
Advanced Glycation End Products: New Clinical and Molecular Perspectives

 ,
,  , , ,
, , , 
 , ,
, ,  and
and
Abstract
:1. Introduction
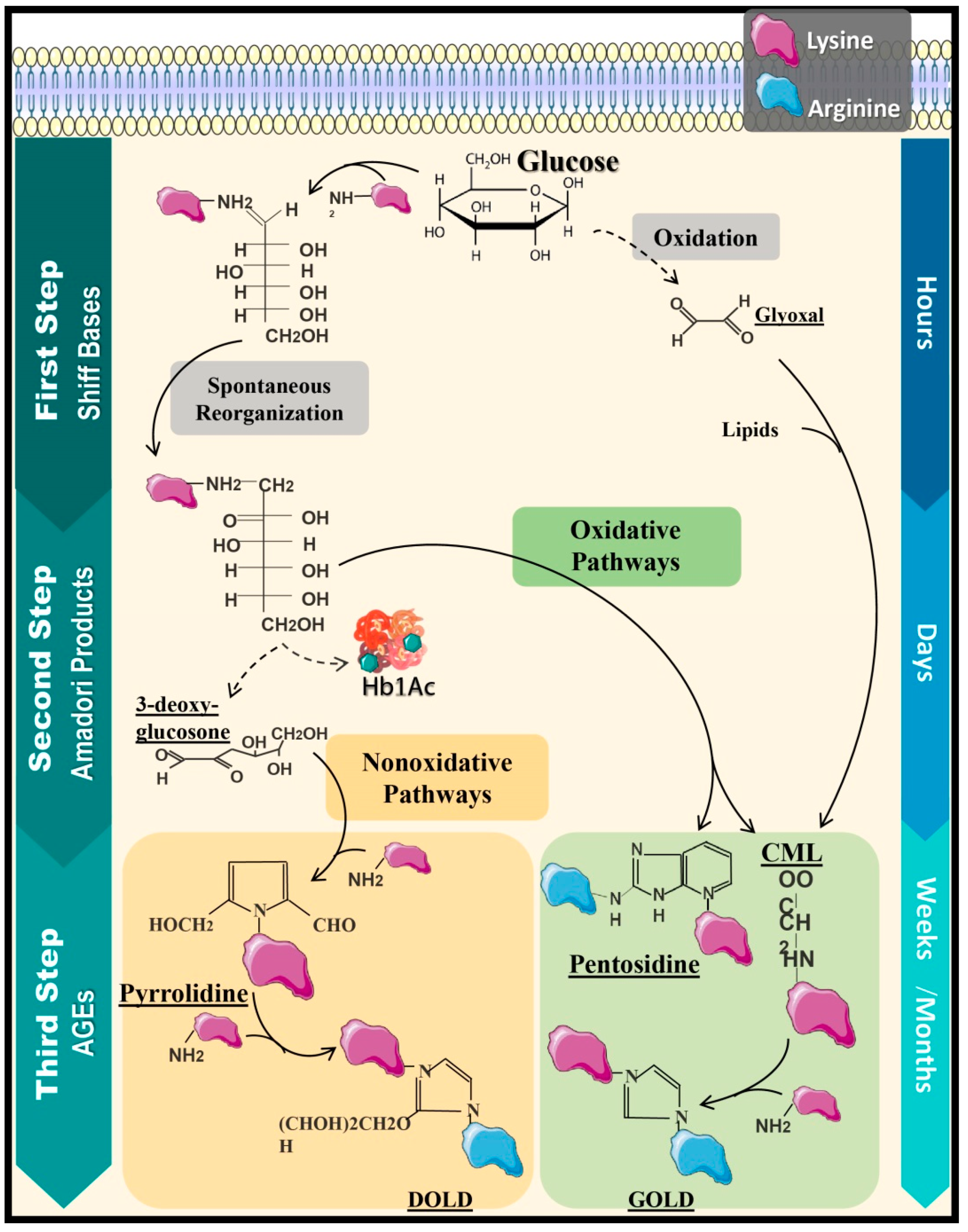
2. Protein Glycation and Formation of Advanced Glycation End Products
3. AGEs and Their Implication in Chronic Complications of Diabetes Mellitus
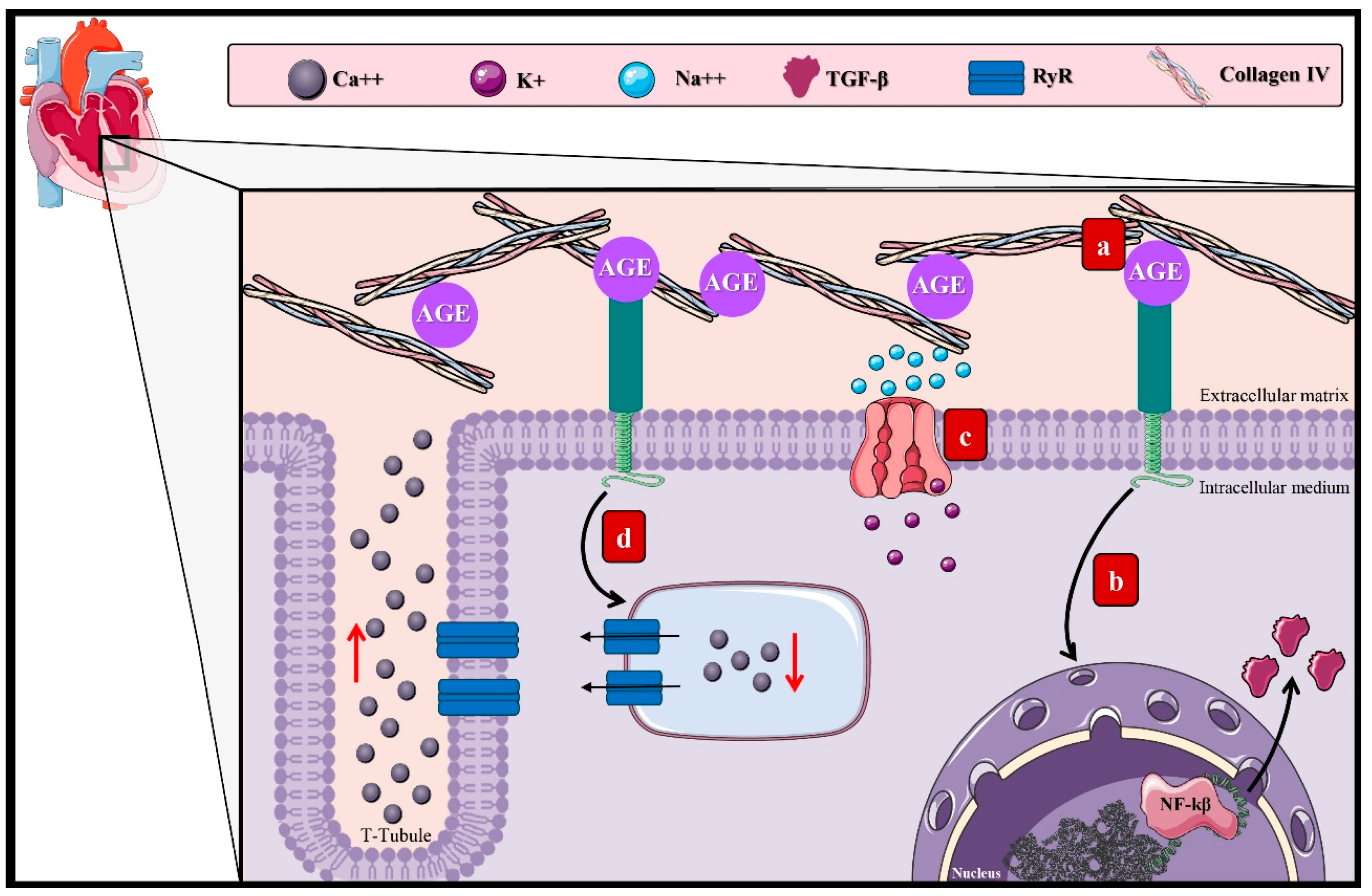
3.1. Molecular Mechanisms of AGEs in Microvascular Complications of DM
3.2. AGEs and the Macrovascular Alterations in DM
4. Progression of Measurement Techniques of AGEs in Patients with DM
5. Measurement of Skin Fluorescence
6. Therapeutic Strategies: Halting the AGE–RAGE Axis
6.1. Inhibition of the Absorption of Exogenous AGEs
6.2. Inhibition of the Endogenous Formation of AGEs
6.3. Breakage and Reversal of Preformed AGEs
6.4. Antagonism towards RAGE-Binding
7. Future Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kerner, W.; Brückel, J. German Diabetes Association Definition, Classification and Diagnosis of Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2014, 122, 384–386. [Google Scholar] [CrossRef] [Green Version]
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes Mellitus Statistics on Prevalence and Mortality: Facts and Fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation (IDF). IDF Diabetes Atlas, 7th ed.; International Diabetes Federation: Brussels, Belgium, 2015; Available online: https://www.desang.net/2017/11/idf-diabetes-atlas-7th-edition/ (accessed on 7 May 2021).
