Environmental and Endogenous Acids Can Trigger Allergic-Type Airway Reactions


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Outdoor Acid Air Pollutants: Chemical and Toxicological Characteristics
2.2. Biochemical Effects of Cellular Acidification in Epithelial Tissues
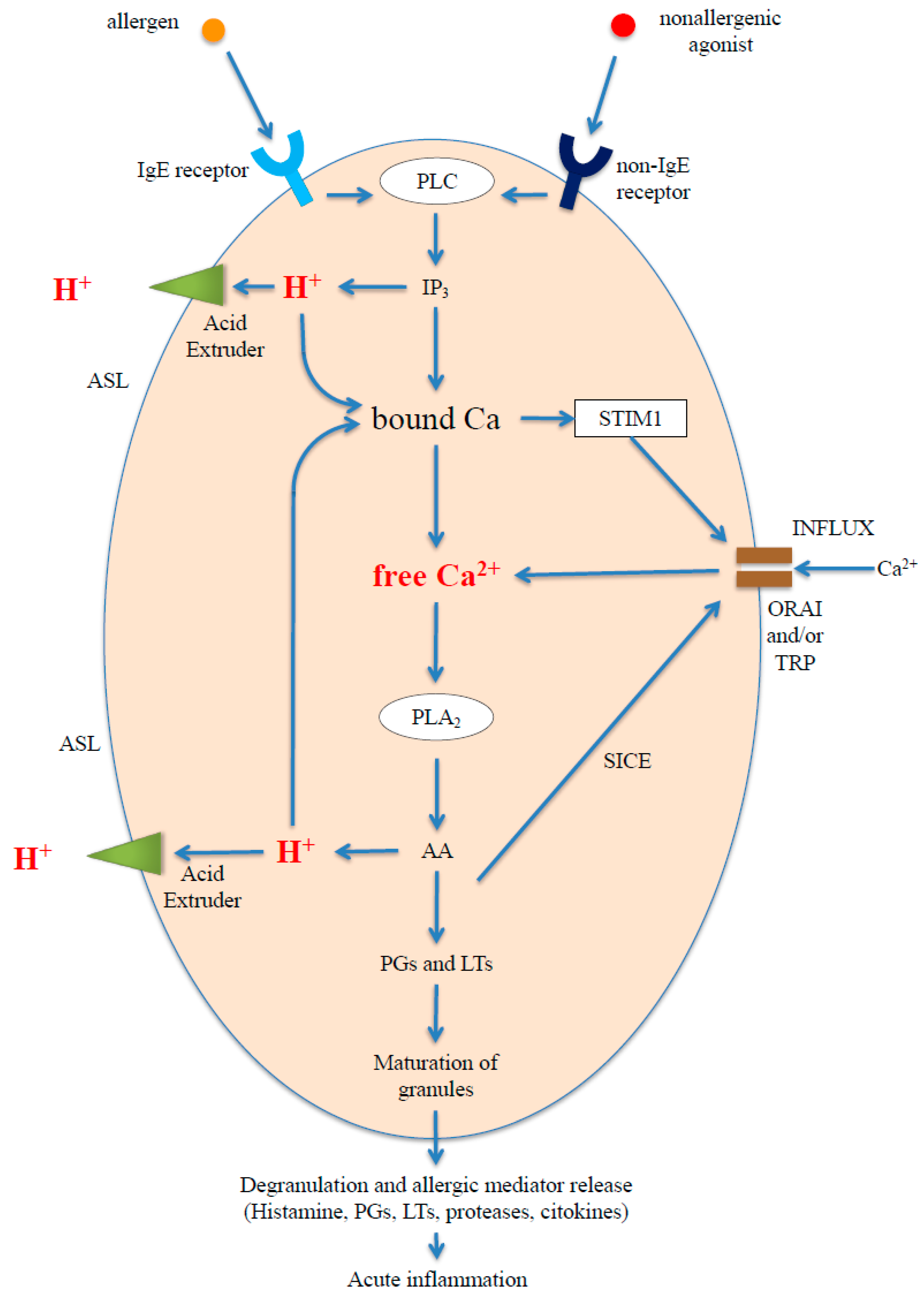
2.3. Intracellular H+: Intracellular Acidification May Be Caused by the Action of Phospholipases in the Cytosol or by Protons Entering the Cell through the Plasma Membrane
- (a)
- The stimulation of the receptor, both of the FcεRI and GPCR types, activates phospholipase C (PLC) [85,86,87,88] and hence the hydrolysis of phosphatidylinositol 4,5-biphosphate (PIP2) on the inner wall of the plasma membrane, generating and releasing IP3, a protonated acid salt [62,89], in the cytosol;
- (b)
- (c)
- The increase in (Ca2+)c activates numerous calcium-dependent enzymes, including phospholipase A2 (PLA2), which produces arachidonic acid (AA) [91,92], which in turn dissociates releasing more H+ and inducing the release of more Ca2+ [56,58,93]; from the AA hundreds of derivatives (eicosanoids cascade) are formed, including leukotrienes (LTs) and prostaglandins (PGs) [94,95]. Both leukotrienes and prostaglandins are known to play a pivotal role in inflammatory and allergic reactions;
- (d)
- The store depletion stimulates the entry of more Ca2+ from the extracellular space (calcium influx) via the mechanism known as Store Operated Calcium Entry (SOCE), in which, from the surface of the Endoplasmic Reticulum (ER), Stromal Interaction Molecule1 (STIM1) activates the opening of ORAI1 and Transient Receptor Potential Cation Canonical (TRPC) [96,97,98,99] channels on the plasma membrane;
- (e)
- The calcium influx further stimulates PLA2 activity and fosters the maturing of the granules and subsequent degranulation and release [100,101,102,103] of mediators [94,104,105], including histamine, PGs, LTs, cytokine, tryptase, and chymase, which promote the acute phase of allergic inflammation. The cysteinyl LTs are thought to be responsible for the increase in the basal tone of the ASM and in bronchoconstriction in asthma [6,106].
