
Vasculitis, Autoimmunity, and Cytokines: How the Immune System Can Harm the Brain

 , ,
, , 
Abstract
:1. Introduction
2. From Monogenic to Multifactorial: Interferon-Related Brain Disorders
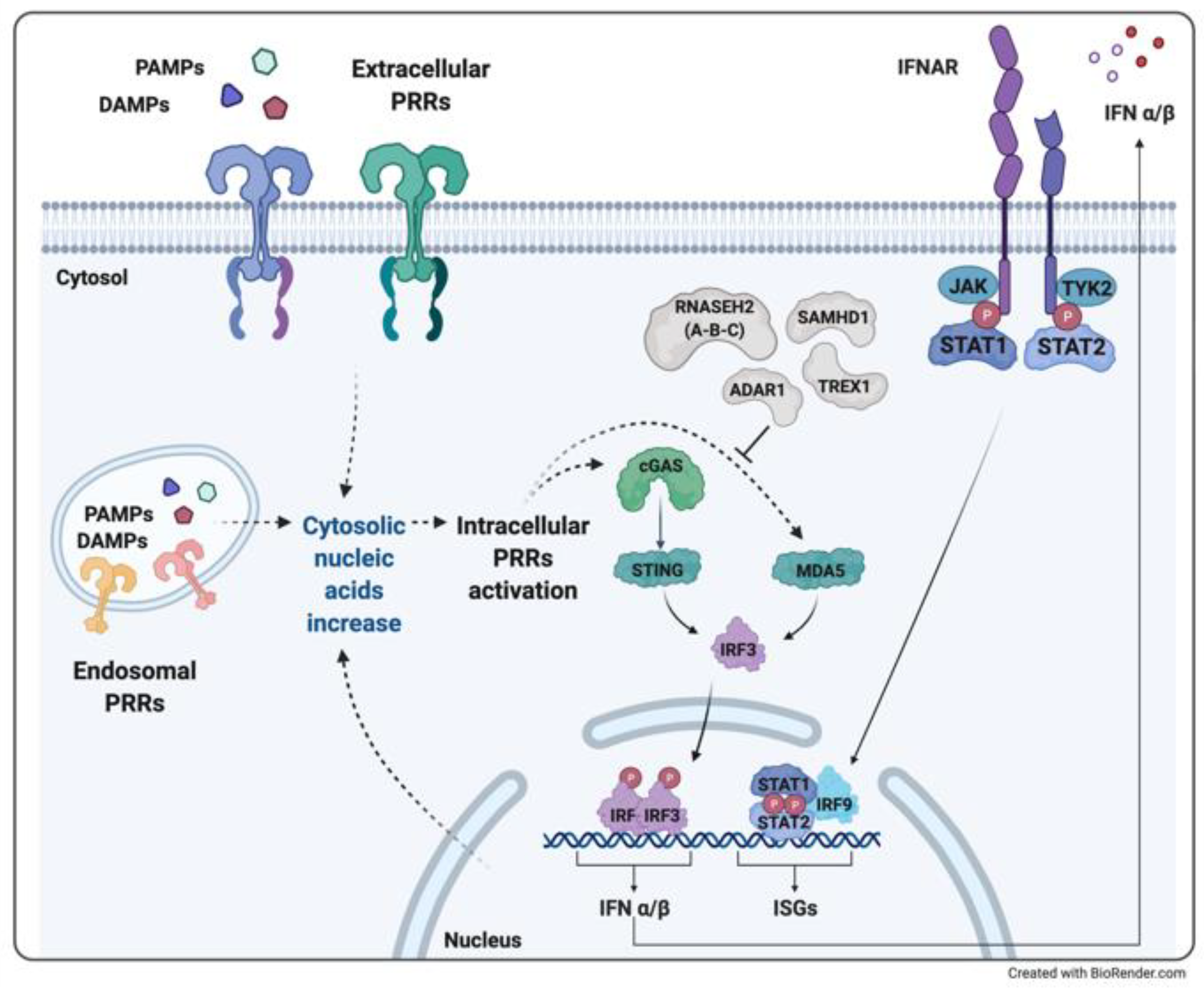
2.1. The Role of Type I Interferon: The Immune Response to Nucleic Acids
2.2. Aicardi-Goutières Syndrome and Other Interferonopathies with Brain Involvement
2.3. Therapeutic Experiences
2.4. A Clinical Experience of Monogenic Interferonopathy: Aicardi-Goutières Syndrome
2.5. Relevance to Multifactorial Disorders: Neuropsychiatric Systemic Lupus Erythematosus
3. From Monogenic to Multifactorial: TNFα-Related Brain Disorders
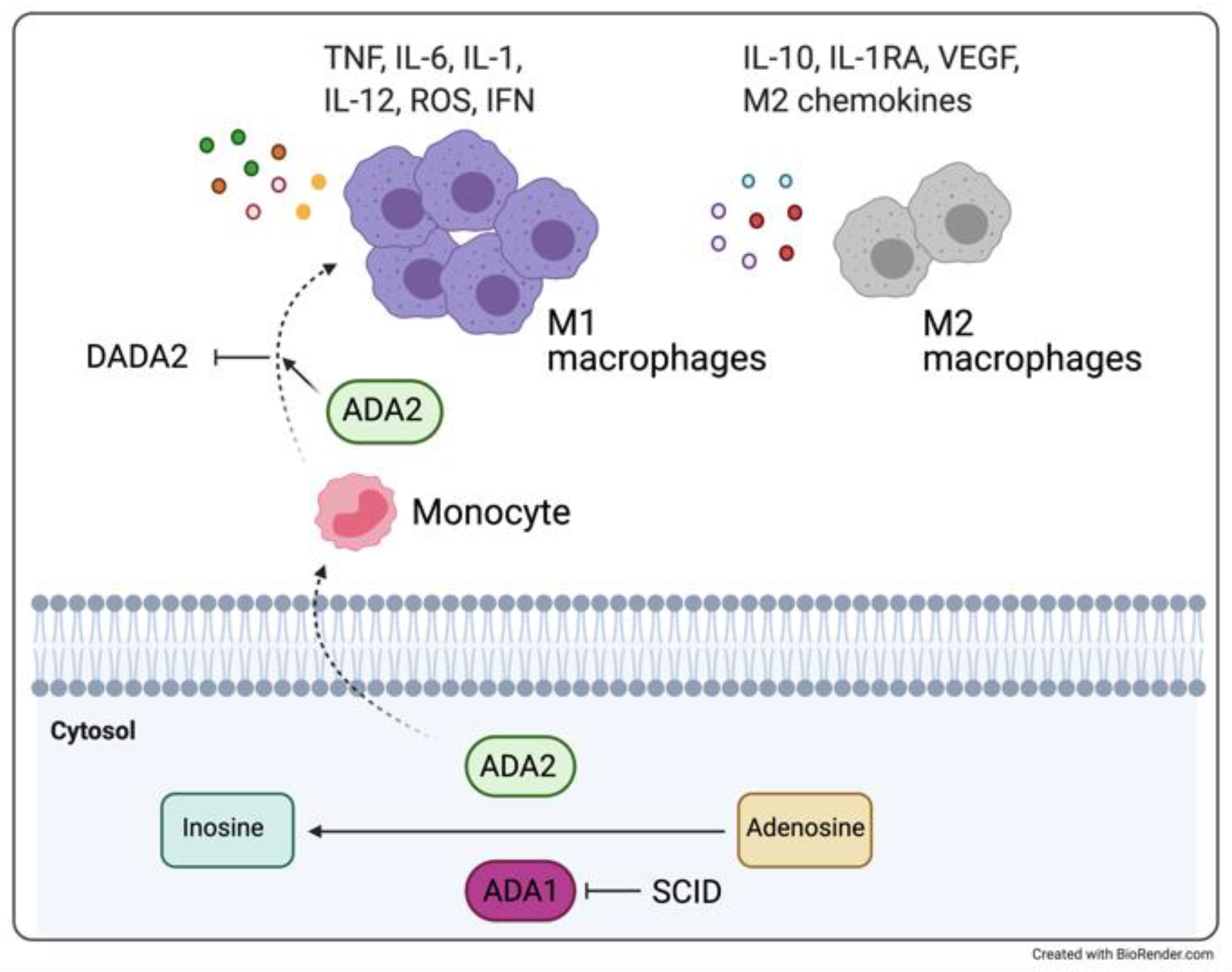
3.1. DADA2 and Monogenic Vasculitis Syndromes with Cerebral Involvement
3.2. Therapeutic Experiences
3.3. A Clinical Experience of a Monogenic Disorder Associated with TNF-Related Inflammation: DADA2
3.4. Relevance to Multifactorial Models: Behçet’s Disease-Associated Vasculitis
4. From Monogenic to Multifactorial: IL-1β-Related Brain Disorders
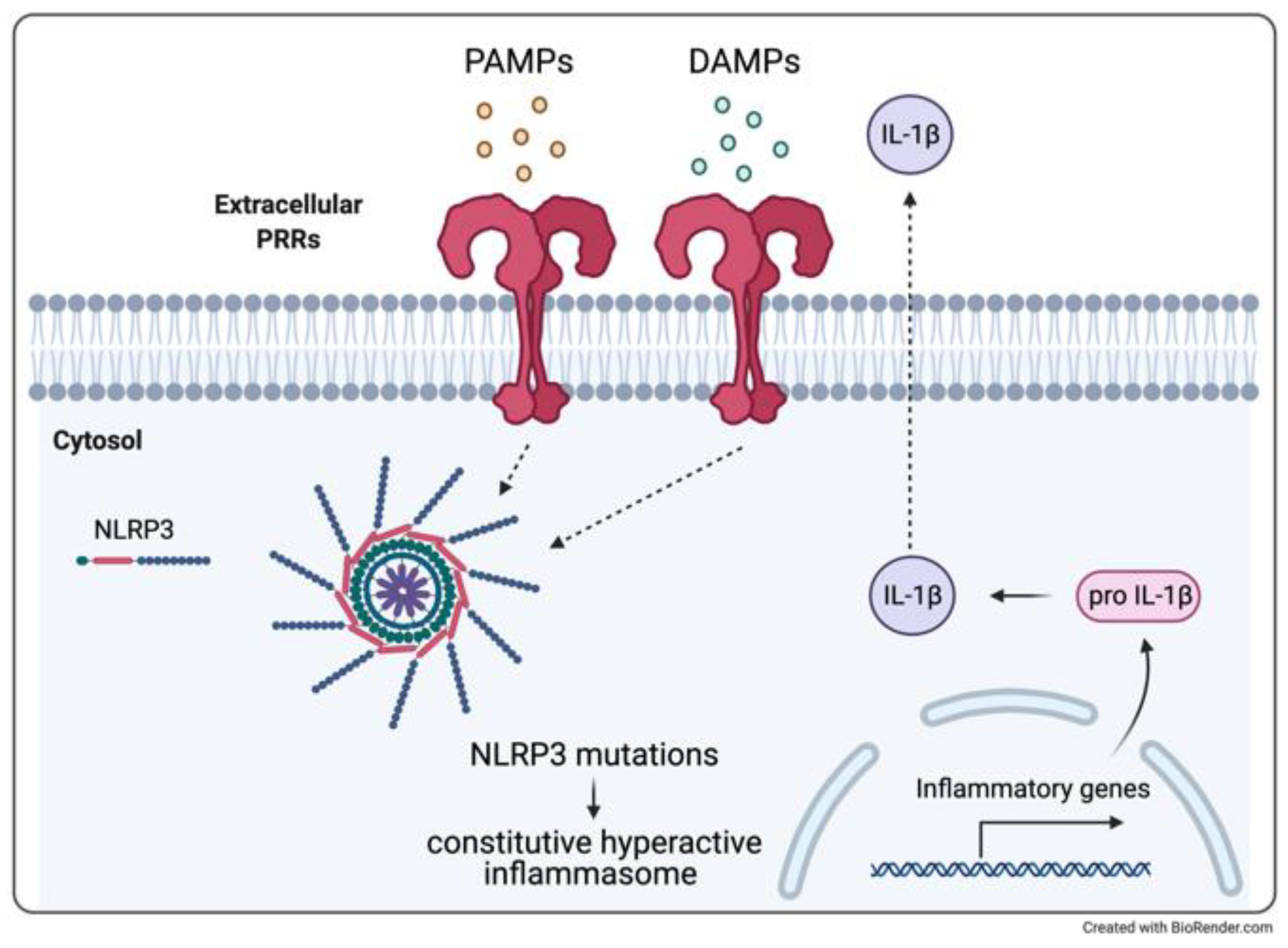
4.1. Cryopyrin-Associated Periodic Syndrome and Aseptic Meningitis
4.2. Therapeutic Experiences
4.3. Clinical Experience of Monogenic Model: CINCA
4.4. Relevance to Multifactorial Neurologic Inflammatory Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gregson, A.; Thompson, K.; Tsirka, S.E.; Selwood, D.L. Emerging small-molecule treatments for multiple sclerosis: Focus on B cells. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santisteban, M.M.; Ahn, S.J.; Lane, D.; Faraco, G.; Garcia-Bonilla, L.; Racchumi, G.; Poon, C.; Schaeffer, S.; Segarra, S.G.; Körbelin, J.; et al. Endothelium-Macrophage Crosstalk Mediates Blood-Brain Barrier Dysfunction in Hypertension. Hypertension 2020, 76, 795–807. [Google Scholar] [CrossRef]
- Kalucka, J.; Bierhansl, L.; Wielockx, B.; Carmeliet, P.; Eelen, G. Interaction of endothelial cells with macrophages-linking molecular and metabolic signaling. Pflug. Arch. 2017, 469, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Bettiol, A.; Lopalco, G.; Emmi, G.; Cantarini, L.; Urban, M.L.; Vitale, A.; Denora, N.; Lopalco, A.; Cutrignelli, A.; Lopedota, A.; et al. Unveiling the Efficacy, Safety, and Tolerability of Anti-Interleukin-1 Treatment in Monogenic and Multifactorial Autoinflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 1898. [Google Scholar] [CrossRef] [Green Version]
- Solís Marquínez, M.N.; García Fernández, E.; Morís de la Tassa, J. Periodic fever: From Still’s disease to Muckle-Wells syndrome. Reumatol. Clin. 2019, 15, e39–e40. [Google Scholar] [CrossRef] [PubMed]
- Montealegre Sanchez, G.A.; Hashkes, P.J. Neurological manifestations of the Mendelian-inherited autoinflammatory syndromes. Dev. Med. Child Neurol. 2009, 51, 420–428. [Google Scholar] [CrossRef]
- Pain, C.E. Juvenile-onset Behcet’s syndrome and mimics. Clin. Immunol. 2020, 214, 108381. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, C.; Omoyinmi, E.; Standing, A.; Pain, C.E.; Booth, C.; D’Arco, F.; Gilmour, K.; Buckland, M.; Eleftheriou, D.; Brogan, P.A. Monogenic mimics of Behcet’s disease in the young. Rheumatology 2019, 58, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Aicardi, J.; Goutières, F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann. Neurol. 1984, 15, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J. Aicardi-Goutieres syndrome. Handb. Clin. Neurol. 2013, 113, 1629–1635. [Google Scholar] [CrossRef]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Sanchez, G.A.M.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Yang, D.; Ombrello, A.K.; Zavialov, A.V.; Toro, C.; Stone, D.L.; Chae, J.J.; Rosenzweig, S.D.; Bishop, K.; Barron, K.S.; et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N. Engl. J. Med. 2014, 370, 911–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caorsi, R.; Penco, F.; Grossi, A.; Insalaco, A.; Omenetti, A.; Alessio, M.; Conti, G.; Marchetti, F.; Picco, P.; Tommasini, A.; et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: A multicentre national study. Ann. Rheum. Dis. 2017, 76, 1648–1656. