute sclerosing panencephalitis. J. Child. Neurol. 2007, 22, 341–343. [Google Scholar] [CrossRef]
- Hutton, J. Does rubella cause autism: A 2015 reappraisal? Front. Hum. Neurosci. 2016, 10, 25. [Google Scholar] [CrossRef]
- Barger, B.D.; Campbell, J.M.; McDonough, J.D. Prevalence and onset of regression within autism spectrum disorders: A meta-analytic review. J. Autism Dev. Disord. 2013, 43, 817–828. [Google Scholar] [CrossRef]
- Thompson, L.; Gillberg, C.; Landberg, S.; Kantzer, A.K.; Miniscalco, C.; Barnevik Olsson, M.; Eriksson, M.A.; Fernell, E. Autism with and without regression: A two-year prospective longitudinal study in two population-derived Swedish cohorts. J. Autism Dev. Disord. 2019, 49, 2281–2290. [Google Scholar] [CrossRef]
- CDC. Vaccine Safety. 2015. Available online: https://www.cdc.gov/vaccinesafety/ensuringsafety/history/index.html (accessed on 1 January 2019).
- Whitney, C.G.; Zhou, F.; Singleton, J.; Schuchat, A. Benefits from immunization during the vaccines for children program era—United States, 1994–2013. Morb. Mortal. Weekly Rep. 2014, 63, 352–355. [Google Scholar]
- Taylor, L.E.; Swerdfeger, A.L.; Eslick, G.D. Vaccines are not associated with autism: An evidence-based meta-analysis of case-control and cohort studies. Vaccine 2014, 32, 3623–3629. [Google Scholar] [CrossRef]
- Offit, P.A. Vaccines and autism in primate model. Proc. Natl. Acad. Sci. USA 2015, 112, 12236–12237. [Google Scholar] [CrossRef] [Green Version]
- Hviid, A.; Hansen, J.V.; Frisch, M.; Melbye, M. Measles, Mumps, Rubella Vaccination and Autism: A Nationwide Cohort Study. Ann. Intern. Med. 2019, 170, 513–520. [Google Scholar] [CrossRef]
- Parker, A. Testing new hypotheses of neurological and immunological outcomes with aluminum-containing vaccines is warranted. J. Trace Elem. Med. Biol. 2019, 51, 28–30. [Google Scholar] [CrossRef]
- Maglione, M.A.; Das, L.; Raaen, L.; Smith, A.; Chari, R.; Newberry, S.; Shanman, R.; Perry, T.; Goetz, M.B.; Gidengil, C. Safety of vaccines used for routine immunization of U.S. children: A systematic review. Pediatrics 2014, 134, 325–337. [Google Scholar] [CrossRef]
- Mawson, A.R.; Croft, A.M. Gulf War Illness: Unifying hypothesis for a continuing health problem. Int. J. Environ. Res. Public Health 2019, 16, 111. [Google Scholar] [CrossRef]
- National Vaccine Information Center. Available online: https://www.nvic.org/ (accessed on 17 September 2019).
- Shaw, C.A. Aluminum as a CNS and Immune System Toxin Across the Life Span. In Neurotoxicity of Aluminum; Advances in Experimental Medicine and Biology; Niu, Q., Ed.; Springer: Singapore, 2018; Volume 1091. [Google Scholar]
- CDC. Recommended Immunization Schedules for Persons Aged 0 Through 18 Years, United States. 2018. Available online: https://www.cdc.gov/vaccines/schedules/downloads/child/0-18yrs-child-combined-schedule.pdf (accessed on 26 December 2018).
- CDC. Recommended Schedule for Active Immunization of Normal Infants and Children. 1983. Available online: https://wonder.cdc.gov/wonder/prevguid/p0000206/p0000206.asp (accessed on 26 December 2018).
- IOM (Institute of Medicine). Adverse Effects of Vaccines: Evidence and Causality; The National Academies Press: Washington, DC, USA, 2012; Available online: https://www.nap.edu/read/13164/chapter/1 (accessed on 1 January 2019).