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced Glycation End Products and Diabetic Complications. Korean J. Physiol. Pharm. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butalia, S.; Patel, A.B.; Johnson, J.A.; Ghali, W.A.; Rabi, D.M. Geographic Clustering of Acute Complications and Sociodemographic Factors in Adults with Type 1 Diabetes. Can. J. Diabetes 2017, 41, 132–137. [Google Scholar] [CrossRef]
- Elgart, J.F.; Caporale, J.E.; Asteazarán, S.; De La Fuente, J.L.; Camilluci, C.; Brown, J.B.; González, C.D.; Gagliardino, J.J. Association between Socioeconomic Status, Type 2 Diabetes and Its Chronic Complications in Argentina. Diabetes Res. Clin. Pract. 2014, 104, 241–247. [Google Scholar] [CrossRef]
- Shi, Y.; Vanhoutte, P.M. Macro- and Microvascular Endothelial Dysfunction in Diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomis, S.J.; Chen, Y.; Sacks, D.B.; Christenson, E.S.; Christenson, R.H.; Rebholz, C.M.; Selvin, E. Cross-Sectional Analysis of AGE-CML, SRAGE, and EsRAGE with Diabetes and Cardiometabolic Risk Factors in a Community-Based Cohort. Clin. Chem. 2017, 63, 980–989. [Google Scholar] [CrossRef] [Green Version]
- Vélayoudom-Céphise, F.-L.; Rajaobelina, K.; Helmer, C.; Nov, S.; Pupier, E.; Blanco, L.; Hugo, M.; Farges, B.; Astrugue, C.; Gin, H.; et al. Skin Autofluorescence Predicts Cardio-Renal Outcome in Type 1 Diabetes: A Longitudinal Study. Cardiovasc. Diabetol. 2016, 15, 127. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.C.; Woodward, M.; Neal, B.; Li, Q.; Pickering, R.; Marre, M.; Williams, B.; Perkovic, V.; Cooper, M.E.; Zoungas, S.; et al. Relationship between Levels of Advanced Glycation End Products and Their Soluble Receptor and Adverse Outcomes in Adults with Type 2 Diabetes. Diabetes Care 2015, 38, 1891–1897. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, A.; Mirasole, C.; Zolla, L. Haemoglobin Glycation (Hb1Ac) Increases during Red Blood Cell Storage: A MALDI-TOF Mass-Spectrometry-Based Investigation. Vox Sang. 2013, 105, 177–180. [Google Scholar] [CrossRef]
- Cho, Y.H.; Craig, M.E.; Januszewski, A.S.; Benitez-Aguirre, P.; Hing, S.; Jenkins, A.J.; Donaghue, K.C. Higher Skin Autofluorescence in Young People with Type 1 Diabetes and Microvascular Complications. Diabet. Med. 2017, 34, 543–550. [Google Scholar] [CrossRef]
- Botros, N.; Sluik, D.; van Waateringe, R.P.; de Vries, J.H.M.; Geelen, A.; Feskens, E.J.M. Advanced Glycation End-Products (AGEs) and Associations with Cardio-Metabolic, Lifestyle, and Dietary Factors in a General Population: The NQplus Study. Diabetes Metab. Res. Rev. 2017, 33. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondaca-Navarro, B.A.; Ávila-Villa, L.A.; González-Córdova, A.F.; López-Cervantes, J.; Sánchez-Machado, D.I.; Campas-Baypoli, O.N.; Rodríguez-Ramírez, R. Antioxidant and Chelating Capacity of Maillard Reaction Products in Amino Acid-Sugar Model Systems: Applications for Food Processing. J. Sci. Food Agric. 2017, 97, 3522–3529. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M.; Vlassara, H.; Cerami, A. Nonenzymatic Glycosylation and the Pathogenesis of Diabetic Complications. Ann. Intern. Med. 1984, 101, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.L.; Yaylayan, V.A. Post-Schiff Base Chemistry of the Maillard Reaction: Mechanism of Imine Isomerization. Ann. N. Y. Acad. Sci. 2008, 1126, 30–37. [Google Scholar] [CrossRef]
- Johnson, K.L.; Williams, J.G.; Maleki, S.J.; Hurlburt, B.K.; London, R.E.; Mueller, G.A. Enhanced Approaches for Identifying Amadori Products: Application to Peanut Allergens. J. Agric. Food Chem. 2016, 64, 1406–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucala, R.; Model, P.; Cerami, A. Modification of DNA by Reducing Sugars: A Possible Mechanism for Nucleic Acid Aging and Age-Related Dysfunction in Gene Expression. Proc. Natl. Acad. Sci. USA 1984, 81, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, N.A.; Moinuddin, null; Mir, A.R.; Habib, S.; Alam, K.; Ali, A.; Khan, R.H. Role of Early Glycation Amadori Products of Lysine-Rich Proteins in the Production of Autoantibodies in Diabetes Type 2 Patients. Cell Biochem. Biophys. 2014, 70, 857–865. [Google Scholar] [CrossRef]
- Olar, L.; Razvan, Ștefan; Berce, C.; Ciobanu, D.; Papuc, I. Bulletin of University of Agricultural Sciences and Veterinary Medicine Cluj-Napoca. Vet. Med. 2015, 65, 358. [Google Scholar]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular Effects of Advanced Glycation Endproducts: Clinical Effects and Molecular Mechanisms. Mol. Metab. 2014, 3, 94–108. [Google Scholar] [CrossRef]
- Macías-Cervantes, M.H.; Rodríguez-Soto, J.M.D.; Uribarri, J.; Díaz-Cisneros, F.J.; Cai, W.; Garay-Sevilla, M.E. Effect of an Advanced Glycation End Product-Restricted Diet and Exercise on Metabolic Parameters in Adult Overweight Men. Nutrition 2015, 31, 446–451. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of Advanced Glycation End Products Improves Insulin Resistance in Human Type 2 Diabetes: Potential Role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef] [Green Version]
- Angoorani, P.; Ejtahed, H.-S.; Mirmiran, P.; Mirzaei, S.; Azizi, F. Dietary Consumption of Advanced Glycation End Products and Risk of Metabolic Syndrome. Int. J. Food Sci. Nutr. 2016, 67, 170–176. [Google Scholar] [CrossRef]
- Saha, A.; Poojary, P.; Chan, L.; Chauhan, K.; Nadkarni, G.; DO, S.C.; Uribarri, J. Increased Odds of Metabolic Syndrome with Consumption of High Dietary Advanced Glycation End Products in Adolescents. Diabetes Metab. 2017, 43, 469–471. [Google Scholar] [CrossRef]
- Lv, X.; Lv, G.-H.; Dai, G.-Y.; Sun, H.-M.; Xu, H.-Q. Food-Advanced Glycation End Products Aggravate the Diabetic Vascular Complications via Modulating the AGEs/RAGE Pathway. Chin. J. Nat. Med. 2016, 14, 844–855. [Google Scholar] [CrossRef]
- Li, Z.; Wang, G.; Zhu, Y.-J.; Li, C.-G.; Tang, Y.-Z.; Jiang, Z.-H.; Yang, M.; Ni, C.-L.; Chen, L.-M.; Niu, W.-Y. The Relationship between Circulating Irisin Levels and Tissues AGE Accumulation in Type 2 Diabetes Patients. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Chawla, D.; Bansal, S.; Banerjee, B.D.; Madhu, S.V.; Kalra, O.P.; Tripathi, A.K. Role of Advanced Glycation End Product (AGE)-Induced Receptor (RAGE) Expression in Diabetic Vascular Complications. Microvasc. Res. 2014, 95, 1–6. [Google Scholar] [CrossRef]
- Xue, J.; Ray, R.; Singer, D.; Böhme, D.; Burz, D.S.; Rai, V.; Hoffmann, R.; Shekhtman, A. The Receptor for Advanced Glycation End Products (RAGE) Specifically Recognizes Methylglyoxal-Derived AGEs. Biochemistry 2014, 53, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE Mediates a Novel Proinflammatory Axis: A Central Cell Surface Receptor for S100/Calgranulin Polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Grimm, S.; Ott, C.; Hörlacher, M.; Weber, D.; Höhn, A.; Grune, T. Advanced-Glycation-End-Product-Induced Formation of Immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem. J. 2012, 448, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.Q.; Yang, C.; Wang, Y.Y.; Wang, P.; Chen, H.L.; Zhang, X.D.; Liu, R.; Li, W.L.; Qin, X.J.; Liang, X.; et al. RAGE Upregulation and Nuclear Factor-KappaB Activation Associated with Ageing Rat Cardiomyocyte Dysfunction. Gen. Physiol. Biophys. 2008, 27, 152–158. [Google Scholar]
- Ohashi, K.; Takahashi, H.K.; Mori, S.; Liu, K.; Wake, H.; Sadamori, H.; Matsuda, H.; Yagi, T.; Yoshino, T.; Nishibori, M.; et al. Advanced Glycation End Products Enhance Monocyte Activation during Human Mixed Lymphocyte Reaction. Clin. Immunol. 2010, 134, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Yao, T.; Zhou, Z.; Zhu, J.; Zhang, S.; Hu, W.; Shen, C. Advanced Glycation End Products Enhance Macrophages Polarization into M1 Phenotype through Activating RAGE/NF-ΚB Pathway. Biomed. Res. Int. 2015, 2015, 732450. [Google Scholar] [CrossRef] [Green Version]
- Jhun, J.; Lee, S.; Kim, H.; Her, Y.-M.; Byun, J.K.; Kim, E.-K.; Lee, S.K.; Cho, M.-L.; Choi, J.Y. HMGB1/RAGE Induces IL-17 Expression to Exaggerate Inflammation in Peripheral Blood Cells of Hepatitis B Patients. J. Transl. Med. 2015, 13. [Google Scholar] [CrossRef] [Green Version]