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Nowak, M.; Mallah, M.; Chae, J.J.; Watford, W.T.; Hofmann, S.R.; Stein, L.; Russo, R.; Goldsmith, D.; Dent, P.; et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): A new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002, 46, 3340–3348. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.R.; Leslie, K.S. Cryopyrin-associated periodic syndrome: An update on diagnosis and treatment response. Curr. Allergy Asthma Rep. 2011, 11, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Janeway, C.A. Approaching the asymptote? Evolution and revolution in immunology. Cold spring harb symp quant biol. J. Immunol. 2013, 191, 4475–4487. [Google Scholar]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Auerbuch, V.; Brockstedt, D.G.; Meyer-Morse, N.; O’Riordan, M.; Portnoy, D.A. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 2004, 200, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrero, J.A.; Calderon, B.; Unanue, E.R. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 2004, 200, 535–540. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Saha, S.K.; Vaidya, S.A.; Bruhn, K.W.; Miranda, G.A.; Zarnegar, B.; Perry, A.K.; Nguyen, B.O.; Lane, T.F.; Taniguchi, T.; et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 2004, 200, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Schafer, S.L.; Lin, R.; Moore, P.A.; Hiscott, J.; Pitha, P.M. Regulation of type I interferon gene expression by interferon regulatory factor-3. J. Biol. Chem. 1998, 273, 2714–2720. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef]
- De Weerd, N.A.; Samarajiwa, S.A.; Hertzog, P.J. Type I interferon receptors: Biochemistry and biological functions. J. Biol. Chem. 2007, 282, 20053–20057. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Henrickson, M.; Wang, H. Tocilizumab reverses cerebral vasculopathy in a patient with homozygous SAMHD1 mutation. Clin. Rheumatol. 2017, 36, 1445–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinchieri, G. Type I interferon: Friend or foe? J. Exp. Med. 2010, 207, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Palucka, A.K.; Blanck, J.P.; Chalouni, C.; Pascual, V.; Banchereau, J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 2003, 19, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Rizza, P.; Moretti, F.; Belardelli, F. Recent advances on the immunomodulatory effects of IFN-alpha: Implications for cancer immunotherapy and autoimmunity. Autoimmunity 2010, 43, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Coffman, R.L.; Barrat, F.J. Signalling pathways leading to IFN-alpha production in human plasmacytoid dendritic cell and the possible use of agonists or antagonists of TLR7 and TLR9 in clinical indications. J. Intern. Med. 2009, 265, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Akwa, Y.; Hassett, D.E.; Eloranta, M.L.; Sandberg, K.; Masliah, E.; Powell, H.; Whitton, J.L.; Bloom, F.E.; Campbell, I.L. Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J. Immunol. 1998, 161, 5016–5026. [Google Scholar] [PubMed]
- Campbell, I.L.; Krucker, T.; Steffensen, S.; Akwa, Y.; Powell, H.C.; Lane, T.; Carr, D.J.; Gold, L.H.; Henriksen, S.J.; Siggins, G.R. Structural and functional neuropathology in transgenic mice with CNS expression of IFN-alpha. Brain Res. 1999, 835, 46–61. [Google Scholar] [CrossRef]