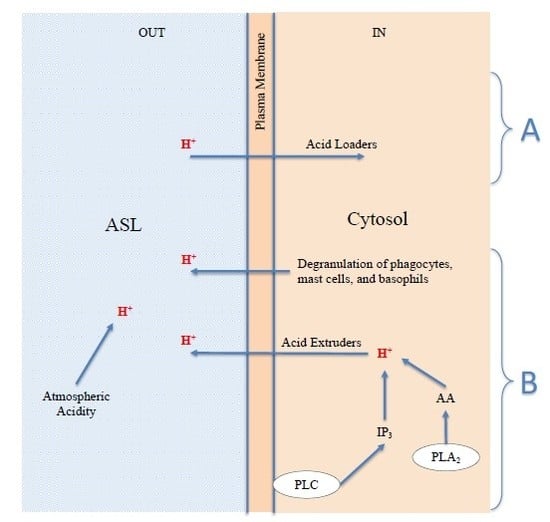
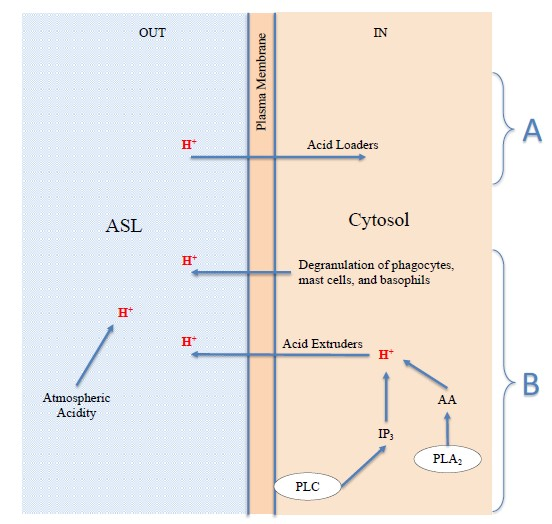
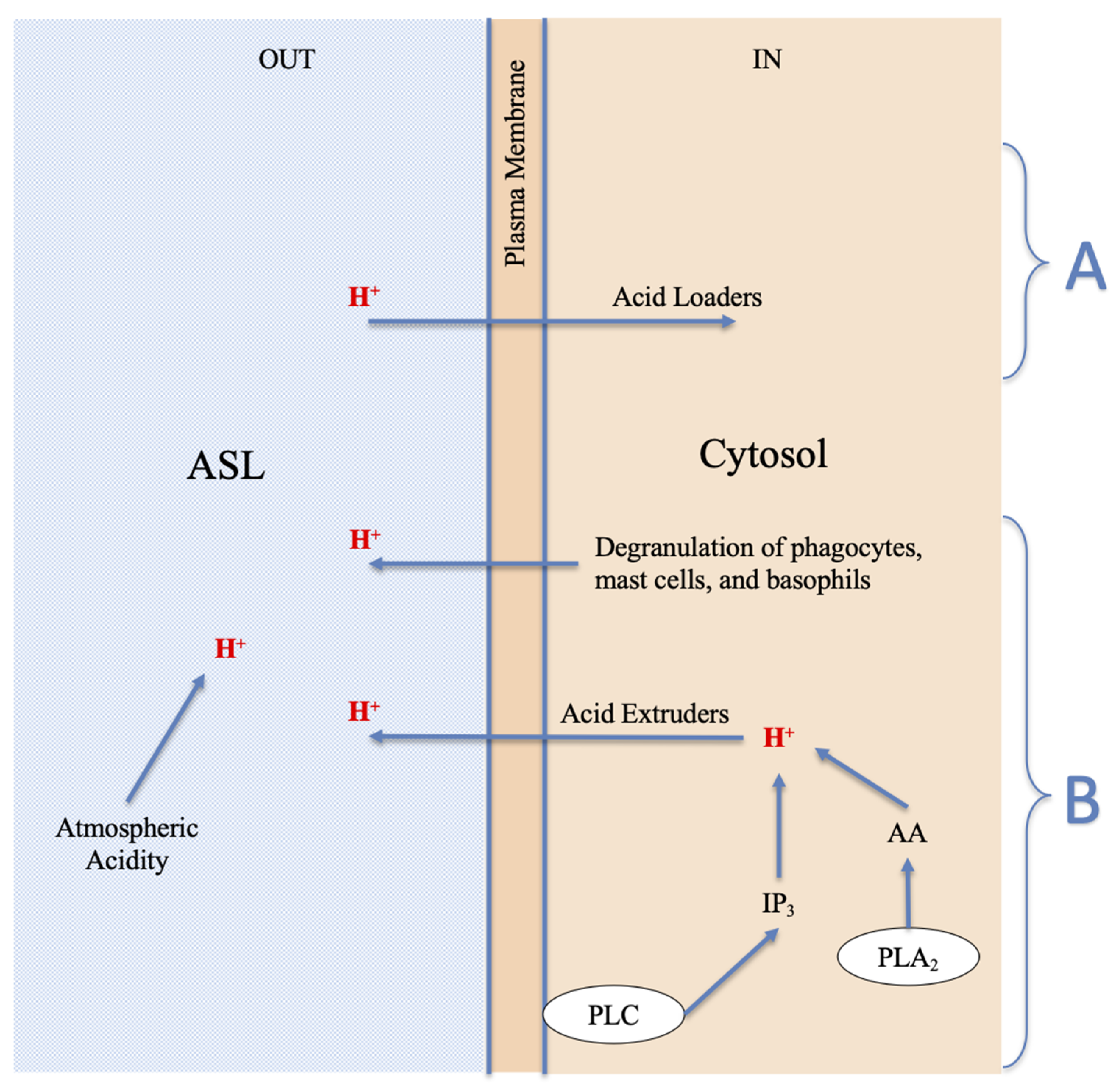
2.4. Extracellular H+: The Acidification of the Surfaces of the Respiratory Airways May Be Due to Environmental Acid Pollutants or Endogenous Acids
- (a)
- H+ derived from the physiological process of restoring prestimulus conditions, carried out by all cells through the expulsion of excess protons, generated by acidifying enzymes, to return to the steady state; cells can use acid extruders as exchangers and channels to transfer H+ externally; the Na+-H+ exchanger (NHE) in some cells is the major acid-extruder, also the Cystic fibrosis transmembrane conductance regulator (CFTR) plays an important role in the acidification of the ASL [117]; in addition, the excess protons in the cytosol may exit the cell via voltage-gated proton channels (Hv1), TRP channels, plasma membrane vacuolar V-type H+-ATPase [126,132,133,134,135,136], and diffusion [125];
- (b)
- The degranulation of phagocytes, such as macrophages and granulocyte neutrophils and eosinophils [69,135,137,138], produced as a defensive inflammatory action [24,126] in response to the stimulus. This acidifying action may be significant and long lasting, and is therefore the basis for chronic disease;
- (c)
- The degranulation of mast cells and basophils, caused by the stimulus, the basis of the acute allergic response [77,78,80,84,138], as described above in Figure 1. It is known that, like phagocytes, basophils and mast cells [138] can produce and secrete acids and phospholipolytic enzymes with the contents of their cytosolic granules and vesicles. Examples of secreted acids are lactic, hypochlorous, uric, phosphoric acid, and fatty acids. Examples of enzymes are the cytosolic and secretory phospholipases A2, which produce fatty acids such as AA through hydrolysis of cellular triglycerides and phospholipids [139]. Each of the secreted acids can contribute to the release of protons and thus act as new stimuli for cellular responses;
- (d)
- In addition to the endogenous acids described above in point a, band c, which are transferred by the cells to the ASL by means of expulsion, extrusion, and/or degranulation, the acidification of the ASL may be due to exogenous acids, and hence, possibly, to the presence and direct action of atmospheric acid pollutants.