- Sheth, S.K.S.; Li, Y.; Shaw, C.A. Is exposure to aluminium adjuvants associated with social impairments in mice? A pilot study. J. Inorg. Biochem. 2018, 181, 96–103. [Google Scholar] [CrossRef]
- Principi, N.; Esposito, S. Aluminum in vaccines: Does it create a safety problem? Vaccine 2018, 36, 5825–5831. [Google Scholar] [CrossRef]
- Exley, C. The toxicity of aluminium in humans. Morphology 2016, 100, 51–55. [Google Scholar] [CrossRef]
- Morris, G.; Puri, B.K.; Frye, R.E. The putative role of environmental aluminium in the development of chronic neuropathology in adults and children. How strong is the evidence and what could be the mechanisms involved? Metab. Brain Dis. 2017, 32, 1335–1355. [Google Scholar] [CrossRef] [Green Version]
- Crépeaux, G.; Eidi, H.; David, M.-O.; Baba-Amer, Y.; Tzavara, E.; Giros, B.; Authier, F.-J.; Exley, C.; Shaw, C.A.; Cadusseau, J.; et al. Non-linear dose-response of aluminium hydroxyde adjuvant particles: Selective low dose neurotoxicity. Toxicology 2017, 375, 48–75. [Google Scholar] [CrossRef]
- Mold, M.; Umar, D.; King, A.; Exley, C. Aluminium in brain tissue in autism. J. Trace Elem. Med. Biol. 2018, 46, 76–82. [Google Scholar] [CrossRef]
- Sienkiewicz, D.; Kulak, W.; Okurowska-Zawada, B.; Paszko-Pateg, G. Neurologic adverse events following vaccination. Prog. Health Sci. 2012, 2, 129–141. [Google Scholar]
- U.S. Food and Drug Administration. Measles, Mumps and Rubella Virus Vaccine, Live. 2 February 2018. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/measles-mumps-and-rubella-virus-vaccine-live (accessed on 11 May 2019).
- The College of Physicians of Philadelphia. The History of Vaccines: An Educational Resource. Available online: https://www.historyofvaccines.org/content/articles/vaccine-side-effects-and-adverse-events (accessed on 13 May 2019).
- Goldman, G.S.; Miller, N.Z. Relative trends in hospitalizations and mortality among infants by the number of vaccine doses and age, based on the Vaccine Adverse Event Reporting System (VAERS), 1990–2010. Hum. Exp. Toxicol. 2012, 31, 1012–1021. [Google Scholar] [CrossRef]
- Institute of Medicine. The Childhood Immunization Schedule and Safety: Stakeholder Concerns, Scientific Evidence, and Future Studies; The National Academies Press: Washington, DC, USA, 2013. [Google Scholar]
- Mawson, A.R.; Ray, B.D.; Bhuiyan, A.R.; Jacob, B. Pilot comparative study on the health of vaccinated and unvaccinated 6- to 12-year-old U.S. children. J. Transl. Sci. 2017, 3, 1–12. [Google Scholar] [CrossRef]
- Mawson, A.R.; Bhuiyan, A.; Jacob, B.; Ray, B.D. Preterm birth, vaccination and neurodevelopmental disorders: A cross-sectional study of 6- to 12-year-old vaccinated and unvaccinated children. J. Transl. Sci. 2017, 3, 1–8. [Google Scholar] [CrossRef]
- Mawson, A.R. A role for the liver in parturition and preterm birth. J. Transl. Sci. 2016, 2, 154–159. [Google Scholar] [CrossRef]
- Zhuang, X.; Cui, A.-M.; Wang, Q.; Cheng, X.-Y.; Shen, Y.; Cai, W.-H.; Li, Ha.; Zhang, S.; Qin, G. Liver dysfunction during pregnancy and its association with preterm birth in China: A prospective cohort study. EBioMedicine 2017, 26, 152–156. [Google Scholar] [CrossRef]
- Kidd, P.M. Autism, an extreme challenge to integrative medicine. Part 1: The knowledge base. Altern. Med. Rev. 2002, 7, 292–316. [Google Scholar]
- Horvath, K.; Perman, J.A. Autistic disorder and gastrointestinal disease. Curr. Opin. Pediatr. 2002, 14, 583–587. [Google Scholar] [CrossRef]