- Crow, Y.J.; Lebon, P.; Casanova, J.L.; Gresser, I. A Brief Historical Perspective on the Pathological Consequences of Excessive Type I Interferon Exposure In vivo. J. Clin. Immunol. 2018, 38, 694–698. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, E.; Jansen, M.H.; Anink, J.; De Filippis, L.; Vescovi, A.L.; Watts, C.; Aronica, E.; Hol, E.M.; Kuijpers, T.W. Chronic exposure of astrocytes to interferon-α reveals molecular changes related to Aicardi-Goutieres syndrome. Brain 2013, 136, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.I.; Forte, G.M.; Szynkiewicz, M.; Chase, D.S.; Aeby, A.; Abdel-Hamid, M.S.; Ackroyd, S.; Allcock, R.; Bailey, K.M.; Balottin, U.; et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet Neurol. 2013, 12, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- Staels, F.; Betrains, A.; Doubel, P.; Willemsen, M.; Cleemput, V.; Vanderschueren, S.; Corveleyn, A.; Meyts, I.; Sprangers, B.; Crow, Y.J.; et al. Adult-Onset ANCA-Associated Vasculitis in SAVI: Extension of the Phenotypic Spectrum, Case Report and Review of the Literature. Front. Immunol. 2020, 11, 575219. [Google Scholar] [CrossRef]
- Saldanha, R.G.; Balka, K.R.; Davidson, S.; Wainstein, B.K.; Wong, M.; Macintosh, R.; Loo, C.K.C.; Weber, M.A.; Kamath, V.; Moghaddas, F.; et al. A Mutation Outside the Dimerization Domain Causing Atypical STING-Associated Vasculopathy With Onset in Infancy. Front. Immunol. 2018, 9, 1535. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.; Kang, J.A.; Suh, D.I.; Park, E.B.; Lee, C.R.; Choi, S.A.; Kim, S.Y.; Kim, Y.; Park, S.H.; Ye, M.; et al. Tofacitinib relieves symptoms of stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy caused by 2 de novo variants in TMEM173. J. Allergy Clin. Immunol. 2017, 139, 1396–1399.e1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pin, A.; Tesser, A.; Pastore, S.; Moressa, V.; Valencic, E.; Arbo, A.; Maestro, A.; Tommasini, A.; Taddio, A. Biological and Clinical Changes in a Pediatric Series Treated with Off-Label JAK Inhibitors. Int. J. Mol. Sci. 2020, 21, 7767. [Google Scholar] [CrossRef]
- Sanchez, G.A.M.; Reinhardt, A.; Ramsey, S.; Wittkowski, H.; Hashkes, P.J.; Berkun, Y.; Schalm, S.; Murias, S.; Dare, J.A.; Brown, D.; et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J. Clin. Investig. 2018, 128, 3041–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderver, A.; Adang, L.; Gavazzi, F.; McDonald, K.; Helman, G.; Frank, D.B.; Jaffe, N.; Yum, S.W.; Collins, A.; Keller, S.R.; et al. Janus Kinase Inhibition in the Aicardi-Goutières Syndrome. N. Engl. J. Med. 2020, 383, 986–989. [Google Scholar] [CrossRef]
- Frémond, M.L.; Rodero, M.P.; Jeremiah, N.; Belot, A.; Jeziorski, E.; Duffy, D.; Bessis, D.; Cros, G.; Rice, G.I.; Charbit, B.; et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J. Allergy Clin. Immunol. 2016, 138, 1752–1755. [Google Scholar] [CrossRef] [Green Version]
- Volpi, S.; Insalaco, A.; Caorsi, R.; Santori, E.; Messia, V.; Sacco, O.; Terheggen-Lagro, S.; Cardinale, F.; Scarselli, A.; Pastorino, C.; et al. Efficacy and Adverse Events During Janus Kinase Inhibitor Treatment of SAVI Syndrome. J. Clin. Immunol. 2019, 39, 476–485. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Woodward, J.J.; Sasaki, T.; Minie, M.; Elkon, K.B. Cutting edge: Antimalarial drugs inhibit IFN-β production through blockade of cyclic GMP-AMP synthase-DNA interaction. J. Immunol. 2015, 194, 4089–4093. [Google Scholar] [CrossRef] [Green Version]
- Garau, J.; Sproviero, D.; Dragoni, F.; Piscianz, E.; Santonicola, C.; Tonduti, D.; Carelli, S.; Tesser, A.; Zuccotti, G.V.; Tommasini, A.; et al. Hydroxychloroquine modulates immunological pathways activated by RNA:DNA hybrids in Aicardi-Goutieres syndrome patients carrying RNASEH2 mutations. Cell Mol. Immunol. 2021. [Google Scholar] [CrossRef]