3. Discussion
3.1. Difficulties to Overcome
3.2. Possible Deductions
- (a)
- Environmental acidity increases the sensitivity of epithelial surfaces and promotes AHR;
- (b)
- Exogenous and endogenous acids contribute to both the decrease in ASL pH and the increase in ASM basal tone, thus favoring bronchoconstriction;
- (c)
- The excess of temporary intracellular acidification is at the origin of acute manifestations of an allergic kind;
- (d)
- Recurrent or continuous acidification is the biochemical basis of airway inflammation, hyper-responsiveness, tissue remodeling, and chronicity.
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thomsen, S.F. Epidemiology and natural history of atopic diseases. Eur. Clin. Respir. J. 2015, 2, 506. [Google Scholar] [CrossRef] [Green Version]
- Bønnelykke, K.; Ober, C. Leveraging gene-environment interactions and endotypes for asthma gene discovery. J. Allergy Clin. Immunol. 2016, 137, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Bowatte, G.; Lodge, C.; Knibbs, L.D.; Lowe, A.; Erbas, B.; Dennekamp, M.; Marks, G.; Giles, G.G.; Morrison, S.; Thompson, B.; et al. Traffic-related air pollution exposure is associated with allergic sensitization, asthma, and poor lung function in middle age. J. Allergy Clin. Immunol. 2017, 139, 122–129.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acevedo, N.; Zakzuk, J.; Caraballo, L. House Dust Mite Allergy Under Changing Environments. Allergy Asthma Immunol. Res. 2019, 11, 450–469. [Google Scholar] [CrossRef] [PubMed]
- Damialis, A.; Traidl-Hoffmann, C.; Treudler, R. Climate Change and Pollen Allergies. In Biodiversity and Health in the Face of Climate Change; Marselle, M., Stadler, J., Korn, H., Irvine, K., Bonn, A., Eds.; Springer Science and Business Media LLC: Cham, Switzerland, 2019; Chapter 3; pp. 47–66. [Google Scholar]
- Bousquet, J.; Khaltaev, N.; Cruz, A.A.; Denburg, J.; Fokkens, W.J.; Togias, A.; Zuberbier, T.; Baena-Cagnani, C.E.; Canonica, G.W.; van Weel, C.; et al. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 Update. Allergy 2008, 63, 8–160. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, G.; Holgate, S.; Pawankar, R.; Ledford, D.; Cecchi, L.; Al-Ahmad, M.; Al-Enezi, F.; Al-Muhsen, S.; Ansotegui, I.; Baena-Cagnani, C.E.; et al. Meteorological conditions, climate change, new emerging factors, and asthma and related allergic disorders. A statement of the World Allergy Organization. World Allergy Organ. J. 2015, 8, 1–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Kang, M.-G.; Chang, Y.-S.; Cho, S.-H. Epidemiology of adult asthma in Asia: Toward a better understanding. Asia Pac. Allergy 2014, 4, 75–85. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. WHO Guidelines for indoor air quality. In WHO Housing and Health Guidelines; World Health Organization: Geneva, Switzerland, 2018; pp. 90–96, ISBN-13:978-92-4-155037-6. [Google Scholar]
- Nicolaou, N.; Siddique, N.; Custovic, A. Allergic disease in urban and rural populations: Increasing prevalence with increasing urbanization. Allergy 2005, 60, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rueda, D.; Turner, M.C. Commentary: Diesel, Cars, and Public Health. Epidemiology 2016, 27, 159–162. [Google Scholar] [PubMed]
- Gehring, U.; Gruzieva, O.; Agius, R.M.; Beelen, R.; Custovic, A.; Cyrys, J.; Eeftens, M.; Flexeder, C.; Fuertes, E.; Heinrich, J.; et al. Air Pollution Exposure and Lung Function in Children: The ESCAPE Project. Environ. Health Perspect. 2013, 121, 1357–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO (World Health Organization), WHO Regional Office for Europe. Review of Evidence on Health Aspects of Air Pollution–REVIHAAP Project; Technical Report; WHO: Geneva, Switzerland, 2013; Available online: http://www.euro.who.int/__data/assets/pdf_file/0004/193108/REVIHAAP-Final-technical-report.pdf (accessed on 27 June 2020).