- Bangert, A.; Andrassy, M.; Müller, A.-M.; Bockstahler, M.; Fischer, A.; Volz, C.H.; Leib, C.; Göser, S.; Korkmaz-Icöz, S.; Zittrich, S.; et al. Critical Role of RAGE and HMGB1 in Inflammatory Heart Disease. Proc. Natl. Acad. Sci. USA 2016, 113, E155–E164. [Google Scholar] [CrossRef] [Green Version]
- Detzen, L.; Cheng, B.; Chen, C.-Y.; Papapanou, P.N.; Lalla, E. Soluble Forms of the Receptor for Advanced Glycation Endproducts (RAGE) in Periodontitis. Sci. Rep. 2019, 9, 8170. [Google Scholar] [CrossRef]
- Egaña-Gorroño, L.; López-Díez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights From Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef]
- Schmidt, A.M. Soluble RAGEs Prospects for Treating & Tracking Metabolic and Inflammatory Disease. Vasc. Pharm. 2015, 72, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Farhan, S.S.; Hussain, S.A. Advanced Glycation End Products (AGEs) and Their Soluble Receptors (SRAGE) as Early Predictors of Reno-Vascular Complications in Patients with Uncontrolled Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 2457–2461. [Google Scholar] [CrossRef]
- Gerrits, E.G.; Lutgers, H.L.; Kleefstra, N.; Graaff, R.; Groenier, K.H.; Smit, A.J.; Gans, R.O.; Bilo, H.J. Skin Autofluorescence: A Tool to Identify Type 2 Diabetic Patients at Risk for Developing Microvascular Complications. Diabetes Care 2008, 31, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Zerbini, G.; Maestroni, S.; Turco, V.; Secchi, A. The Eye as a Window to the Microvascular Complications of Diabetes. Dev. Ophthalmol. 2017, 60, 6–15. [Google Scholar] [CrossRef]
- Sun, H.; Yuan, Y.; Sun, Z. Update on Mechanisms of Renal Tubule Injury Caused by Advanced Glycation End Products. Biomed. Res. Int. 2016, 2016, e5475120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Ward, M.; Madden, A.; Yong, P.H.; Limb, G.A.; Curtis, T.M.; Stitt, A.W. Hyperglycaemia-Induced pro-Inflammatory Responses by Retinal Müller Glia Are Regulated by the Receptor for Advanced Glycation End-Products (RAGE). Diabetologia 2010, 53, 2656–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Tatsunami, R.; Yama, K.; Tampo, Y. Glycolaldehyde Induces Cytotoxicity and Increases Glutathione and Multidrug-Resistance-Associated Protein Levels in Schwann Cells. Biol. Pharm. Bull. 2013, 36, 1111–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Liu, N.; Wang, F. Epigenetic Regulations in Diabetic Nephropathy. J. Diabetes Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Espinel, E.; Agraz, I.; Ibernon, M.; Ramos, N.; Fort, J.; Serón, D. Renal Biopsy in Type 2 Diabetic Patients. J. Clin. Med. 2015, 4, 998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, P.Y.; Yu, Q.; Fang, W.; Uribarri, J.; He, J.C. Advanced Glycation Endproducts Induce Podocyte Apoptosis by Activation of the FOXO4 Transcription Factor. Kidney Int. 2007, 72, 965–976. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Feng, L.; Zhu, M.; Gu, J.; Jiang, J.; Cheng, X.; Ding, S.; Wu, C.; Jia, X. The Anti-Inflammation Effect of Moutan Cortex on Advanced Glycation End Products-Induced Rat Mesangial Cells Dysfunction and High-Glucose-Fat Diet and Streptozotocin-Induced Diabetic Nephropathy Rats. J. Ethnopharmacol. 2014, 151, 591–600. [Google Scholar] [CrossRef]
- Ki, H.-J.; Kim, S.Y.; Lee, S.H.; Moon, J.-Y.; Jeong, K.H.; Lee, T.W.; Ihm, C.G.; Kim, S.K.; Chung, J.-H.; Kang, S.W.; et al. Transforming Growth Factor-β Receptor 2 Gene Polymorphisms Are Associated with End-Stage Renal Disease. Kidney Res. Clin. Pract. 2015, 34, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Miura, J.; Yamagishi, S.I.; Uchigata, Y.; Takeuchi, M.; Yamamoto, H.; Makita, Z.; Iwamoto, Y. Serum Levels of Non-Carboxymethyllysine Advanced Glycation Endproducts Are Correlated to Severity of Microvascular Complications in Patients with Type 1 Diabetes. J. Diabetes Complicat. 2003, 17, 16–21. [Google Scholar] [CrossRef]
- Liu, J.; Huang, K.; Cai, G.-Y.; Chen, X.-M.; Yang, J.-R.; Lin, L.-R.; Yang, J.; Huo, B.-G.; Zhan, J.; He, Y.-N. Receptor for Advanced Glycation End-Products Promotes Premature Senescence of Proximal Tubular Epithelial Cells via Activation of Endoplasmic Reticulum Stress-Dependent P21 Signaling. Cell Signal 2014, 26, 110–121. [Google Scholar] [CrossRef]
- Li, Y.; Ma, W.; Xie, C.; Zhang, M.; Yin, X.; Wang, F.; Xu, J.; Shi, B. Identification of Genes and Signaling Pathways Associated with Diabetic Neuropathy Using a Weighted Correlation Network Analysis: A Consort Study. Medicine 2016, 95, e5443. [Google Scholar] [CrossRef] [PubMed]
- Araszkiewicz, A.; Gandecka, A.; Nowicki, M.; Uruska, A.; Malińska, A.; Kowalska, K.; Wierusz-Wysocka, B.; Zozulińska-Ziółkiewicz, D. Association between Small Fiber Neuropathy and Higher Skin Accumulation of Advanced Glycation End Products in Patients with Type 1 Diabetes. Pol. Arch. Med. Wewn. 2016, 126, 847–853. [Google Scholar] [CrossRef] [Green Version]
- Duran-Jimenez, B.; Dobler, D.; Moffatt, S.; Rabbani, N.; Streuli, C.H.; Thornalley, P.J.; Tomlinson, D.R.; Gardiner, N.J. Advanced Glycation End Products in Extracellular Matrix Proteins Contribute to the Failure of Sensory Nerve Regeneration in Diabetes. Diabetes 2009, 58, 2893–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loske, C.; Neumann, A.; Cunningham, A.M.; Nichol, K.; Schinzel, R.; Riederer, P.; Münch, G. Cytotoxicity of Advanced Glycation Endproducts Is Mediated by Oxidative Stress. J. Neur. Transm 1998, 105, 1005–1015. [Google Scholar] [CrossRef]
- Yu, T.; Li, L.; Chen, T.; Liu, Z.; Liu, H.; Li, Z. Erythropoietin Attenuates Advanced Glycation Endproducts-Induced Toxicity of Schwann Cells in Vitro. Neurochem. Res. 2015, 40, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Guitart, K.; Loers, G.; Schachner, M.; Kleene, R. Prion Protein Regulates Glutathione Metabolism and Neural Glutamate and Cysteine Uptake via Excitatory Amino Acid Transporter 3. J. Neurochem. 2015, 133, 558–571. [Google Scholar] [CrossRef] [Green Version]
- Bus, S.A.; Haspels, R.; Busch-Westbroek, T.E. Evaluation and Optimization of Therapeutic Footwear for Neuropathic Diabetic Foot Patients Using In-Shoe Plantar Pressure Analysis. Diabetes Care 2011, 34, 1595–1600. [Google Scholar] [CrossRef] [Green Version]
- Vouillarmet, J.; Maucort-Boulch, D.; Michon, P.; Thivolet, C. Advanced Glycation End Products Assessed by Skin Autofluorescence: A New Marker of Diabetic Foot Ulceration. Diabetes Technol. 2013, 15, 601–605. [Google Scholar] [CrossRef]
- American Diabetes Association. Screening Guidelines for Diabetic Retinopathy: Clinical Guideline. Ophthalmology 1992, 99, 1626–1628. [Google Scholar] [CrossRef]
- Frank, R.N. Diabetic Retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef]
- Tracey, M.L.; McHugh, S.M.; Fitzgerald, A.P.; Buckley, C.M.; Canavan, R.J.; Kearney, P.M. Trends in Blindness Due to Diabetic Retinopathy among Adults Aged 18-69years over a Decade in Ireland. Diabetes Res. Clin. Pract. 2016, 121, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A. Effect of Advanced Glycation End Products on Accelerated Apoptosis of Retinal Capillary Cells under in Vitro Conditions. Life Sci. 2005, 76, 1051–1060. [Google Scholar] [CrossRef]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller Cells in the Healthy and Diseased Retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef]
- Cheng, L.; Bu, H.; Portillo, J.-A.C.; Li, Y.; Subauste, C.S.; Huang, S.S.; Kern, T.S.; Lin, F. Modulation of Retinal Müller Cells by Complement Receptor C5aR. Invest. Ophthalmol. Vis. Sci. 2013, 54, 8191–8198. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Sato, T.; Takeuchi, M. Potential Utility of Statins, 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibitors in Diabetic Retinopathy. Med. Hypotheses 2006, 66, 1019–1021. [Google Scholar] [CrossRef]
- AI, J.; LIU, Y.; SUN, J.-H. Advanced Glycation End-Products Stimulate Basic Fibroblast Growth Factor Expression in Cultured Müller Cells. Mol. Med. Rep. 2013, 7, 16–20. [Google Scholar] [CrossRef]