- Piscianz, E.; Cuzzoni, E.; Sharma, R.; Tesser, A.; Sapra, P.; Tommasini, A. Reappraisal of Antimalarials in Interferonopathies: New Perspectives for Old Drugs. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; McDermott, M.F. A proposed classification of the immunological diseases. PLoS Med. 2006, 3, e297. [Google Scholar] [CrossRef] [Green Version]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, C.C.; Lau, C.S. Pathogenesis of systemic lupus erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Dörner, T.; Giesecke, C.; Lipsky, P.E. Mechanisms of B cell autoimmunity in SLE. Arthritis Res. Ther. 2011, 13, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsokos, G.C.; Lo, M.S.; Costa Reis, P.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Moulton, V.R.; Suarez-Fueyo, A.; Meidan, E.; Li, H.; Mizui, M.; Tsokos, G.C. Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol. Med. 2017, 23, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune interferon in the circulation of patients with autoimmune disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef]
- Rhiannon, J.J. Systemic lupus erythematosus involving the nervous system: Presentation, pathogenesis, and management. Clin. Rev. Allergy Immunol. 2008, 34, 356–360. [Google Scholar] [CrossRef]
- Kivity, S.; Agmon-Levin, N.; Zandman-Goddard, G.; Chapman, J.; Shoenfeld, Y. Neuropsychiatric lupus: A mosaic of clinical presentations. BMC Med. 2015, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, M.; Horn, M.; Schmidt, F.; Schmid-Wendtner, M.H.; Volkenandt, M.; Ackenheil, M.; Mueller, N.; Schwarz, M.J. Correlation between sICAM-1 and depressive symptoms during adjuvant treatment of melanoma with interferon-alpha. Brain Behav. Immun. 2004, 18, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L. Pathogenesis of the antiphospholipid syndrome: An additional example of the mosaic of autoimmunity. J. Autoimmun. 2008, 30, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Bialas, A.R.; Presumey, J.; Das, A.; van der Poel, C.E.; Lapchak, P.H.; Mesin, L.; Victora, G.; Tsokos, G.C.; Mawrin, C.; Herbst, R.; et al. Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature 2017, 546, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Popescu, A.; Kao, A.H. Neuropsychiatric systemic lupus erythematosus. Curr. Neuropharmacol. 2011, 9, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, N.; Stock, A.D.; Putterman, C. Neuropsychiatric lupus: New mechanistic insights and future treatment directions. Nat. Rev. Rheumatol. 2019, 15, 137–152. [Google Scholar] [CrossRef]
- Collins, M.P.; Hadden, R.D. The nonsystemic vasculitic neuropathies. Nat. Rev. Neurol. 2017, 13, 302–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elefante, E.; Bond, M.; Monti, S.; Lepri, G.; Cavallaro, E.; Felicetti, M.; Calabresi, E.; Posarelli, C.; Talarico, R.; Quartuccio, L.; et al. One year in review 2018: Systemic vasculitis. Clin. Exp. Rheumatol. 2018, 36, 12–32. [Google Scholar]
- Lintermans, L.L.; Stegeman, C.A.; Heeringa, P.; Abdulahad, W.H. T cells in vascular inflammatory diseases. Front. Immunol. 2014, 5, 504. [Google Scholar] [CrossRef] [Green Version]
- Versari, D.; Daghini, E.; Virdis, A.; Ghiadoni, L.; Taddei, S. Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br. J. Pharmacol. 2009, 157, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Taddei, S.; Ghiadoni, L.; Virdis, A.; Versari, D.; Salvetti, A. Mechanisms of endothelial dysfunction: Clinical significance and preventive non-pharmacological therapeutic strategies. Curr. Pharm. Des. 2003, 9, 2385–2402. [Google Scholar] [CrossRef]
- Meroni, P.; Ronda, N.; Raschi, E.; Borghi, M.O. Humoral autoimmunity against endothelium: Theory or reality? Trends Immunol. 2005, 26, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Varela, C.; de Haro, J.; Bleda, S.; Esparza, L.; de Maturana, I.L.; Acin, F. Anti-endothelial cell antibodies are associated with peripheral arterial disease and markers of endothelial dysfunction and inflammation. Interact. Cardiovasc. Thorac. Surg. 2011, 13, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Tesser, A.; de Carvalho, L.M.; Sandrin-Garcia, P.; Pin, A.; Pastore, S.; Taddio, A.; Roberti, L.R.; de Paula Queiroz, R.G.; Ferriani, V.P.L.; Crovella, S.; et al. Higher interferon score and normal complement levels may identify a distinct clinical subset in children with systemic lupus erythematosus. Arthritis Res. Ther. 2020, 22, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domiciano, D.S.; Carvalho, J.F.; Shoenfeld, Y. Pathogenic role of anti-endothelial cell antibodies in autoimmune rheumatic diseases. Lupus 2009, 18, 1233–1238. [Google Scholar] [CrossRef]