- Bowatte, G.; Lodge, C.; Lowe, A.J.; Erbas, B.; Perret, J.; Abramson, M.J.; Matheson, M.C.; Dharmage, S.C. The influence of childhood traffic-related air pollution exposure on asthma, allergy and sensitization: A systematic review and a meta-analysis of birth cohort studies. Allergy 2015, 70, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, J.; Anto, J.M.; Akdis, M.; Auffray, C.; Keil, T.; Momas, I.; Postma, D.; Valenta, R.; Wickman, M.; Cambon-Thomsen, A.; et al. Paving the way of systems biology and precision medicine in allergic diseases: The MeDALL success story. Mechanisms of the Development of Allergy. Allergy 2016, 71, 1513–1525. [Google Scholar] [CrossRef] [Green Version]
- Anto, J.M.; Bousquet, J.; Akdis, M.; Auffray, C.; Keil, T.; Momas, I.; Postma, D.S.; Valenta, R.; Wickman, M.; Cambon-Thomsen, A.; et al. Mechanisms of the Development of Allergy (MeDALL): Introducing novel concepts in allergy phenotypes. J. Allergy Clin. Immunol. 2017, 139, 388–399. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, G.; Ortega, O.P.M.; Annesi-Maesano, I.; D’Amato, M. Prevention of Allergic Asthma with Allergen Avoidance Measures and the Role of Exposome. Curr. Allergy Asthma Rep. 2020, 20, 8. [Google Scholar] [CrossRef]
- Simon, D. Recent Advances in Clinical Allergy and Immunology. Int. Arch. Allergy Immunol. 2018, 177, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Adams, O.J.; von Gunten, S. Recent Advances in Experimental Allergy. Int. Arch. Allergy Immunol. 2018, 177, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Pinart, M.; Benet, M.; Annesi-Maesano, I.; von Berg, A.; Berdel, D.; Carlsen, K.C.L.; Carlsen, K.-H.; Bindslev-Jensen, C.; Eller, E.; Fantini, M.P.; et al. Comorbidity of eczema, rhinitis, and asthma in IgE-sensitised and non-IgE-sensitised children in MeDALL: A population-based cohort study. Lancet Respir. Med. 2014, 2, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Apel, K.; Costet, N.; Chapron, A.; Cordier, S.; Monfort, C.; Chevrier, C.; Pelé, F. Home environment: Respiratory and allergic phenotypes from birth to age six in the PELAGIE cohort. NPJ Prim. Care Respir. Med. 2019, 29, 1–8. [Google Scholar] [CrossRef]
- Liu, L.; Poon, R.; Chen, L.; Frescura, A.M.; Montuschi, P.; Ciabattoni, G.; Wheeler, A.; Dales, R. Acute Effects of Air Pollution on Pulmonary Function, Airway Inflammation, and Oxidative Stress in Asthmatic Children. Environ. Health Perspect. 2009, 117, 668–674. [Google Scholar] [CrossRef]
- Balmes, J.R.; Fine, J.M.; Gordon, T.; Sheppard, D. Potential Bronchoconstrictor Stimuli in Acid Fog. Environ. Health Perspect. 1989, 79, 163–166. [Google Scholar] [CrossRef]
- Ricciardolo, F.L.; Gaston, B.; Hunt, J. Acid stress in the pathology of asthma. J. Allergy Clin. Immunol. 2004, 113, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Héroux, M.E.; Anderson, H.R.; Atkinson, R.; Brunekreef, B.; Cohen, A.; Forastiere, F.; Hurley, F.; Katsouyanni, K.; Krewski, D.; Krzyzanowski, M.; et al. Quantifying the health impacts of ambient air pollutants: Recommendations of a WHO/Europe project. Int. J. Public Health 2015, 60, 619–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, S.G.; Ma, Y.X.; Shang, K.Z.; Cheng, Y.F.; Li, X.; Ning, G.C.; Zhao, W.J.; Li, N.R. Association between Ambient Air Pollution and Hospital Emergency Admissions for Respiratory and Cardiovascular Diseases in Beijing: A Time Series Study. Biomed. Environ. Sci. 2015, 28, 352–363. [Google Scholar] [PubMed]
- Paulin, L.M.; Hansel, N. Particulate air pollution and impaired lung function. F1000Research 2016, 5, F1000. [Google Scholar] [CrossRef] [PubMed]
- Endeward, V.; Al-Samir, S.; Itel, F.; Gros, G. How does carbon dioxide permeate cell membranes? A discussion of concepts, results and methods. Front. Physiol. 2014, 4, 382. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Zhang, T.; Mao, J. Haze facilitates sensitization to house dust mites in children. Environ. Geochem. Health 2019. [Google Scholar] [CrossRef]
- Koehler, C.; Paulus, M.; Ginzkey, C.; Hackenberg, S.; Scherzad, A.; Ickrath, P.; Hagen, R.; Kleinsasser, N. The Proinflammatory Potential of Nitrogen Dioxide and Its Influence on the House Dust Mite Allergen Der p 1. Int. Arch. Allergy Immunol. 2016, 171, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Reno, A.L.; Brooks, E.G.; Ameredes, B.T. Mechanisms of Heightened Airway Sensitivity and Responses to Inhaled SO2 in Asthmatics. Environ. Health Insights 2015, 9, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Yorifuji, T.; Suzuki, E.; Kashima, S. Hourly differences in air pollution and risk of respiratory disease in the elderly: A time-stratified case-crossover study. Environ. Health 2014, 13, 67. [Google Scholar] [CrossRef] [Green Version]
- Vally, H.; Misso, N.L.A. Adverse reactions to the sulphite additives. Gastroenterol. Hepatol. Bed Bench 2012, 5, 16–23. [Google Scholar]
- Schlesinger, R.B.; Chen, L.C.; Finkelsein, I.; Zelikoff, J.T. Comparative potency of inhaled acidic sulphates: Speciation and the role of hydrogen ion. Environ. Res. 1990, 52, 210–224. [Google Scholar] [CrossRef]
- Fine, J.M.; Gordon, T.; Sheppard, D. The Roles of pH and Ionic Species in Sulfur Dioxide- and Sulfite-Induced Bronchoconstriction. Am. Rev. Respir. Dis. 1987, 136, 1122–1126. [Google Scholar] [CrossRef]
- Mirić, M.; Plavec, D. Risk of Acute Bronchospasm and Bronchial Hyperreactivity from Inhaled Acid Aerosol in 79 Healthy Subjects: Randomized, Double-blind Controlled Trial. Croat. Med. J. 2004, 45, 709–714. [Google Scholar] [PubMed]
- Kamide, Y.; Ishizuka, T.; Tobo, M.; Tsurumaki, H.; Aoki, H.; Mogi, C.; Nakakura, T.; Yatomi, M.; Ono, A.; Koga, Y.; et al. Acidic environment augments FcεRI-mediated production of IL-6 and IL-13 in mast cells. Biochem. Biophys. Res. Commun. 2015, 464, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Trevani, A.S.; Andonegui, G.; Giordano, M.; López, D.H.; Gamberale, R.; Minucci, F.; Geffner, J. Extracellular Acidification Induces Human Neutrophil Activation. J. Immunol. 1999, 162, 4849–4857. [Google Scholar] [PubMed]