- Shimizu, F.; Sano, Y.; Haruki, H.; Kanda, T. Advanced Glycation End-Products Induce Basement Membrane Hypertrophy in Endoneurial Microvessels and Disrupt the Blood-Nerve Barrier by Stimulating the Release of TGF-β and Vascular Endothelial Growth Factor (VEGF) by Pericytes. Diabetologia 2011, 54, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New Type of Cardiomyopathy Associated with Diabetic Glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Abd-El Aziz, F.M.; Abdelghaffar, S.; Hussien, E.M.; Fattouh, A.M. Evaluation of Cardiac Functions in Children and Adolescents with Type 1 Diabetes. J. Cardiovasc. Ultrasound. 2017, 25, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Gao, H.; Dong, R.; Wu, Y.-Q. Sequential Changes of Endoplasmic Reticulum Stress and Apoptosis in Myocardial Fibrosis of Diabetes Mellitus-Induced Rats. Mol. Med. Rep. 2016, 13, 5037–5044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa, U.; Arauna, D.; Moran, M.; Nuñez, M.; Zagmutt, S.; Saldivia, S.; Valdes, C.; Villaseñor, J.; Zambrano, C.G.; Gonzalez, D.R. High-Intensity Exercise Reduces Cardiac Fibrosis and Hypertrophy but Does Not Restore the Nitroso-Redox Imbalance in Diabetic Cardiomyopathy. Oxid. Med. Cell Longev. 2017, 2017, 7921363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Chen, J.; Chen, Y.; Chen, X.; Liu, P. Advanced Glycation End Products Promote Heart Failure through Inducing the Immune Maturation of Dendritic Cells. Appl. Biochem. Biotechnol. 2014, 172, 4062–4077. [Google Scholar] [CrossRef]
- Zerif, E.; Maalem, A.; Gaudreau, S.; Guindi, C.; Ramzan, M.; Véroneau, S.; Gris, D.; Stankova, J.; Rola-Pleszczynski, M.; Mourad, W.; et al. Constitutively Active Stat5b Signaling Confers Tolerogenic Functions to Dendritic Cells of NOD Mice and Halts Diabetes Progression. J. Autoimmun. 2017, 76, 63–74. [Google Scholar] [CrossRef]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory Role of Dendritic Cells in Postinfarction Healing and Left Ventricular Remodeling. Circulation 2012, 125, 1234–1245. [Google Scholar] [CrossRef] [Green Version]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A Molecular Basis for Familial Hypertrophic Cardiomyopathy: A Beta Cardiac Myosin Heavy Chain Gene Missense Mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Herrmann, K.L.; McCulloch, A.D.; Omens, J.H. Glycated Collagen Cross-Linking Alters Cardiac Mechanics in Volume-Overload Hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1277–H1284. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, S.; Hartog, J.W.L.; Hummel, Y.M.; van Ruijven, M.H.I.; van der Horst, I.C.C.; van Veldhuisen, D.J.; Voors, A.A. Tissue Advanced Glycation End Products Are Associated with Diastolic Function and Aerobic Exercise Capacity in Diabetic Heart Failure Patients. Eur. J. Heart Fail 2011, 13, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Wang, J.; Li, S.; Guo, Y. Advanced Glycation End-Products Accelerate the Cardiac Aging Process through the Receptor for Advanced Glycation End-Products/Transforming Growth Factor-β-Smad Signaling Pathway in Cardiac Fibroblasts. Geriatr. Gerontol. Int. 2016, 16, 522–527. [Google Scholar] [CrossRef]
- Kawashima, T.; Inuzuka, Y.; Okuda, J.; Kato, T.; Niizuma, S.; Tamaki, Y.; Iwanaga, Y.; Kawamoto, A.; Narazaki, M.; Matsuda, T.; et al. Constitutive SIRT1 Overexpression Impairs Mitochondria and Reduces Cardiac Function in Mice. J. Mol. Cell Cardiol. 2011, 51, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.S.; Wang, Z.B.; Ye, Z.; Lei, J.P.; Li, L.; Su, D.F.; Zheng, X. Resveratrol, an Activator of SIRT1, Upregulates AMPK and Improves Cardiac Function in Heart Failure. Genet. Mol. Res. 2014, 13, 323–335. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhou, Q.-Y.; Liu, D.; Yu, L.; Zhan, L.; Li, X.-J.; Peng, H.-Y.; Zhang, X.-L.; Yuan, X.-C. Advanced Glycation End-Products Impair Na+/K+-ATPase Activity in Diabetic Cardiomyopathy: Role of the Adenosine Monophosphate-Activated Protein Kinase/Sirtuin 1 Pathway. Clin. Exp. Pharm. Physiol. 2014, 41, 127–133. [Google Scholar] [CrossRef]
- Yan, D.; Luo, X.; Li, Y.; Liu, W.; Deng, J.; Zheng, N.; Gao, K.; Huang, Q.; Liu, J. Effects of Advanced Glycation End Products on Calcium Handling in Cardiomyocytes. CRD 2014, 129, 75–83. [Google Scholar] [CrossRef]
- Niggli, E. The Cardiac Sarcoplasmic Reticulum. Circ. Res. 2007, 100, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.H.; Herting, J.; Tirilomis, T.; Renner, A.; Neef, S.; Toischer, K.; Ellenberger, D.; Förster, A.; Schmitto, J.D.; Gummert, J.; et al. Ca2+/Calmodulin-Dependent Protein Kinase II and Protein Kinase A Differentially Regulate Sarcoplasmic Reticulum Ca2+ Leak in Human Cardiac Pathology. Circulation 2013, 128, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S.-I. RAGE-Aptamer Blocks the Development and Progression of Experimental Diabetic Nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [Green Version]
- de la Hoz, C.L.; Cheng, C.; Fernyhough, P.; Zochodne, D.W. A Model of Chronic Diabetic Polyneuropathy: Benefits from Intranasal Insulin Are Modified by Sex and RAGE Deletion. Am. J. Physiol.Endocrinol. Metab. 2017, 312, E407–E419. [Google Scholar] [CrossRef]
- Yamagishi, S.-I.; Matsui, T. Pathologic Role of Dietary Advanced Glycation End Products in Cardiometabolic Disorders, and Therapeutic Intervention. Nutrition 2016, 32, 157–165. [Google Scholar] [CrossRef]
- Monnier, V.M.; Bautista, O.; Kenny, D.; Sell, D.R.; Fogarty, J.; Dahms, W.; Cleary, P.A.; Lachin, J.; Genuth, S. Skin Collagen Glycation, Glycoxidation, and Crosslinking Are Lower in Subjects with Long-Term Intensive versus Conventional Therapy of Type 1 Diabetes: Relevance of Glycated Collagen Products versus HbA1c as Markers of Diabetic Complications. DCCT Skin Collagen Ancillary Study Group. Diabetes Control and Complications Trial. Diabetes 1999, 48, 870–880. [Google Scholar] [CrossRef]
- Meerwaldt, R.; Links, T.; Graaff, R.; Thorpe, S.R.; Baynes, J.W.; Hartog, J.; Gans, R.; Smit, A. Simple Noninvasive Measurement of Skin Autofluorescence. Ann. N. Y. Acad. Sci. 2005, 1043, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Siriopol, D.; Hogas, S.; Veisa, G.; Mititiuc, I.; Volovat, C.; Apetrii, M.; Onofriescu, M.; Busila, I.; Oleniuc, M.; Covic, A. Tissue Advanced Glycation End Products (AGEs), Measured by Skin Autofluorescence, Predict Mortality in Peritoneal Dialysis. Int. Urol. Nephrol. 2015, 47, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, S.; Hartog, J.W.L.; Heiner-Fokkema, M.R.; van Veldhuisen, D.J.; Voors, A.A. Advanced Glycation End-Products, a Pathophysiological Pathway in the Cardiorenal Syndrome. Heart Fail Rev. 2012, 17, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lapidos, K.A.; Gal-Moscovici, A.; Sprague, S.M.; Ameer, G.A. A Receptor-Based Bioadsorbent to Target Advanced Glycation End Products in Chronic Kidney Disease. Artif. Organs 2014, 38, 474–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, M.; Knani, I.; Bouzidi, H.; Berriche, O.; Hammami, M.; Kerkeni, M. Plasma Levels of Pentosidine, Carboxymethyl-Lysine, Soluble Receptor for Advanced Glycation End Products, and Metabolic Syndrome: The Metformin Effect. Dis. Markers 2016, 2016, 6248264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNair, E.; Qureshi, M.; Prasad, K.; Pearce, C. Atherosclerosis and the Hypercholesterolemic AGE–RAGE Axis. Int. J. Angiol. 2016, 25, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Scheijen, J.L.J.M.; van de Waarenburg, M.P.H.; Stehouwer, C.D.A.; Schalkwijk, C.G. Measurement of Pentosidine in Human Plasma Protein by a Single-Column High-Performance Liquid Chromatography Method with Fluorescence Detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 610–614. [Google Scholar] [CrossRef]
- Bird, I.M. High Performance Liquid Chromatography: Principles and Clinical Applications. BMJ 1989, 299, 783–787. [Google Scholar] [CrossRef] [Green Version]
- Vogeser, M.; Parhofer, K.G. Liquid Chromatography Tandem-Mass Spectrometry (LC-MS/MS)--Technique and Applications in Endocrinology. Exp. Clin. Endocrinol. Diabetes 2007, 115, 559–570. [Google Scholar] [CrossRef]