- Shedlock, K.; Susi, A.; Gorman, G.H.; Hisle-Gorman, E.; Erdie-Lalena, C.R.; Nylund, C.M. Autism spectrum disorders and metabolic complications of obesity. J. Pediatr. 2016, 178, 183–187. [Google Scholar] [CrossRef]
- Lund, N.; Biering-Sørensen, S.; Andersen, A.; Monteiro, I.; Camala, L.; Jørgensen, M.J.; Aaby, P.; Benn, C.S. Neonatal vitamin A supplementation associated with a cluster of deaths and poor early growth in a randomised trial among low-birth-weight boys of vitamin A versus oral polio vaccine at birth. BMC Pediatr. 2014, 14, 214. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Tanaka, H.; Akutsu, T.; Koide Kentaro Sakuma, M.; Okazaki, M.; Ida, H.; Urashima, M. Prenatal vitamin A supplementation associated with adverse child behavior at 3 years in a prospective birth cohort in Japan. Pediatr. Internat. 2016, 58, 855–861. [Google Scholar] [CrossRef]
- Benn, C.S.; Aaby, P.; Arts, R.J.; Jensen, K.J.; Netea, M.G.; Fisker, A.B. An enigma: Why vitamin A supplementation does not always reduce mortality even though vitamin A deficiency is associated with increased mortality. Int. J. Epidemiol. 2015, 44, 906–918. [Google Scholar] [CrossRef]
- Beaton, G.H.; Martorell, R.; Aronson, K.J.; Edmonston, B.; McCabe, G.; Ross, A.C.; Harvey, B. Effectiveness of Vitamin a Supplementation in the Control. of Young Child. Morbidity and Mortality in Developing Countries; Administrative Committee on Coordination—Subcommittee on Nutrition (ACC/SCN): Geneva, Switzerland, 1993. [Google Scholar]
- Latham, M. The great vitamin A fiasco. World Nutr. May 2010, 1, 12–45. [Google Scholar]
- Bhattacharya, S.; Singh, A. Time to revisit the strategy of massive vitamin A prophylaxis dose administration to the under five children in India—An analysis of available evidence. Clin. Nutr. ESPEN 2017, 21, 26–30. [Google Scholar] [CrossRef]
- Greiner, T.; Mason, J.; Benn, C.S.; Sachdev, H.P.S. Does India Need a Universal High-Dose Vitamin A Supplementation Program? Indian J. Pediatr. 2019, 84, 1–4. [Google Scholar] [CrossRef]
- Imdad, A.; Yakoob, M.Y.; Sudfeld, C.; Haider, B.A.; Black, R.E.; Bhutta, Z.A. Impact of vitamin A supplementation on infant and childhood mortality. BMC Public Health 2011, 11, 20. [Google Scholar] [CrossRef]
- Bello, S.; Meremikwu, M.M.; Ejemot-Nwadiaro, R.I.; Oduwole, O. Routine vitamin A supplementation for the prevention of blindness due to measles infection in children. Cochrane Database Syst. Rev. 2016, CD007719. [Google Scholar] [CrossRef]
- Iannotti, L.L.; Trehan, I.; Manary, M.J. Review of the safety and efficacy of vitamin A supplementation in the treatment of children with severe acute malnutrition. Nutr. J. 2013, 12, 125. [Google Scholar] [CrossRef]
- Benn, C.S. Combining vitamin A and vaccines: Convenience or conflict? Dan. Med. J. 2012, 59, 4378. [Google Scholar]
- Loughrill, E.; Govinden, P.; Zand, N. Vitamins A and E content of commercial infant foods in the UK: A cause for concern? Food Chem. 2016, 210, 56–62. [Google Scholar] [CrossRef]
- Schultz, S.T.; Klonoff-Cohen, H.S.; Wingard, D.L.; Akshoomoff, N.A.; Macera, C.A.; Ji, M.; Bacher, C. Breastfeeding, infant formula supplementation, and autistic disorder: The results of a parent survey. Int. Breastfeed. J. 2006, 1, 16. [Google Scholar] [CrossRef]
- U.S. Department of Agriculture, Agricultural Research Service. Nutrient Intakes from Food and Beverages: Mean Amounts Consumed per Individual, by Gender and Age, What We Eat in America, NHANES 2011–2012. 2014. Available online: http://www.ars.usda.gov/SP2UserFiles/Place/80400530/pdf/1112/Table_1_NIN_GEN_11.pdf (accessed on 4 August 2018).