- Herrmann, J.M.; Kantarci, A.; Long, H.; Bernardo, J.; Hasturk, H.; Wray, L.V., Jr.; Simons, E.R.; Van Dyke, T.E. Simultaneous measurements of cytoplasmic Ca2+ responses and intracellular pH in neutrophils of localized aggressive periodontitis (LAP) patients. J. Leukoc. Biol. 2005, 78, 612–619. [Google Scholar] [CrossRef] [Green Version]
- Martinez, D.; Vermeulen, M.; Trevani, A.; Ceballos, A.; Sabatté, J.; Gamberale, R.; Alvarez, M.E.; Salamone, G.; Tanos, T.; Coso, O.A.; et al. Extracellular Acidosis Induces Neutrophil Activation by a Mechanism Dependent on Activation of Phosphatidylinositol 3-Kinase/Akt and ERK Pathways. J. Immunol. 2006, 176, 1163–1171. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, M.; Giordano, M.; Trevani, A.S.; Sedlik, C.; Gamberale, R.; Fernández-Calotti, P.; Salamone, G.; Raiden, S.; Sanjurjo, J.; Geffner, J.R. Acidosis improves uptake of antigens and MHC class I-restricted presentation by dendritic cells. J. Immunol. 2004, 172, 3196–3204. [Google Scholar] [CrossRef] [Green Version]
- Kottyan, L.C.; Collier, A.R.; Cao, K.H.; Niese, K.A.; Hedgebeth, M.; Radu, C.G.; Witte, O.N.; Hershey, G.K.K.; Rothenberg, M.E.; Zimmermann, N. Eosinophil viability is increased by acidic pH in a cAMP- and GPR65-dependent manner. Blood 2009, 114, 2774–2782. [Google Scholar] [CrossRef] [Green Version]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T Cell Activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Wang, X.; Hatatani, K.; Sun, Y.; Fukamachi, T.; Saito, H.; Kobayashi, H. TCR Signaling via ZAP-70 Induced by CD 3 Stimulation is More Active Under Acidic Conditions. J. Cell Sci. Ther. 2012, S15, 002. [Google Scholar]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; alTuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Narita, A.; Yawata, K.; Nagata, M.; et al. Effects of Extracellular Acidification on Intracellular pH and ATP-Induced Calcium Mobilization in Rabbit Lens Epithelial Cells. Yonago Acta Med. 1999, 42, 51–59. [Google Scholar]
- Chin, W.-C.; Quesada, I.; Nguyen, T.; Verdugo, P. Oscillations of pH inside the secretory granule control the gain of Ca2+ release for signal transduction in goblet cell exocytosis. Novartis Found. Symp. 2002, 248, 132–141. [Google Scholar] [PubMed]
- Ichimonji, I.; Tomura, H.; Mogi, C.; Sato, K.; Aoki, H.; Hisada, T.; Dobashi, K.; Ishizuka, T.; Mori, M.; Okajima, F. Extracellular acidification stimulates IL-6 production and Ca2+ mobilization through proton-sensing OGR1 receptors in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L567–L577. [Google Scholar] [CrossRef]
- Saxena, H.; Deshpande, D.A.; Tiegs, B.C.; Yan, H.; Battafarano, R.J.; Burrows, W.M.; Damera, G.; Panettieri, R.; Duboes, T.D., Jr.; An, S.; et al. The GPCR OGR1 (GPR68) mediates diverse signalling and contraction of airway smooth muscle in response to small reductions in extracellular pH. Br. J. Pharmacol. 2012, 166, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Prakash, Y.S. Airway smooth muscle in airway reactivity and remodeling: What have we learned? Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L912–L933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Liao, J.; Hanrahan, J.W. The buffer capacity of airway epithelial secretions. Front. Physiol. 2014, 5, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilton, D.C. Phospholipases. In Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed.; Vance, D.E., Vance, J.E., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2008; Chapter 11; pp. 305–329. [Google Scholar]
- Hu, Y.L.; Mi, X.; Huang, C.; Wang, H.F.; Song, J.-R.; Shu, Q.; Ni, L.; Chen, J.-G.; Wang, F.; Hu, Z.-L. Multiple H+ sensors mediate the extracellular acidification-induced [Ca2+]i elevation in cultured rat ventricular cardiomyocytes. Sci. Rep. 2017, 7, 44951. [Google Scholar] [CrossRef]
- Huang, J.; Liu, C.H.; Hughes, S.A.; Postma, M.; Schwiening, C.J.; Hardie, R.C. Activation of TRP channels by protons and phosphoinositide depletion in Drosophila photoreceptors. Curr. Biol. 2010, 20, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.B.; Dwyer, S.D.; Smith, L. Lowering Extracellular pH Evokes Inositol Polyphosphate Formation and Calcium Mobilization. J. Biol. Chem. 1989, 264, 8723–8728. [Google Scholar] [PubMed]
- Tsunoda, Y.; Matsuno, K.; Tashiro, Y. Cytosolic acidification leads to Ca2+ mobilization from intracellular stores in single and populational parietal cells and platelets. Exp. Cell Res. 1991, 193, 356–363. [Google Scholar] [CrossRef]
- Donoso, P.; Beltrán, M.; Hidalgo, C. Luminal pH regulated calcium release kinetics in sarcoplasmic reticulum vesicles. Biochemistry 1996, 35, 13419–13425. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-H.; Chen, C.-R.; Yang, K.-T.; Chang, W.-L.; Su, M.-J.; Wu, C.-C.; Wu, M.-L. Arachidonic acid-induced H+ and Ca2+ increases in both the cytoplasm and nucleoplasm of rat cerebellar granule cells. J. Physiol. 2001, 537 (Pt 2), 497–510. [Google Scholar] [CrossRef]
- Bates, R.C.; Fees, C.P.; Holland, W.L.; Winger, C.C.; Batbayar, K.; Ancar, R.; Bergren, T.; Petcoff, D.; Stith, B. Activation of Src and release of intracellular calcium by phosphatidic acid during Xenopus laevis fertilization. Dev. Biol. 2014, 386, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Busa, W.B.; Nuccitelli, R. Metabolic regulation via intracellular pH. Am. J. Physiol. 1984, 246, R409–R438. [Google Scholar] [CrossRef] [Green Version]
- Iida, S.; Potter, J.D. Calcium binding to calmodulin. Cooperativity of the calcium-binding sites. J. Biochem. 1986, 99, 1765–1772. [Google Scholar] [CrossRef]
- Molinari, G.; Nervo, E. Role of Protons in Calcium Signaling. Preprints 2020, 2020030274. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell. Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.J.; Foskett, J.K. Ca2+ signaling and fluid secretion by secretory cells of the airway epithelium. Cell Calcium 2014, 55, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-T.; Beaven, M.A. Regulators of Ca2+ signaling in mast cells: Potential targets for treatment of mast-cell related diseases? Adv. Exp. Med. Biol. 2011, 716, 62–90. [Google Scholar] [PubMed]