- Willemsen, S.; Hartog, J.W.L.; van Veldhuisen, D.J.; van der Meer, P.; Roze, J.F.; Jaarsma, T.; Schalkwijk, C.; van der Horst, I.C.C.; Hillege, H.L.; Voors, A.A. The Role of Advanced Glycation End-Products and Their Receptor on Outcome in Heart Failure Patients with Preserved and Reduced Ejection Fraction. Am. Heart J. 2012, 164, 742–749. [Google Scholar] [CrossRef]
- Vivekanadan-Giri, A.; Wang, J.H.; Byun, J.; Pennathur, S. Mass Spectrometric Quantification of Amino Acid Oxidation Products Identifies Oxidative Mechanisms of Diabetic End-Organ Damage. Rev. Endocr. Metab. Disord. 2008, 9, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic, R.; Futas, J.; Chandoga, J.; Jakus, V. Rapid and Simple Method for Determination of Nepsilon-(Carboxymethyl)Lysine and Nepsilon-(Carboxyethyl)Lysine in Urine Using Gas Chromatography/Mass Spectrometry. Biomed. Chromatogr. 2005, 19, 649–654. [Google Scholar] [CrossRef]
- Thornalley, P.J. Measurement of Protein Glycation, Glycated Peptides, and Glycation Free Adducts. Perit. Dial. Int. 2005, 25, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Maciel, E.; da Silva, R.N.; Simões, C.; Melo, T.; Ferreira, R.; Domingues, P.; Domingues, M.R.M. Liquid Chromatography-Tandem Mass Spectrometry of Phosphatidylserine Advanced Glycated End Products. Chem. Phys. Lipids 2013, 174, 1–7. [Google Scholar] [CrossRef]
- Perkins, B.A.; Rabbani, N.; Weston, A.; Ficociello, L.H.; Adaikalakoteswari, A.; Niewczas, M.; Warram, J.; Krolewski, A.S.; Thornalley, P. Serum Levels of Advanced Glycation Endproducts and Other Markers of Protein Damage in Early Diabetic Nephropathy in Type 1 Diabetes. PLoS ONE 2012, 7, e35655. [Google Scholar] [CrossRef]
- Jaisson, S.; Souchon, P.-F.; Desmons, A.; Salmon, A.-S.; Delemer, B.; Gillery, P. Early Formation of Serum Advanced Glycation End-Products in Children with Type 1 Diabetes Mellitus: Relationship with Glycemic Control. J. Pediatr. 2016, 172, 56–62. [Google Scholar] [CrossRef]
- Meerwaldt, R.; Graaff, R.; Oomen, P.H.N.; Links, T.P.; Jager, J.J.; Alderson, N.L.; Thorpe, S.R.; Baynes, J.W.; Gans, R.O.B.; Smit, A.J. Simple Non-Invasive Assessment of Advanced Glycation Endproduct Accumulation. Diabetologia 2004, 47, 1324–1330. [Google Scholar] [CrossRef] [Green Version]
- Hricik, D.E.; Wu, Y.C.; Schulak, A.; Friedlander, M.A. Disparate Changes in Plasma and Tissue Pentosidine Levels after Kidney and Kidney-Pancreas Transplantation. Clin. Transpl. 1996, 10, 568–573. [Google Scholar]
- Monnier, V.M.; Sell, D.R.; Strauch, C.; Sun, W.; Lachin, J.M.; Cleary, P.A.; Genuth, S. The Association between Skin Collagen Glucosepane and Past Progression of Microvascular and Neuropathic Complications in Type 1 Diabetes. J. Diabetes Complicat. 2013, 27, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Januszewski, A.S.; Sachithanandan, N.; Karschimkus, C.; O’Neal, D.N.; Yeung, C.K.; Alkatib, N.; Jenkins, A.J. Non-Invasive Measures of Tissue Autofluorescence Are Increased in Type 1 Diabetes Complications and Correlate with a Non-Invasive Measure of Vascular Dysfunction. Diabet. Med. 2012, 29, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Genuth, S.; Sun, W.; Cleary, P.; Sell, D.R.; Dahms, W.; Malone, J.; Sivitz, W.; Monnier, V.M. Glycation and Carboxymethyllysine Levels in Skin Collagen Predict the Risk of Future 10-Year Progression of Diabetic Retinopathy and Nephropathy in the Diabetes Control and Complications Trial and Epidemiology of Diabetes Interventions and Complications Participants With Type 1 Diabetes. Diabetes 2005, 54, 3103–3111. [Google Scholar]
- van der Heyden, J.C.; Birnie, E.; Mul, D.; Bovenberg, S.; Veeze, H.J.; Aanstoot, H.-J. Increased Skin Autofluorescence of Children and Adolescents with Type 1 Diabetes despite a Well-Controlled HbA1c: Results from a Cohort Study. BMC Endocr. Disord. 2016, 16, 49. [Google Scholar] [CrossRef] [Green Version]
- van Waateringe, R.P.; Slagter, S.N.; van der Klauw, M.M.; van Vliet-Ostaptchouk, J.V.; Graaff, R.; Paterson, A.D.; Lutgers, H.L.; Wolffenbuttel, B.H.R. Lifestyle and Clinical Determinants of Skin Autofluorescence in a Population-Based Cohort Study. Eur. J. Clin. Investig. 2016, 46, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Rajaobelina, K.; Helmer, C.; Vélayoudom-Céphise, F.-L.; Nov, S.; Farges, B.; Pupier, E.; Blanco, L.; Hugo, M.; Gin, H.; Rigalleau, V. Progression of Skin Autofluorescence of AGEs over 4 Years in Patients with Type 1 Diabetes. Diabetes/Metab. Res. Rev. 2017, 33, e2917. [Google Scholar] [CrossRef]
- Kouidrat, Y.; Zaitouni, A.; Amad, A.; Diouf, M.; Desailloud, R.; Loas, G.; Lalau, J.-D. Skin Autofluorescence (a Marker for Advanced Glycation End Products) and Erectile Dysfunction in Diabetes. J. Diabetes Complicat. 2017, 31, 108–113. [Google Scholar] [CrossRef]
- Temma, J.; Matsuhisa, M.; Horie, T.; Kuroda, A.; Mori, H.; Tamaki, M.; Endo, I.; Aihara, K.; Abe, M.; Matsumoto, T. Non-Invasive Measurement of Skin Autofluorescence as a Beneficial Surrogate Marker for Atherosclerosis in Patients with Type 2 Diabetes. J. Med. Investig. 2015, 62, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzijl, N.; DeGroot, J.; Thorpe, S.R.; Bank, R.A.; Shaw, J.N.; Lyons, T.J.; Bijlsma, J.W.; Lafeber, F.P.; Baynes, J.W.; TeKoppele, J.M. Effect of Collagen Turnover on the Accumulation of Advanced Glycation End Products. J. Biol. Chem. 2000, 275, 39027–39031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugisawa, E.; Miura, J.; Iwamoto, Y.; Uchigata, Y. Skin Autofluorescence Reflects Integration of Past Long-Term Glycemic Control in Patients with Type 1 Diabetes. Diabetes Care 2013, 36, 2339–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-C.; Wang, Y.-C.; Wang, G.-J.; Shen, M.-Y.; Chang, Y.-L.; Liou, S.-Y.; Chen, H.-C.; Lee, A.-S.; Chang, K.-C.; Chen, W.-Y.; et al. Skin Autofluorescence Is Associated with Inappropriate Left Ventricular Mass and Diastolic Dysfunction in Subjects at Risk for Cardiovascular Disease. Cardiovasc. Diabetol. 2017, 16, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koetsier, M.; Nur, E.; Chunmao, H.; Lutgers, H.L.; Links, T.P.; Smit, A.J.; Rakhorst, G.; Graaff, R. Skin Color Independent Assessment of Aging Using Skin Autofluorescence. Opt. Express 2010, 18, 14416–14429. [Google Scholar] [CrossRef] [Green Version]
- Brenner, M.; Hearing, V.J. The Protective Role of Melanin Against UV Damage in Human Skin. Photochem. Photobiol. 2008, 84, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Báez, E.A.; Shah, S.; Felipe, D.; Maynard, J.; Chalew, S. Correlation of Advanced Glycation Endproducts Estimated From Skin Fluorescence in First-Degree Relatives. J. Diabetes Sci. Technol. 2014, 9, 278–281. [Google Scholar] [CrossRef] [Green Version]
- Nenna, A.; Nappi, F.; Avtaar Singh, S.S.; Sutherland, F.W.; Di Domenico, F.; Chello, M.; Spadaccio, C. Pharmacologic Approaches Against Advanced Glycation End Products (AGEs) in Diabetic Cardiovascular Disease. Res. Cardiovasc. Med. 2015, 4. [Google Scholar] [CrossRef]
- Prasad, C.; Davis, K.E.; Imrhan, V.; Juma, S.; Vijayagopal, P. Advanced Glycation End Products and Risks for Chronic Diseases: Intervening Through Lifestyle Modification. Am. J. Lifestyle Med. 2019, 13, 384–404. [Google Scholar] [CrossRef] [PubMed]
- van Waateringe, R.P.; Mook-Kanamori, M.J.; Slagter, S.N.; van der Klauw, M.M.; van der Vliet-Ostaptchouk, J.V.; Graaff, R.; Lutgers, H.L.; Suhre, K.; Selim, M.M.E.-D.; Mook-Kanamori, D.O.; et al. The Association between Various Smoking Behaviors, Cotinine Biomarkers and Skin Autofluorescence, a Marker for Advanced Glycation End Product Accumulation. PLoS ONE 2017, 12, e0179330. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Dhar, I.; Caspar-Bell, G. Role of Advanced Glycation End Products and Its Receptors in the Pathogenesis of Cigarette Smoke-Induced Cardiovascular Disease. Int. J. Angiol. 2015, 24, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.-S.; Park, S.; Kim, J. The Role of Glycation in the Pathogenesis of Aging and Its Prevention through Herbal Products and Physical Exercise. J. Exerc. Nutr. Biochem. 2017, 21, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Borg, D.J.; Forbes, J.M. Targeting Advanced Glycation with Pharmaceutical Agents: Where Are We Now? Glycoconj. J. 2016, 33, 653–670. [Google Scholar] [CrossRef]