- Rawish, E.; Nording, H.; Münte, T.; Langer, H.F. Platelets as Mediators of Neuroinflammation and Thrombosis. Front. Immunol. 2020, 11, 548631. [Google Scholar] [CrossRef] [PubMed]
- Leiter, O.; Walker, T.L. Platelets: The missing link between the blood and brain? Prog. Neurobiol. 2019, 183, 101695. [Google Scholar] [CrossRef]
- Graf, J.; Imboden, J. Vasculitis and peripheral neuropathy. Curr. Opin. Rheumatol. 2019, 31, 40–45. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, D.; Tan, G. Neurological Involvement in Primary Systemic Vasculitis. Front. Neurol. 2019, 10, 430. [Google Scholar] [CrossRef]
- Meyts, I.; Aksentijevich, I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. J. Clin. Immunol. 2018, 38, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Kaljas, Y.; Liu, C.; Skaldin, M.; Wu, C.; Zhou, Q.; Lu, Y.; Aksentijevich, I.; Zavialov, A.V. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell Mol. Life Sci. 2017, 74, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Navon Elkan, P.; Pierce, S.B.; Segel, R.; Walsh, T.; Barash, J.; Padeh, S.; Zlotogorski, A.; Berkun, Y.; Press, J.J.; Mukamel, M.; et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N. Engl. J. Med. 2014, 370, 921–931. [Google Scholar] [CrossRef]
- Zavialov, A.V.; Gracia, E.; Glaichenhaus, N.; Franco, R.; Lauvau, G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J. Leukoc. Biol. 2010, 88, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Caorsi, R.; Penco, F.; Schena, F.; Gattorno, M. Monogenic polyarteritis: The lesson of ADA2 deficiency. Pediatr. Rheumatol. Online J. 2016, 14, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Rivera, C.; Khaznadar, S.S.; Shwin, K.W.; Irizarry-Caro, J.A.; O’Neil, L.J.; Liu, Y.; Jacobson, K.A.; Ombrello, A.K.; Stone, D.L.; Tsai, W.L.; et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood 2019, 134, 395–406. [Google Scholar] [CrossRef]
- Kendall, J.L.; Springer, J.M. The Many Faces of a Monogenic Autoinflammatory Disease: Adenosine Deaminase 2 Deficiency. Curr. Rheumatol. Rep. 2020, 22, 64. [Google Scholar] [CrossRef]
- Martinon, F.; Aksentijevich, I. New players driving inflammation in monogenic autoinflammatory diseases. Nat. Rev. Rheumatol. 2015, 11, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S.; Omyinmi, E.; Hong, Y.; Papadopoulou, C.; Harper, L.; Al-Abadi, E.; Goel, R.; Dubey, S.; Wood, M.; Jolles, S.; et al. Anti-tumour necrosis factor treatment for the prevention of ischaemic events in patients with deficiency of adenosine deaminase 2 (DADA2). Rheumatology 2021. [Google Scholar] [CrossRef]
- Insalaco, A.; Moneta, G.M.; Pardeo, M.; Caiello, I.; Messia, V.; Bracaglia, C.; Passarelli, C.; De Benedetti, F. Variable Clinical Phenotypes and Relation of Interferon Signature with Disease Activity in ADA2 Deficiency. J. Rheumatol. 2019, 46, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Skrabl-Baumgartner, A.; Plecko, B.; Schmidt, W.M.; König, N.; Hershfield, M.; Gruber-Sedlmayr, U.; Lee-Kirsch, M.A. Autoimmune phenotype with type I interferon signature in two brothers with ADA2 deficiency carrying a novel CECR1 mutation. Pediatr. Rheumatol. Online J. 2017, 15, 67. [Google Scholar] [CrossRef] [Green Version]