- Chapman, D.G.; Irvin, C.G. Mechanisms of airway hyper-responsiveness in asthma: The past, present and yet to come. Clin. Exp. Allergy 2015, 45, 706–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozier, A.; Allard, B.; Bara, I.; Girodet, P.-O.; Trian, T.; Marthan, R.; Berger, P. The Pivotal Role of Airway Smooth Muscle in Asthma Pathophysiology. J. Allergy 2011, 742710. [Google Scholar] [CrossRef] [Green Version]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vines, C.M.; Prossnitz, E.R. Mechanisms of G protein-coupled receptor-mediated degranulation. FEMS Microbiol. Lett. 2004, 236, 1–6. [Google Scholar] [CrossRef]
- Maurer, M.; Pucillo, C. What we know (and don′t know) about the biology and functions of mast cells and basophils. Immunol. Rev. 2018, 282, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; Karasuyama, H. Emerging roles of basophils in allergic inflammation. Allergol. Int. 2017, 66, 382-–391. [Google Scholar] [CrossRef]
- Olivera, A.; Beaven, M.A.; Metcalfe, D.D. Mast cells signal their importance in health and disease. J. Allergy Clin. Immunol. 2018, 142, 381–393. [Google Scholar] [CrossRef] [Green Version]
- Gilfillan, A.M.; Peavy, R.D.; Metcalfe, D.D. Amplification mechanisms for the enhancement of antigen mediated mast cell activation. Immunol. Res. 2009, 43, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, R. The Emerging Picture of Mast Cell Activation: The Complex Regulatory Network of High-Affinity Receptor for Immunoglobulin E Signaling. Biol. Pharm. Bull. 2017, 40, 1828–1832. [Google Scholar] [CrossRef] [Green Version]
- Kuehn, H.S.; Gilfillan, A.M. G protein-coupled receptors and the modification of FcεRI-mediated mast cell activation. Immunol. Lett. 2007, 113, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, H.; Gupta, K.; Ali, H. Roles of MAS-related G protein coupled receptor-X2 (MRGPRX2) on mast cell-mediated host defense, pseudoallergic drug reactions and chronic inflammatory diseases. J. Allergy Clin. Immunol. 2016, 138, 700–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudenzio, N.; Sibilano, R.; Marichal, T.; Starkl, P.; Reber, L.L.; Cenac, N.; McNeil, B.D.; Dong, X.; Hernandez, J.D.; Sagi-Eisenberg, R.; et al. Different activation signals induce distinct mast cell degranulation strategies. J. Clin. Invest. 2016, 126, 3981–3998. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Jacobson, K.A. Purinergic Signaling in Mast Cell Degranulation and Asthma. Front. Pharmacol. 2017, 8, 947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jairaman, A.; Maguire, C.H.; Schleimer, R.P.; Prakriya, M. Allergens stimulate store-operated calcium entry and cytokine production in airway epithelial cells. Sci. Rep. 2016, 6, 32311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, M.; Ito, K.; Yuno, T.; Soma, N.; Aburakawa, S.; Kasai, K.; Nakamura, T.; Takami, H. UDP/P2Y6 receptor signaling regulates IgE-dependent degranulation in human basophils. Allergol. Int. 2017, 66, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Mogi, C.; Okajima, F. Ionotropic and Metabotropic Proton-Sensing Receptors Involved in Airway Inflammation in Allergic Asthma. Mediators Inflamm. 2014, 2014, 712962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, S.P.H.; Fabbro, R.; Kelly, E.; Marrion, N.V.; A Peters, J.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; et al. The concise guide to pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174, S17–S129. [Google Scholar] [CrossRef]
- Seifert, R. How do basic secretagogues activate mast cells? Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 279–281. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Chang, Y.-C.; Chang, H.-A.; Lin, Y.-S.; Tsao, C.-W.; Shen, M.-R.; Chiu, W.-T. Differential Ca2+ mobilization and mast cell degranulation by FcεRI- and GPCR-mediated signaling. Cell Calcium 2017, 67, 31–39. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Tkaczyk, C. Integrated signalling pathways for mast-cell activation. Nat. Rev. Immunol. 2006, 6, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Itsuki, K.; Imai, Y.; Hase, H.; Okamura, Y.; Inoue, R.; Mori, M.X. PLC-mediated PI(4,5)P2 hydrolysis regulates activation and inactivation of TRPC6/7 channels. J. Gen. Physiol. 2014, 143, 183–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocco, L.; Follo, M.Y.; Manzoli, L.; Suh, P.-G. Phosphoinositide-specific phospholipase C in health and disease. J. Lip. Res. 2015, 53, 1853–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Fukami, K. Regulation and physiological functions of mammalian phospholipase C. J. Biochem. 2017, 161, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Molinari, G. Is hydrogen ion (H(+)) the real second messenger in calcium signalling? Cell. Signal. 2015, 27, 1392–1397. [Google Scholar] [CrossRef]
- Berridge, M.J. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [Green Version]
- Fujishima, H.; Sanchez Mejia, R.O.; Bingham, C.O.; Lam, B.K.; Sapirstein, A.; Bonventre, J.V.; Austen, K.F.; Arm, J.P. Cytosolic phospholipase A2 is essential for both the immediate and the delayed phases of eicosanoid generation in mouse bone marrow-derived mast cells. Proc. Natl. Acad. Sci. USA 1999, 96, 4803–4807. [Google Scholar] [CrossRef] [Green Version]
- Leslie, C.C. Cytosolic phospholipase A2: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [Green Version]
- Damron, D.S.; Van Wagoner, D.R.; Moravec, C.S.; Bond, M. Arachidonic acid and endothelin potentiate Ca2+ transients in rat cardiac myocytes via inhibition of distinct K+ channels. J. Biol. Chem. 1993, 268, 27335–27344. [Google Scholar]
- Dennis, E.A.; Norris, P.C. Eicosanoid Storm in Infection and Inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef]