- Garg, S.; Syngle, A.; Vohra, K. Efficacy and Tolerability of Advanced Glycation End-Products Inhibitor in Osteoarthritis: A Randomized, Double-Blind, Placebo-Controlled Study. Clin. J. Pain. 2013, 29, 717–724. [Google Scholar] [CrossRef]
- Mirmiranpour, H.; Mousavizadeh, M.; Noshad, S.; Ghavami, M.; Ebadi, M.; Ghasemiesfe, M.; Nakhjavani, M.; Esteghamati, A. Comparative Effects of Pioglitazone and Metformin on Oxidative Stress Markers in Newly Diagnosed Type 2 Diabetes Patients: A Randomized Clinical Trial. J. Diabetes Complicat. 2013, 27, 501–507. [Google Scholar] [CrossRef]
- Derosa, G.; Bonaventura, A.; Romano, D.; Bianchi, L.; Fogari, E.; D’Angelo, A.; Maffioli, P. Enalapril/Lercanidipine Combination on Markers of Cardiovascular Risk: A Randomized Study. J. Am. Soc. Hypertens. 2014, 8, 422–428. [Google Scholar] [CrossRef]
- Contreras, C.L.; Guzman-Rosiles, I.; Castillo, D.D.; Gomez-Ojeda, A.; Garay-Sevilla, M.E. Advanced Glycation End Products (AGEs) and SRAGE Levels after Benfotiamine Treatment in Diabetes Mellitus Type 2. FASEB J. 2017, 31, 646.32. [Google Scholar] [CrossRef]
- Fujimoto, N.; Hastings Jeffrey, L.; Carrick-Ranson, G.; Shafer Keri, M.; Shibata, S.; Bhella Paul, S.; Abdullah Shuaib, M.; Barkley Kyler, W.; Adams-Huet, B.; Boyd Kara, N.; et al. Cardiovascular Effects of 1 Year of Alagebrium and Endurance Exercise Training in Healthy Older Individuals. Circ. Heart Fail. 2013, 6, 1155–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudegeest-Sander, M.H.; Rikkert, M.G.M.O.; Smits, P.; Thijssen, D.H.J.; van Dijk, A.P.J.; Levine, B.D.; Hopman, M.T.E. The Effect of an Advanced Glycation End-Product Crosslink Breaker and Exercise Training on Vascular Function in Older Individuals: A Randomized Factorial Design Trial. Exp. Gerontol. 2013, 48, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yubero-Serrano, E.M.; Woodward, M.; Poretsky, L.; Vlassara, H.; Striker, G.E. AGE-less Study Group Effects of Sevelamer Carbonate on Advanced Glycation End Products and Antioxidant/pro-Oxidant Status in Patients with Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2015, 10, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Ueda, S.; Yamagishi, S.; Takeuchi, M.; Kohno, K.; Shibata, R.; Matsumoto, Y.; Kaneyuki, U.; Fujimura, T.; Hayashida, A.; Okuda, S. Oral Adsorbent AST–120 Decreases Serum Levels of AGEs in Patients with Chronic Renal Failure. Mol. Med. 2006, 12, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Inoue, H.; Takeuchi, M. Oral Administration of AST-120 (Kremezin) Is a Promising Therapeutic Strategy for Advanced Glycation End Product (AGE)-Related Disorders. Med. Hypotheses 2007, 69, 666–668. [Google Scholar] [CrossRef]
- Habbous, S.; Przech, S.; Acedillo, R.; Sarma, S.; Garg, A.X.; Martin, J. The Efficacy and Safety of Sevelamer and Lanthanum versus Calcium-Containing and Iron-Based Binders in Treating Hyperphosphatemia in Patients with Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Nephrol. Dial. Transpl. 2017, 32, 111–125. [Google Scholar] [CrossRef] [Green Version]
- Vlassara, H.; Uribarri, J.; Cai, W.; Goodman, S.; Pyzik, R.; Post, J.; Grosjean, F.; Woodward, M.; Striker, G.E. Effects of Sevelamer on HbA1c, Inflammation, and Advanced Glycation End Products in Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, D.; Brownlee, M. Mechanistic Studies of Advanced Glycosylation End Product Inhibition by Aminoguanidine. Diabetes 1992, 41, 26–29. [Google Scholar] [CrossRef]
- Brownlee, M.; Vlassara, H.; Kooney, A.; Ulrich, P.; Cerami, A. Aminoguanidine Prevents Diabetes-Induced Arterial Wall Protein Cross-Linking. Science 1986, 232, 1629–1632. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, M.M.; Bavkar, L.N.; Sistla, S.; Arvindekar, A.U. Effective Inhibition of Protein Glycation by Combinatorial Usage of Limonene and Aminoguanidine through Differential and Synergistic Mechanisms. Int. J. Biol. Macromol. 2017, 99, 563–569. [Google Scholar] [CrossRef]
- Thornalley, P.J. Use of Aminoguanidine (Pimagedine) to Prevent the Formation of Advanced Glycation Endproducts. Arch Biochem Biophys 2003, 419, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Soulis-Liparota, T.; Cooper, M.; Papazoglou, D.; Clarke, B.; Jerums, G. Retardation by Aminoguanidine of Development of Albuminuria, Mesangial Expansion, and Tissue Fluorescence in Streptozocin-Induced Diabetic Rat. Diabetes 1991, 40, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Yagihashi, S.; Kamijo, M.; Baba, M.; Yagihashi, N.; Nagai, K. Effect of Aminoguanidine on Functional and Structural Abnormalities in Peripheral Nerve of STZ-Induced Diabetic Rats. Diabetes 1992, 41, 47–52. [Google Scholar] [CrossRef]
- Sampath, C.; Zhu, Y.; Sang, S.; Ahmedna, M. Bioactive Compounds Isolated from Apple, Tea, and Ginger Protect against Dicarbonyl Induced Stress in Cultured Human Retinal Epithelial Cells. Phytomedicine 2016, 23, 200–213. [Google Scholar] [CrossRef]
- Tanaka, Y.; Uchino, H.; Shimizu, T.; Yoshii, H.; Niwa, M.; Ohmura, C.; Mitsuhashi, N.; Onuma, T.; Kawamori, R. Effect of Metformin on Advanced Glycation Endproduct Formation and Peripheral Nerve Function in Streptozotocin-Induced Diabetic Rats. Eur. J. Pharm. 1999, 376, 17–22. [Google Scholar] [CrossRef]
- Engelen, L.; Stehouwer, C.D.A.; Schalkwijk, C.G. Current Therapeutic Interventions in the Glycation Pathway: Evidence from Clinical Studies. Diabetes Obes. Metab. 2013, 15, 677–689. [Google Scholar] [CrossRef]
- Jang, D.S.; Kim, J.M.; Kim, J.; Yoo, J.L.; Kim, Y.S.; Kim, J.S. Effects of Compounds Isolated from the Fruits of Rumex Japonicus on the Protein Glycation. Chem. Biodivers. 2008, 5, 2718–2723. [Google Scholar] [CrossRef]
- Starowicz, M.; Zieliński, H. Inhibition of Advanced Glycation End-Product Formation by High Antioxidant-Leveled Spices Commonly Used in European Cuisine. Antioxidants 2019, 8, 100. [Google Scholar] [CrossRef] [Green Version]
- Gugliucci, A.; Bastos, D.H.M.; Schulze, J.; Souza, M.F.F. Caffeic and Chlorogenic Acids in Ilex Paraguariensis Extracts Are the Main Inhibitors of AGE Generation by Methylglyoxal in Model Proteins. Fitoterapia 2009, 80, 339–344. [Google Scholar] [CrossRef]
- Perez Gutierrez, R.M.; Flores Cotera, L.B.; Gonzalez, A.M.N. Evaluation of the Antioxidant and Anti-Glication Effects of the Hexane Extract from Piper Auritum Leaves in Vitro and Beneficial Activity on Oxidative Stress and Advanced Glycation End-Product-Mediated Renal Injury in Streptozotocin-Treated Diabetic Rats. Molecules 2012, 17, 1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, W.-J.; Hsia, S.-M.; Lee, W.-H.; Wu, C.-H. Polyphenols with Antiglycation Activity and Mechanisms of Action: A Review of Recent Findings. J. Food Drug Anal. 2017, 25, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, R.; Yao, H.; Wu, Y.; Pan, W.; Jia, A.-Q. Inhibiting the Formation of Advanced Glycation End-Products by Three Stilbenes and the Identification of Their Adducts. Food Chem. 2019, 295, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E. The Target of Metformin in Type 2 Diabetes. N. Engl. J. Med. 2014, 371, 1547–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Morales, N.; Rovira-Llopis, S.; Bañuls, C.; Lopez-Domenech, S.; Escribano-Lopez, I.; Veses, S.; Jover, A.; Rocha, M.; Hernandez-Mijares, A.; Victor, V.M. Does Metformin Protect Diabetic Patients from Oxidative Stress and Leukocyte-Endothelium Interactions? Antioxid. Redox Signal 2017, 27, 1439–1445. [Google Scholar] [CrossRef]
- Esteghamati, A.; Eskandari, D.; Mirmiranpour, H.; Noshad, S.; Mousavizadeh, M.; Hedayati, M.; Nakhjavani, M. Effects of Metformin on Markers of Oxidative Stress and Antioxidant Reserve in Patients with Newly Diagnosed Type 2 Diabetes: A Randomized Clinical Trial. Clin. Nutr. 2013, 32, 179–185. [Google Scholar] [CrossRef]
- Ruggiero-Lopez, D.; Lecomte, M.; Moinet, G.; Patereau, G.; Lagarde, M.; Wiernsperger, N. Reaction of Metformin with Dicarbonyl Compounds. Possible Implication in the Inhibition of Advanced Glycation End Product Formation. Biochem. Pharm. 1999, 58, 1765–1773. [Google Scholar] [CrossRef]