- Hashem, H.; Kumar, A.R.; Müller, I.; Babor, F.; Bredius, R.; Dalal, J.; Hsu, A.P.; Holland, S.M.; Hickstein, D.D.; Jolles, S.; et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, and vascular phenotype in DADA2. Blood 2017, 130, 2682–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanthapisal, S.; Murphy, C.; Omoyinmi, E.; Hong, Y.; Standing, A.; Berg, S.; Ekelund, M.; Jolles, S.; Harper, L.; Youngstein, T.; et al. Deficiency of Adenosine Deaminase Type 2: A Description of Phenotype and Genotype in Fifteen Cases. Arthritis Rheumatol. 2016, 68, 2314–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Human, A.; Pagnoux, C. Diagnosis and management of ADA2 deficient polyarteritis nodosa. Int. J. Rheum. Dis. 2019, 22, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Koné-Paut, I. Behçet’s disease in children, an overview. Pediatr. Rheumatol. Online J. 2016, 14, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, B.; Liu, X.; Xiao, J.; Su, G. Immunopathogenesis of Behcet’s Disease. Front. Immunol. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, A.; De Virgilio, A.; Ralli, M.; Ciofalo, A.; Mancini, P.; Attanasio, G.; de Vincentiis, M.; Lambiase, A. Behçet’s disease: New insights into pathophysiology, clinical features and treatment options. Autoimmun. Rev. 2018, 17, 567–575. [Google Scholar] [CrossRef]
- Li, G.; Li, Y.; Liu, H.; Shi, Y.; Guan, W.; Zhang, T.; Yao, W.; Wu, B.; Xu, H.; Sun, L. Genetic heterogeneity of pediatric systemic lupus erythematosus with lymphoproliferation. Medicine 2020, 99, e20232. [Google Scholar] [CrossRef]
- Tsuchida, N.; Kirino, Y.; Soejima, Y.; Onodera, M.; Arai, K.; Tamura, E.; Ishikawa, T.; Kawai, T.; Uchiyama, T.; Nomura, S.; et al. Haploinsufficiency of A20 caused by a novel nonsense variant or entire deletion of TNFAIP3 is clinically distinct from Behçet’s disease. Arthritis Res. Ther. 2019, 21, 137. [Google Scholar] [CrossRef] [Green Version]
- Emmi, G.; Becatti, M.; Bettiol, A.; Hatemi, G.; Prisco, D.; Fiorillo, C. Behçet’s Syndrome as a Model of Thrombo-Inflammation: The Role of Neutrophils. Front. Immunol. 2019, 10, 1085. [Google Scholar] [CrossRef] [PubMed]
- Becatti, M.; Emmi, G.; Silvestri, E.; Bruschi, G.; Ciucciarelli, L.; Squatrito, D.; Vaglio, A.; Taddei, N.; Abbate, R.; Emmi, L.; et al. Neutrophil Activation Promotes Fibrinogen Oxidation and Thrombus Formation in Behçet Disease. Circulation 2016, 133, 302–311. [Google Scholar] [CrossRef]
- Mege, J.L.; Dilsen, N.; Sanguedolce, V.; Gul, A.; Bongrand, P.; Roux, H.; Ocal, L.; Inanç, M.; Capo, C. Overproduction of monocyte derived tumor necrosis factor alpha, interleukin (IL) 6, IL-8 and increased neutrophil superoxide generation in Behçet’s disease. A comparative study with familial Mediterranean fever and healthy subjects. J. Rheumatol. 1993, 20, 1544–1549. [Google Scholar] [PubMed]
- Park, J.; Cheon, J.H. Anti-Tumor Necrosis Factor Therapy in Intestinal Behçet’s Disease. Gut Liver 2018, 12, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Emmi, G.; Silvestri, E.; Bella, C.D.; Grassi, A.; Benagiano, M.; Cianchi, F.; Squatrito, D.; Cantarini, L.; Emmi, L.; Selmi, C.; et al. Cytotoxic Th1 and Th17 cells infiltrate the intestinal mucosa of Behcet patients and exhibit high levels of TNF-α in early phases of the disease. Medicine 2016, 95, e5516. [Google Scholar] [CrossRef]
- Wechsler, B.; Sbaï, A.; Du-Boutin, L.T.; Duhaut, P.; Dormont, D.; Piette, J.C. Neurological manifestations of Behçet’s disease. Rev. Neurol. 2002, 158, 926–933. [Google Scholar]
- Kalra, S.; Silman, A.; Akman-Demir, G.; Bohlega, S.; Borhani-Haghighi, A.; Constantinescu, C.S.; Houman, H.; Mahr, A.; Salvarani, C.; Sfikakis, P.P.; et al. Diagnosis and management of Neuro-Behçet’s disease: International consensus recommendations. J. Neurol. 2014, 261, 1662–1676. [Google Scholar] [CrossRef] [Green Version]
- Borhani-Haghighi, A.; Kardeh, B.; Banerjee, S.; Yadollahikhales, G.; Safari, A.; Sahraian, M.A.; Shapiro, L. Neuro-Behcet’s disease: An update on diagnosis, differential diagnoses, and treatment. Mult. Scler. Relat. Disord. 2019, 39, 101906. [Google Scholar] [CrossRef] [PubMed]