- Bodnar, D.; Chung, W.Y.; Yang, D.; Hong, J.H.; Jha, A.; Muallem, S. STIM-TRP Pathways and Microdomain Organization: Ca2+ Influx Channels: The Orai-STIM1-TRPC Complexes. Adv. Exp. Med. Biol. 2017, 993, 139–157. [Google Scholar] [CrossRef]
- Parenti, A.; De Logu, F.; Geppetti, P.; Benemei, S. What is the evidence for the role of TRP channels in inflammatory and immune cells? Br. J. Pharmacol. 2016, 173, 953–969. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 2017, 63, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrner, M.; Stadlbauer, M.; Muik, M.; Rathner, P.; Stathopulos, P.; Ikura, M.; Mueller, N.; Romanin, C. A dual mechanism promotes switching of the Stormorken STIM1 R304W mutant into the activated state. Nat. Commun. 2018, 9, 825. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, G.; Maggio, K.; Thirion, S.; Horoyan, M.; Keicher, E. The calcium loading of secretory granules. A possible key event in stimulus-secretion coupling. Biol. Cell 1992, 75, 89–99. [Google Scholar] [CrossRef]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Yamamoto, K.; Miki, Y.; Murase, R.; Sato, H.; Taketomi, Y. The Roles of the Secreted Phospholipase A2 Gene Family in Immunology. Adv. Immunol. 2016, 132, 91–134. [Google Scholar]
- Pejler, G.; Frisk, J.M.; Sjöström, D.; Paivandy, A.; Ohrvik, H. Acidic pH is essential for maintaining mast cell secretory granule homeostasis. Cell Death Dis. 2017, 8, 22785. [Google Scholar] [CrossRef]
- Galli, S.J.; Tsai, M.; Marichal, T.; Tchougounova, E.; Reber, L.L.; Pejler, G. Approaches for Analyzing the Roles of Mast Cells and Their Proteases In Vivo. Adv. Immunol. 2015, 126, 45–127. [Google Scholar]
- Kormelink, T.G.; Arkesteijn, G.J.A.; van de Lest, C.H.A.; Geerts, W.J.C.; Goerdayal, S.S.; Altelaar, M.; Redegeld, F.A.; Nolte-’t Hoen, E.N.M.; Wauben, M.H.M. Mast Cell Degranulation Is Accompanied by the Release of a Selective Subset of Extracellular Vesicles That Contain Mast Cell–Specific Proteases. J. Immunol. 2016, 197, 3382–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulkhaleq, L.A.; Assi, M.A.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.H.; Hezmee, M. The crucial roles of inflammatory mediators in inflammation: A review. Vet. World 2018, 11, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Swietach, P.; Youm, J.B.; Saegusa, N.; Leem, C.H.; Spitzer, K.W.; Vaughan-Jones, R.D. Coupled Ca2+/H+ transport by cytoplasmic buffers regulates local Ca2+ and H+ ion signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E2064–E2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Gueguinou, M.; Trebak, M. Store-Independent Orai Channels Regulated by STIM. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Jr., Eds.; Taylor & Francis Group: Abingdon, UK, 2018; Chapter 11; pp. 197–213. [Google Scholar]
- Hellwig, N.; Plant, T.D.; Janson, W.; Schäfer, M.; Schultz, G.; Schaefer, M. TRPV1 acts as proton channel to induce acidification in nociceptive neurons. J. Biol. Chem. 2004, 279, 34553–34561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Li, M.; Yue, L. Potentiation of TRPM7 inward currents by protons. J. Gen. Physiol. 2005, 126, 137–150. [Google Scholar] [CrossRef] [Green Version]
- White, J.P.M.; Cibelli, M.; Urban, L.; Nilius, B.; McGeown, J.G.; Nagy, I. TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol. Rev. 2016, 96, 911–973. [Google Scholar] [CrossRef] [Green Version]
- Starkus, J.G.; Fleig, A.; Penner, R. The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular acidification. J. Physiol. 2010, 588 (Pt 8), 1227–1240. [Google Scholar] [CrossRef]
- Cairns, S.P.; Westerblad, H.; Allen, D.G. Changes in myoplasmic pH and calcium concentration during exposure to lactate in isolated rat ventricular Myocytes. J. Physiol. 1993, 464, 561–574. [Google Scholar] [CrossRef] [Green Version]
- Marin, M.; Sellier, C.; Paul-Antoine, A.F.; Cailliau, K.; Browaeys-Poly, E.; Bodart, J.F.; Vilain, J.P. Calcium Dynamics During Physiological Acidification in Xenopus Oocyte. J. Membr. Biol. 2010, 236, 233–245. [Google Scholar] [CrossRef]
- Lee, S.E.; Lee, S.H. Skin Barrier and Calcium. Ann. Dermatol. 2018, 30, 265–275. [Google Scholar] [CrossRef]
- Brune, K.; Frank, J.A.; Schwingshackl, A.; Finigan, J.; Venkataramana, K.S. Pulmonary epithelial barrier function: Some new players and mechanisms. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L731–L745. [Google Scholar] [CrossRef] [Green Version]
- De Rose, V.; Molloy, K.; Gohy, S.; Pilette, C.; Greene, C.M. Airway Epithelium Dysfunction in Cystic Fibrosis and COPD. Mediat. Inflamm. 2018, 8, 1309746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alka, K.; Casey, J.R. Bicarbonate Transport in Health and Disease. IUBMB Life 2014, 66, 596–615. [Google Scholar] [CrossRef]
- Liu, X.; Li, T.; Tuo, B. Physiological and Pathophysiological Relevance of the Anion Transporter Slc26a9 in Multiple Organs. Front. Physiol. 2018, 9, 1197. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Hong, J.H. The Fundamental Role of Bicarbonate Transporters and Associated Carbonic Anhydrase Enzymes in Maintaining Ion and pH Homeostasis in Non-Secretory Organs. Int. J. Mol. Sci. 2020, 21, 339. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Carafoli, E. The plasma membrane Ca²+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb Perspect Biol. 2011, 3, a004168. [Google Scholar] [CrossRef] [PubMed]
- Boscardin, E.; Alijevic, O.; Hummler, E.; Frateschi, S.; Kellenberger, S. The function and regulation of acid-sensingion channels (ASICs) and the epithelial Na+ channel (ENaC): IUPHAR Review 19. Br. J. Pharmacol. 2016, 173, 2671–2701. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.S.; Yue, Z.; Feng, J.; Yue, L. Regulation of Orai/STIM Channels by pH. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Jr., Eds.; CRC Press/Taylor & Francis Group: Boca Raton, FL, USA; Abingdon, UK , 2018; Chapter 9; pp. 161–176. [Google Scholar]
- DeCoursey, T.E. Voltage-Gated Proton Channels and Other Proton Transfer Pathways. Physiol. Rev. 2003, 83, 475–579. [Google Scholar] [CrossRef] [Green Version]