- Veniaminova, E.; Cespuglio, R.; Cheung, C.W.; Umriukhin, A.; Markova, N.; Shevtsova, E.; Lesch, K.P.; Anthony, D.C.; Strekalova, T. Autism-like behaviours and memory deficits result from a western diet in mice. Neural. Plast. 2017, 2017, 9498247. [Google Scholar] [CrossRef]
- Herbert, M.R.; Buckley, J.A. Autism and dietary therapy: Case report and review of the literature. J. Child. Neurol. 2013, 28, 975–982. [Google Scholar] [CrossRef]
- Piwowarczyk, A.; Horvath, A.; Łukasik, J.; Pisula, E.; Szajewska, H. Gluten- and casein-free diet and autism spectrum disorders in children: A systematic review. Eur. J. Nutr. 2018, 57, 433–440. [Google Scholar] [CrossRef]
- Adams, J.B.; Audhya, T.; Geis, E.; Gehn, E.; Fimbres, V.; Pollard, E.L.; Mitchell, J.; Ingram, J.; Hellmers, R.; Laake, D.; et al. Comprehensive Nutritional and Dietary Intervention for Autism Spectrum Disorder—A Randomized, Controlled 12-Month Trial. Nutrients 2018, 10, 369. [Google Scholar] [CrossRef]
- David, E.; Pucci, A.; Amoroso, A.; Salizzoni, M. Liver transplantation in defects of cholesterol biosynthesis: The case of lathosterolosis. Am. J. Transplant. 2014, 14, 960–965. [Google Scholar] [CrossRef]
- Landrigan, P.J.; Belpoggi, F. The need for independent research on the health effects of glyphosate-based herbicides. Environ. Health 2018, 17, 51. [Google Scholar] [CrossRef]
- Von Ehrenstein, O.S.; Ling, C.; Cui, X.; Cockburn, M.; Park, A.S.; Yu, F.; Wu, J.; Ritz, B. Prenatal and infant exposure to ambient pesticides and autism spectrum disorder in children: Population based case-control study. BMJ 2019, 364, l962. [Google Scholar] [CrossRef]
- Samsel, A.; Seneff, S. Glyphosate pathways to modern diseases VI: Prions, amyloidoses and autoimmune neurological diseases. J. Biol. Phys. Chem. 2017, 17, 8–32. [Google Scholar] [CrossRef]
- Roy, N.M.; Carneiro, B.; Ochs, J. Glyphosate induces neurotoxicity in zebrafish. Environ. Toxicol. Pharmacol. 2016, 42, 45–54. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Symptoms/Signs | Rubella | Hypervitaminosis A |
|---|---|---|
| Mild fever | + | + |
| Arthralgia | + | + |
| Polyarthritis | + | + |
| Myalgia | + | + |
| Headache | + | + |
| Flu-like symptoms | + | + |
| Conjunctivitis | + | + |
| Lymphadenopathy | + | + |
| Maculopapular rash | + | + |
| Pruritus | + | + |
| Fatigue | + | + |
| Anorexia | + | + |
| Slight desquamation | + | + |
| Encephalitis/Encephalopathy | + | + |
| Splenomegaly | + | + |
| Miscarriage | + | + |
| Thrombocytopenic purpura | + | + |
| Guillain–Barré syndrome | + | + |
| Features | Congenital Rubella Syndrome | Hypervitaminosis A-Associated Effects |
|---|---|---|
| Cataract | + | + |
| Microphthalmia | + | + |
| Retinopathy | + | + |
| Bulging fontanelle | + | + |
| Intrauterine growth restriction | + | + |
| Seizures | + | + |
| Hearing defects | + | + |
| Heart defects, including Patent Ductus Arteriosus | + | + |
| Microcephaly | + | + |
| Cognitive deficits, behavioral and speech disorders | + | + |
| Hepatitis/jaundice | + | + |
| Hepatosplenomegaly | + | + |
| Bone lesions/osteoporosis | + | + |
| Type 1 diabetes mellitus | + | + |
| Thyroiditis | + | + |
| Encephalitis/Encephalopathy | + | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mawson, A.R.; Croft, A.M. Rubella Virus Infection, the Congenital Rubella Syndrome, and the Link to Autism. Int. J. Environ. Res. Public Health 2019, 16, 3543. https://doi.org/10.3390/ijerph16193543
Mawson AR, Croft AM. Rubella Virus Infection, the Congenital Rubella Syndrome, and the Link to Autism. International Journal of Environmental Research and Public Health. 2019; 16(19):3543. https://doi.org/10.3390/ijerph16193543
Chicago/Turabian StyleMawson, Anthony R., and Ashley M. Croft. 2019. "Rubella Virus Infection, the Congenital Rubella Syndrome, and the Link to Autism" International Journal of Environmental Research and Public Health 16, no. 19: 3543. https://doi.org/10.3390/ijerph16193543
APA StyleMawson, A. R., & Croft, A. M. (2019). Rubella Virus Infection, the Congenital Rubella Syndrome, and the Link to Autism. International Journal of Environmental Research and Public Health, 16(19), 3543. https://doi.org/10.3390/ijerph16193543