- Sfikakis, P.P.; Markomichelakis, N.; Alpsoy, E.; Assaad-Khalil, S.; Bodaghi, B.; Gul, A.; Ohno, S.; Pipitone, N.; Schirmer, M.; Stanford, M.; et al. Anti-TNF therapy in the management of Behcet’s disease--review and basis for recommendations. Rheumatology 2007, 46, 736–741. [Google Scholar] [CrossRef] [Green Version]
- Vallet, H.; Riviere, S.; Sanna, A.; Deroux, A.; Moulis, G.; Addimanda, O.; Salvarani, C.; Lambert, M.; Bielefeld, P.; Seve, P.; et al. Efficacy of anti-TNF alpha in severe and/or refractory Behçet’s disease: Multicenter study of 124 patients. J. Autoimmun. 2015, 62, 67–74. [Google Scholar] [CrossRef]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kuemmerle-Deschner, J.B. CAPS--pathogenesis, presentation and treatment of an autoinflammatory disease. Semin. Immunopathol. 2015, 37, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Kuemmerle-Deschner, J.B.; Ozen, S.; Tyrrell, P.N.; Kone-Paut, I.; Goldbach-Mansky, R.; Lachmann, H.; Blank, N.; Hoffman, H.M.; Weissbarth-Riedel, E.; Hugle, B.; et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann. Rheum. Dis. 2017, 76, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol. 2016, 17, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Booshehri, L.M.; Hoffman, H.M. CAPS and NLRP3. J. Clin. Immunol. 2019, 39, 277–286. [Google Scholar] [CrossRef] [PubMed]
- de Torre-Minguela, C.; Mesa Del Castillo, P.; Pelegrín, P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Paim-Marques, L.B.; Cavalcante, A.; Castro, C.; Muskardin, T.L.W.; de Oliveira, J.B.; Niewold, T.B.; Appenzeller, S. Novel mutation in the NRLP3 manifesting as an intermediate phenotype of cryopyrinopathies. Rheumatol. Int. 2021, 41, 219–225. [Google Scholar] [CrossRef]
- Rossi, S.; Motta, C.; Studer, V.; Macchiarulo, G.; Volpe, E.; Barbieri, F.; Ruocco, G.; Buttari, F.; Finardi, A.; Mancino, R.; et al. Interleukin-1β causes excitotoxic neurodegeneration and multiple sclerosis disease progression by activating the apoptotic protein p53. Mol. Neurodegener. 2014, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamoudjy, N.; Maurey, H.; Marie, I.; Koné-Paut, I.; Deiva, K. Neurological outcome of patients with cryopyrin-associated periodic syndrome (CAPS). Orphanet J. Rare Dis. 2017, 12, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldbach-Mansky, R.; Dailey, N.J.; Canna, S.W.; Gelabert, A.; Jones, J.; Rubin, B.I.; Kim, H.J.; Brewer, C.; Zalewski, C.; Wiggs, E.; et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 2006, 355, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachmann, H.J.; Kone-Paut, I.; Kuemmerle-Deschner, J.B.; Leslie, K.S.; Hachulla, E.; Quartier, P.; Gitton, X.; Widmer, A.; Patel, N.; Hawkins, P.N.; et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med. 2009, 360, 2416–2425. [Google Scholar] [CrossRef] [Green Version]
- Lepore, L.; Paloni, G.; Caorsi, R.; Alessio, M.; Rigante, D.; Ruperto, N.; Cattalini, M.; Tommasini, A.; Zulian, F.; Ventura, A.; et al. Follow-up and quality of life of patients with cryopyrin-associated periodic syndromes treated with Anakinra. J. Pediatr. 2010, 157, 310–315.e311. [Google Scholar] [CrossRef]
- Fox, K.; Wells, M.E.; Tennison, M.; Vaughn, B. Febrile Infection-Related Epilepsy Syndrome (FIRES): A Literature Review and Case Study. Neurodiagn. J. 2017, 57, 224–233. [Google Scholar] [CrossRef]
- Hon, K.L.; Leung, A.K.C.; Torres, A.R. Febrile Infection-Related Epilepsy Syndrome (FIRES): An Overview of Treatment and Recent Patents. Recent Pat. Inflamm. Allergy Drug Discov. 2018, 12, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller, S.; Helbig, I.; Stephani, U.; Häusler, M.; Kluger, G.; Bungeroth, M.; Müller, S.; Kuhlenbäumer, G.; van Baalen, A. Febrile infection-related epilepsy syndrome (FIRES) is not caused by SCN1A, POLG, PCDH19 mutations or rare copy number variations. Dev. Med. Child. Neurol. 2012, 54, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, H.; Tanuma, N.; Kuki, I.; Takahashi, Y.; Shiomi, M.; Hayashi, M. Intrathecal overproduction of proinflammatory cytokines and chemokines in febrile infection-related refractory status epilepticus. J. Neurol. Neurosurg. Psychiatry 2015, 86, 820–822. [Google Scholar] [CrossRef] [PubMed]