- Amdursky, N.; Lin, Y.; Aho, N.; Groenhof, G. Exploring fast proton transfer events associated with lateral proton diffusion on the surface of membranes. Proc. Natl. Acad. Sci. USA 2019, 116, 2443–2451. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.; Widdicombe, J.H. Mechanisms of Acid and Base Secretion by the Airway Epithelium. J. Membr. Biol. 2006, 211, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, S.; Song, Y.; Vetrivel, L.; Shankar, L.; Verkman, A.S. Noninvasive in vivo fluorescence measurement of airway-surface liquid depth, salt concentration, and pH. J. Clin. Investig. 2001, 107, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCreanor, J.; Cullinan, P.; Nieuwenhuijsen, M.J.; Stewart-Evans, J.; Malliarou, E.; Jarup, L.; Harrington, R.; Svartengren, M.; Han, I.-K.; Ohman-Strickland, P.; et al. Respiratory Effects of Exposure to Diesel Traffic in Persons with Asthma. N. Engl. J. Med. 2007, 357, 2348–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brant, T.C.; Yoshida, C.T.; Carvalho, T.S.; Nicola, M.L.; Martins, J.A.; Braga, L.M.; De Oliveira, R.C.; Leyton, V.; De André, C.S.; Saldiva, P.H.N.; et al. Mucociliary clearance, airway inflammation and nasal symptoms in urban motorcyclists. Clinics 2014, 69, 867–870. [Google Scholar] [CrossRef]
- Hunt, J.F.; Fang, K.; Malik, R.; Snyder, A.; Malhotra, N.; Platts-Mills, T.A.E.; Gaston, B. Endogenous Airway Acidification Implications for Asthma Pathophysiology. Am. J. Respir. Crit. Care Med. 2000, 161, 694–699. [Google Scholar] [CrossRef]
- Brunetti, L.; Francavilla, R.; Tesse, R.; Strippoli, A.; Polimeno, L.; Loforese, A.; Miniello, V.L.; Armenio, L. Exhaled breath condensate pH measurement in children with asthma, allergic rhinitis and atopic dermatitis. Pediatr. Allergy Immunol. 2006, 17, 422–427. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Matalon, S.; Collawn, J.F. Ion channels of the lung and their role in disease pathogenesis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L859–L872. [Google Scholar] [CrossRef] [PubMed]
- Seifter, J.L.; Chang, H.-Y. Extracellular Acid-Base Balance and Ion Transport Between Body Fluid Compartments. Physiology 2017, 32, 367–379. [Google Scholar] [CrossRef] [PubMed]
- DeCoursey, T.E. Voltage and pH sensing by the voltage-gated proton channel, HV1. J. R. Soc. Interface. 2018, 15, 20180108. [Google Scholar] [CrossRef] [Green Version]
- Sedlyarov, V.; Eichner, R.; Girardi, E.; Essletzbichler, P.; Goldmann, U.; Nunes-Hasler, P.; Srndic, I.; Moskovskich, A.; Heinz, L.X.; Kartnig, F.; et al. The Bicarbonate Transporter SLC4A7 Plays a Key Role in Macrophage Phagosome Acidification. Cell Host Microbe 2018, 23, 766–774.e5. [Google Scholar] [CrossRef] [Green Version]
- Hollenhorst, M.I.; Richter, K.; Fronius, M. Ion Transport by Pulmonary Epithelia. J. Biomed. Biotechnol. 2011, 2011, 174306. [Google Scholar] [CrossRef] [Green Version]
- Weller, P.F.; Spencer, L.A. Functions of tissue-resident eosinophils. Nat. Rev. Immunol. 2017, 17, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Stone, K.D.; Prussin, C.; Metcalfe, D.D. IgE, Mast Cells, Basophils, and Eosinophils. J. Allergy Clin. Immunol. 2010, 125, S73–S80. [Google Scholar] [CrossRef] [PubMed]
- Granata, F.; Nardicchi, V.; Loffredo, S.; Frattini, A.; Staiano, R.I.; Agostini, C.; Triggiani, M. Secreted phospholipases A2: A proinflammatory connection between macrophages and mast cells in the human lung. Immunobiology 2009, 214, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.V.; Alexov, E. Protonation and pK changes in protein-ligand binding. Q. Rev. Biophys. 2013, 46, 181–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Yokomizo, T. The role of leukotrienes in allergic diseases. Allergol. Int. 2015, 64, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, M.J.; Hiebert, P.R.; Park, H.Y.; Stefanowicz, D.; Le, A.; Starkey, M.R.; Deane, A.; Brown, A.C.; Liu, G.; Horvat, J.C.; et al. Mucosal production of uric acid by airway epithelial cells contributes to particulate matter-induced allergic sensitization. Mucosal Immunol. 2016, 9, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Lindeman, K.S.; Croxton, T.L.; Lande, B.; Hirshman, C.A. Hypocapnia-induced contraction of porcine airway smooth muscle. Eur. Respir. J. 1998, 12, 1046–1052. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Hao, B.; Lu, Y.; Yu, P.; Lee, H.-C.; Yue, J. Intracellular Alkalinization Induces Cytosolic Ca2+ Increases by Inhibiting Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA). PLoS ONE 2012, 7, e31905. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Yang, D.; Pan, L.; Shan, J.; Li, H.; Wei, H.; Wang, B.; Huang, J.; Baccarelli, A.A.; Shima, M.; et al. Chemical constituents and sources of ambient particulate air pollution and biomarkers of endothelial function in a panel of healthy adults in Beijing, China. Sci. Total Environ. 2016, 560–561, 141–149. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molinari, G.; Molinari, L.; Nervo, E. Environmental and Endogenous Acids Can Trigger Allergic-Type Airway Reactions. Int. J. Environ. Res. Public Health 2020, 17, 4688. https://doi.org/10.3390/ijerph17134688
Molinari G, Molinari L, Nervo E. Environmental and Endogenous Acids Can Trigger Allergic-Type Airway Reactions. International Journal of Environmental Research and Public Health. 2020; 17(13):4688. https://doi.org/10.3390/ijerph17134688
Chicago/Turabian StyleMolinari, Giuliano, Laura Molinari, and Elsa Nervo. 2020. "Environmental and Endogenous Acids Can Trigger Allergic-Type Airway Reactions" International Journal of Environmental Research and Public Health 17, no. 13: 4688. https://doi.org/10.3390/ijerph17134688
APA StyleMolinari, G., Molinari, L., & Nervo, E. (2020). Environmental and Endogenous Acids Can Trigger Allergic-Type Airway Reactions. International Journal of Environmental Research and Public Health, 17(13), 4688. https://doi.org/10.3390/ijerph17134688