- Adeshara, K.; Tupe, R. Antiglycation and Cell Protective Actions of Metformin and Glipizide in Erythrocytes and Monocytes. Mol. Biol. Rep. 2016, 43, 195–205. [Google Scholar] [CrossRef]
- Metz, T.O.; Alderson, N.L.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine, an Inhibitor of Advanced Glycation and Lipoxidation Reactions: A Novel Therapy for Treatment of Diabetic Complications. Arch. Biochem. Biophys. 2003, 419, 41–49. [Google Scholar] [CrossRef]
- Hammes, H.-P.; Du, X.; Edelstein, D.; Taguchi, T.; Matsumura, T.; Ju, Q.; Lin, J.; Bierhaus, A.; Nawroth, P.; Hannak, D.; et al. Benfotiamine Blocks Three Major Pathways of Hyperglycemic Damage and Prevents Experimental Diabetic Retinopathy. Nat. Med. 2003, 9, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Voziyan, P.A.; Hudson, B.G. Pyridoxamine as a Multifunctional Pharmaceutical: Targeting Pathogenic Glycation and Oxidative Damage. Cell Mol. Life Sci. 2005, 62, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Deluyker, D.; Ferferieva, V.; Driesen, R.B.; Verboven, M.; Lambrichts, I.; Bito, V. Pyridoxamine Improves Survival and Limits Cardiac Dysfunction after MI. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pereira, A.; Fernandes, R.; Crisóstomo, J.; Seiça, R.M.; Sena, C.M. The Sulforaphane and Pyridoxamine Supplementation Normalize Endothelial Dysfunction Associated with Type 2 Diabetes. Sci. Rep. 2017, 7, 14357. [Google Scholar] [CrossRef] [Green Version]
- Nagai, R.; Murray, D.B.; Metz, T.O.; Baynes, J.W. Chelation: A Fundamental Mechanism of Action of AGE Inhibitors, AGE Breakers, and Other Inhibitors of Diabetes Complications. Diabetes 2012, 61, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced Glycoxidation and Lipoxidation End Products (AGEs and ALEs): An Overview of Their Mechanisms of Formation. Free Radic. Res. 2013, 47 (Suppl. 1), 3–27. [Google Scholar] [CrossRef] [Green Version]
- Šebeková, K.; Schinzel, R.; Münch, G.; Krivošíková, Z.; Dzúrik, R.; Heidland, A. Advanced Glycation End-Product Levels in Subtotally Nephrectomized Rats: Beneficial Effects of Angiotensin II Receptor 1 Antagonist Losartan. MEM 1999, 25, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Monacelli, F.; Poggi, A.; Storace, D.; Durante, A.; Traverso, N.; Viviani, G.L.; Odetti, P. Effects of Valsartan Therapy on Protein Glycoxidation. Metabolism 2006, 55, 1619–1624. [Google Scholar] [CrossRef]
- Miyata, T.; van Ypersele de Strihou, C. Angiotensin II Receptor Blockers and Angiotensin Converting Enzyme Inhibitors: Implication of Radical Scavenging and Transition Metal Chelation in Inhibition of Advanced Glycation End Product Formation. Arch. Biochem. Biophys. 2003, 419, 50–54. [Google Scholar] [CrossRef]
- Chang, P.-C.; Tsai, S.-C.; Chong, L.Y.; Kao, M.-J. N-Phenacylthiazolium Bromide Inhibits the Advanced Glycation End Product (AGE)-AGE Receptor Axis to Modulate Experimental Periodontitis in Rats. J. Periodontol. 2014, 85, e268–e276. [Google Scholar] [CrossRef] [PubMed]
- Wolffenbuttel, B.H.R.; Boulanger, C.M.; Crijns, F.R.L.; Huijberts, M.S.P.; Poitevin, P.; Swennen, G.N.M.; Vasan, S.; Egan, J.J.; Ulrich, P.; Cerami, A.; et al. Breakers of Advanced Glycation End Products Restore Large Artery Properties in Experimental Diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 4630–4634. [Google Scholar] [CrossRef] [Green Version]
- Vasan, S.; Zhang, X.; Zhang, X.; Kapurniotu, A.; Bernhagen, J.; Teichberg, S.; Basgen, J.; Wagle, D.; Shih, D.; Terlecky, I.; et al. An Agent Cleaving Glucose-Derived Protein Crosslinks in Vitro and in Vivo. Nature 1996, 382, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Bradke, B.S.; Vashishth, D. N-Phenacylthiazolium Bromide Reduces Bone Fragility Induced by Nonenzymatic Glycation. PLoS ONE 2014, 9, e103199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.Y.; Goddard, T.N.; Sohn, S.; Spiegel, D.A.; Crawford, J.M. Biocatalytic Reversal of Advanced Glycation End Product Modification. Chembiochem 2019, 20, 2402–2410. [Google Scholar] [CrossRef]
- Bakris, G.L.; Bank, A.J.; Kass, D.A.; Neutel, J.M.; Preston, R.A.; Oparil, S. Advanced Glycation End-Product Cross-Link Breakers. A Novel Approach to Cardiovascular Pathologies Related to the Aging Process. Am. J. Hypertens. 2004, 17, 23S–30S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, A.M.D.; Soro-Paavonen, A.; Sheehy, K.; Li, J.; Calkin, A.C.; Koitka, A.; Rajan, S.N.; Brasacchio, D.; Allen, T.J.; Cooper, M.E.; et al. Delayed Intervention with AGE Inhibitors Attenuates the Progression of Diabetes-Accelerated Atherosclerosis in Diabetic Apolipoprotein E Knockout Mice. Diabetologia 2011, 54, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Kranstuber, A.L.; Del Rio, C.; Biesiadecki, B.J.; Hamlin, R.L.; Ottobre, J.; Gyorke, S.; Lacombe, V.A. Advanced Glycation End Product Cross-Link Breaker Attenuates Diabetes-Induced Cardiac Dysfunction by Improving Sarcoplasmic Reticulum Calcium Handling. Front. Physiol. 2012, 3, 292. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; He, K.; Chen, W.; Cheng, X.; Cui, H.; Zhong, W.; Li, S.; Wang, L. Alagebrium (ALT-711) Improves the Anti-Hypertensive Efficacy of Nifedipine in Diabetic-Hypertensive Rats. Hypertens. Res. 2014, 37, 901–907. [Google Scholar] [CrossRef]
- Susic, D.; Varagic, J.; Frohlich, E.D. Cardiovascular and Renal Effects of a Collagen Cross-Link Breaker (ALT 711) in Adult and Aged Spontaneously Hypertensive Rats. Am. J. Hypertens. 2004, 17, 328–333. [Google Scholar] [CrossRef] [Green Version]
- Dozio, E.; Vianello, E.; Bandera, F.; Longhi, E.; Brizzola, S.; Nebuloni, M.; Corsi Romanelli, M.M. Soluble Receptor for Advanced Glycation End Products: A Protective Molecule against Intramyocardial Lipid Accumulation in Obese Zucker Rats? Mediat. Inflamm. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Koyama, H.; Yamamoto, H.; Nishizawa, Y. RAGE and Soluble RAGE: Potential Therapeutic Targets for Cardiovascular Diseases. Mol. Med. 2007, 13, 625–635. [Google Scholar] [CrossRef]
- Xu, L.; Zang, P.; Feng, B.; Qian, Q. Atorvastatin Inhibits the Expression of RAGE Induced by Advanced Glycation End Products on Aortas in Healthy Sprague-Dawley Rats. Diabetol. Metab. Syndr. 2014, 6, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuccurullo, C.; Iezzi, A.; Fazia, M.L.; De Cesare, D.; Di Francesco, A.; Muraro, R.; Bei, R.; Ucchino, S.; Spigonardo, F.; Chiarelli, F.; et al. Suppression of RAGE as a Basis of Simvastatin-Dependent Plaque Stabilization in Type 2 Diabetes. Arter. Thromb. Vasc. Biol. 2006, 26, 2716–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, N.; Walcher, D.; Ivanova, N.; Rautzenberg, K.; Jung, A.; Friedl, R.; Hombach, V.; de Caterina, R.; Basta, G.; Wautier, M.-P.; et al. Thiazolidinediones Reduce Endothelial Expression of Receptors for Advanced Glycation End Products. Diabetes 2004, 53, 2662–2668. [Google Scholar] [CrossRef] [Green Version]
- Sirtori, C.R. The Pharmacology of Statins. Pharm. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the Promise of Insulin Sensitization in Type 2 Diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Li, H.; Wang, G.; Shen, X.; Zhao, S.; Su, W. Atorvastatin Prevents Advanced Glycation End Products (AGEs)-Induced Cardiac Fibrosis via Activating Peroxisome Proliferator-Activated Receptor Gamma (PPAR-γ). Metabolism 2016, 65, 441–453. [Google Scholar] [CrossRef]
- Chiang, M.-C.; Cheng, Y.-C.; Nicol, C.J.; Lin, C.-H. The Neuroprotective Role of Rosiglitazone in Advanced Glycation End Product Treated Human Neural Stem Cells Is PPARgamma-Dependent. Int. J. Biochem. Cell Biol. 2017, 92, 121–133. [Google Scholar] [CrossRef]
- Sabbagh, M.N.; Agro, A.; Bell, J.; Aisen, P.S.; Schweizer, E.; Galasko, D. PF-04494700, an Oral Inhibitor of Receptor For Advanced Glycation End Products (RAGE), in Alzheimer’s Disease. Alzheimer. Dis. Assoc. Disord. 2011, 25, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Burstein, A.H.; Grimes, I.; Galasko, D.R.; Aisen, P.S.; Sabbagh, M.; Mjalli, A.M.M. Effect of TTP488 in Patients with Mild to Moderate Alzheimer’s Disease. BMC Neurol. 2014, 14, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Ward, M.; Stitt, A.W. AGEs, RAGE, and Diabetic Retinopathy. Curr. Diab. Rep. 2011, 11, 244–252. [Google Scholar] [CrossRef]