- Kenney-Jung, D.L.; Vezzani, A.; Kahoud, R.J.; LaFrance-Corey, R.G.; Ho, M.L.; Muskardin, T.W.; Wirrell, E.C.; Howe, C.L.; Payne, E.T. Febrile infection-related epilepsy syndrome treated with anakinra. Ann. Neurol. 2016, 80, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Muscal, E.; Wells, E.; Shukla, N.; Eschbach, K.; Hyeong Lee, K.; Kaliakatsos, M.; Desai, N.; Wickstrom, R.; Viri, M.; et al. Anakinra usage in febrile infection related epilepsy syndrome: An international cohort. Ann. Clin. Transl. Neurol. 2020, 7, 2467–2474. [Google Scholar] [CrossRef]
- Stredny, C.M.; Case, S.; Sansevere, A.J.; Son, M.; Henderson, L.; Gorman, M.P. Interleukin-6 Blockade With Tocilizumab in Anakinra-Refractory Febrile Infection-Related Epilepsy Syndrome (FIRES). Child Neurol. Open 2020, 7, 2329048X20979253. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Medications | Monogenic Disorders (Key Neurological Symptoms) | Multifactorial Disorders |
|---|---|---|---|
| Type I IFN | JAK inhibitors (baricitinib, ruxolitinib, tofacitinib), antimalarials | Type I interferonopathies (seizures, leukodystrophy, headaches) | SLE |
| TNFα | Etanercept, adalimumab, infliximab | DADA2 vasculitis, strokes | Neuro-BD |
| IL-1 receptor | Anakinra | CAPS, aseptic meningitis, pseudotumor cerebri | FIRES |
| Disease | Gene(s) | Dysregulated Signaling Pathway | Principal Clinical Features | Neurological Involvement | Inheritance |
|---|---|---|---|---|---|
| AGS [9,10] | TREX1 RNASEH2 (A, B, C) SAMHD1 ADAR IFIH1 | Type I IFN | Intermittent fevers, hepatosplenomegaly, chilblains | Progressive cerebral atrophy, leukodystrophy, intracranial calcifications, chronic CSF lymphocytosis, progressive psychomotor retardation, seizures | AD/AR |
| SAVI [11] | STING/TMEM173 | Severe skin lesions (face, ears, nose, digits), interstitial lung disease, livedo reticularis, Raynaud phenomenon, recurrent fevers | Developmental delay, brain infarctions | AD | |
| DADA2 [12,13] | CECR1/ADA2 | TNFα | Recurrent fevers, systemic vascular inflammation (skin ulcerations, strokes), Raynaud phenomenon | Neurologic sequelae of stroke, headaches, ataxia | AR |
| CAPS [14,15] | CIAS1/NLRP3 | IL-1β | Fever, rash, arthralgia | Chronic meningitis with headaches, deafness and blindness (partial or complete), mental retardation | AD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesser, A.; Pin, A.; Mencaroni, E.; Gulino, V.; Tommasini, A. Vasculitis, Autoimmunity, and Cytokines: How the Immune System Can Harm the Brain. Int. J. Environ. Res. Public Health 2021, 18, 5585. https://doi.org/10.3390/ijerph18115585
Tesser A, Pin A, Mencaroni E, Gulino V, Tommasini A. Vasculitis, Autoimmunity, and Cytokines: How the Immune System Can Harm the Brain. International Journal of Environmental Research and Public Health. 2021; 18(11):5585. https://doi.org/10.3390/ijerph18115585
Chicago/Turabian StyleTesser, Alessandra, Alessia Pin, Elisabetta Mencaroni, Virginia Gulino, and Alberto Tommasini. 2021. "Vasculitis, Autoimmunity, and Cytokines: How the Immune System Can Harm the Brain" International Journal of Environmental Research and Public Health 18, no. 11: 5585. https://doi.org/10.3390/ijerph18115585
APA StyleTesser, A., Pin, A., Mencaroni, E., Gulino, V., & Tommasini, A. (2021). Vasculitis, Autoimmunity, and Cytokines: How the Immune System Can Harm the Brain. International Journal of Environmental Research and Public Health, 18(11), 5585. https://doi.org/10.3390/ijerph18115585