- Chen, S.; Yin, L.; Xu, Z.; An, F.-M.; Liu, A.-R.; Wang, Y.; Yao, W.-B.; Gao, X.-D. Inhibiting Receptor for Advanced Glycation End Product (AGE) and Oxidative Stress Involved in the Protective Effect Mediated by Glucagon-like Peptide-1 Receptor on AGE Induced Neuronal Apoptosis. Neurosci. Lett. 2016, 612, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-S.; Wu, Z.; Zhang, Z.; Xiong, Z.-Y.; Chen, H.; Huang, Q.-B. Glucagon-like Peptide-1 Inhibits the Receptor for Advanced Glycation Endproducts to Prevent Podocyte Apoptosis Induced by Advanced Oxidative Protein Products. Biochem. Biophys. Res. Commun. 2017, 482, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Dorecka, M.; Siemianowicz, K.; Francuz, T.; Garczorz, W.; Chyra, A.; Klych, A.; Romaniuk, W. Exendin-4 and GLP-1 Decreases Induced Expression of ICAM-1, VCAM-1 and RAGE in Human Retinal Pigment Epithelial Cells. Pharm. Rep. 2013, 65, 884–890. [Google Scholar] [CrossRef]
- Zhan, Y.; Sun, H.; Chen, H.; Zhang, H.; Sun, J.; Zhang, Z.; Cai, D. Glucagon-like Peptide-1 (GLP-1) Protects Vascular Endothelial Cells against Advanced Glycation End Products (AGEs) Induced Apoptosis. Med. Sci. Monit. 2012, 18, BR286–BR291. [Google Scholar] [CrossRef] [Green Version]
- Yi, B.; Hu, X.; Wen, Z.; Zhang, T.; Cai, Y. Exendin-4, a Glucagon-like Peptide-1 Receptor Agonist, Inhibits Hyperglycemia-induced Apoptosis in Myocytes by Suppressing Receptor for Advanced Glycation End Products Expression. Exp. Ther. Med. 2014, 8, 1185–1190. [Google Scholar] [CrossRef] [Green Version]
- Bolton, W.K.; Cattran, D.C.; Williams, M.E.; Adler, S.G.; Appel, G.B.; Cartwright, K.; Foiles, P.G.; Freedman, B.I.; Raskin, P.; Ratner, R.E.; et al. Randomized Trial of an Inhibitor of Formation of Advanced Glycation End Products in Diabetic Nephropathy. Am. J. Nephrol. 2004, 24, 32–40. [Google Scholar] [CrossRef]
- Suji, G.; Sivakami, S. DNA Damage by Free Radical Production by Aminoguanidine. Ann. N. Y. Acad. Sci. 2006, 1067, 191–199. [Google Scholar] [CrossRef]
- Tilton, R.G.; Chang, K.; Hasan, K.S.; Smith, S.R.; Petrash, J.M.; Misko, T.P.; Moore, W.M.; Currie, M.G.; Corbett, J.A.; McDaniel, M.L. Prevention of Diabetic Vascular Dysfunction by Guanidines. Inhibition of Nitric Oxide Synthase versus Advanced Glycation End-Product Formation. Diabetes 1993, 42, 221–232. [Google Scholar] [CrossRef]
- Sakata, K.; Hayakawa, M.; Yano, Y.; Tamaki, N.; Yokota, N.; Eto, T.; Watanabe, R.; Hirayama, N.; Matsuo, T.; Kuroki, K.; et al. Efficacy of Alogliptin, a Dipeptidyl Peptidase-4 Inhibitor, on Glucose Parameters, the Activity of the Advanced Glycation End Product (AGE) Receptor for AGE (RAGE) Axis and Albuminuria in Japanese Type 2 Diabetes. Diabetes Metab. Res. Rev. 2013, 29, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Koyama, H.; Tanaka, S.; Monden, M.; Shoji, T.; Morioka, T.; Fukumoto, S.; Mori, K.; Emoto, M.; Shoji, T.; Fukui, M.; et al. Comparison of Effects of Pioglitazone and Glimepiride on Plasma Soluble RAGE and RAGE Expression in Peripheral Mononuclear Cells in Type 2 Diabetes: Randomized Controlled Trial (PioRAGE). Atherosclerosis 2014, 234, 329–334. [Google Scholar] [CrossRef]
- Liu, J.-S.; Chuang, T.-J.; Chen, J.-H.; Lee, C.-H.; Hsieh, C.-H.; Lin, T.-K.; Hsiao, F.-C.; Hung, Y.-J. Cilostazol Attenuates the Severity of Peripheral Arterial Occlusive Disease in Patients with Type 2 Diabetes: The Role of Plasma Soluble Receptor for Advanced Glycation End-Products. Endocrine 2015, 49, 703–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Park, H.-S.; Tang, L.A.; Horwitz, N.; Lin, L. Generation of SRAGEhigh Transgenic Mice to Study Inflammaging. Front. Biosci. 2019, 24, 555–563. [Google Scholar]
- Wang, B.-J.; Qian, L.; Li, J.; Wang, F.; Yang, Q.-L.; Li, G.; Liang, Y.-L.; Guo, Y.-H. SRAGE Plays a Role as a Protective Factor in the Development of PCOS by Inhibiting Inflammation. Gynecol. Endocrinol. 2020, 36, 148–151. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism of Action | Drug | Methodology | Results | Author [REF] |
|---|---|---|---|---|
| Inhibition of the endogenous formation of AGEs | Benfotiamine and Pyridoxamine | Randomized, double-blinded controlled by placebo trial, which included 30 patients with primary osteoarthritis divided randomly between two groups to receive tablets of inhibitors of AGEs (benfotiamine (50 mg) + PM (50 mg) + methylcobalamin (500 mg)) or placebo tablets, three times a day. | Significant decrease in serum levels and fluorescence of AGEs. Decreased pain and inflammation. Increase in daily activity and mobility in patients with osteoarthritis. | Garg S et al. (2013) [130] |
| Pioglitazone and Metformin | Randomized, open parallel-groups trial, performed in patients recently diagnosticated with DM2, who were given 30 mg/day of pioglitazone (n = 30), 1000 mg/day of metformin (n = 50) or not any drugs (n = 49). | In both treated groups with either pioglitazone or metformin was observed a statistically significant decrease in levels of AOPP and AGEs, besides causing an increase in FRAP (a marker of plasma antioxidant capacity). | Mirmiranpour H et al. (2013) [131] | |
| Enalapril and Lercanidipine | Randomized, double-blinded trial, which included 359 ambulatory patients <65 years of age, first-diagnosed with essential hypertension and without treatment, divided between three groups, who were randomly given: enalapril 20 mg/day (n = 126), lercanidipine 10 mg/day (n = 115), or enalapril + lercanidipine 20/10 mg/day (118), in order to assess their effects in markers of cardiovascular risk. | All treatments showed a significant increase in levels of sRAGE, which was higher in patients treated with enalapril + lercanidipine.Significant reduction of levels of TNF-α and US-CRP in patients treated with enalapril + lercanidipine | Derosa G et al. (2014) [132] | |
| Benfotiamine | Randomized, double-blinded, controlled trial, which included 41 patients with DM2 without complications, who were randomly given 900 mg/day of benfotiamine or 900 mg/day of a placebo, to assess their effect on levels of AGEs and sRAGE. | Patients treated with benfotiamine had a statistically significant decrease in levels of carboxymethyl-lysine. There were no statistically significant differences in levels of sRAGE. | Contreras C et al. (2017) [133] | |
| Breakage and Reversal of preformed AGEs | Alagebrium (ALT-711) | Randomized, double-blinded, controlled by placebo prospective study, performed in 57 healthy subjects over 60 years of age, which were randomly divided between 4 groups: sedentary + placebo, sedentary + alagebrium (200 mg/day), exercise + placebo, and exercise + alagebrium in order to assess their effect in hemodynamics, function, and structure of the left ventricle. | Alagebrium led to a moderate improvement in rigidity of the left ventricle, which was more prominent when it was combined with physical activity. | Fujimoto N et al. (2013) [134] |
| Alagebrium | Randomized, controlled by placebo trial, which included 47 older subjects previously sedentary, who were divided between 4 types of interventions: Exercise + Alagebrium (200 mg/day), Exercise + Placebo, Alagebrium (200 mg/day), and Exercise aiming to examine their effect over endothelial function, arterial stiffness, and cardiovascular risk. | There were no improvements in endothelial function or arterial stiffness in any of the four groups. | Oudegeest-Sander M et al. (2013) [135] | |
| Inhibition of the absorption of exogenous AGEs | Sevelamer carbonate | Randomized, open, single-blinded trial, which included 117 patients with T2DM and diabetic nephropathy in stages 2 to 4, who were given sevelamer carbonate (1600 mg) or calcium carbonate (1200 mg), three times a day, to measure their effects in the AGE–RAGE axis and onto oxidative stress. | Patients treated with sevelamer carbonate showed a significant reduction of both circulating and intracellular AGEs (carboxymethyl-lysine and methylglyoxal).It was also observed a significant increase in antioxidant defenses and reduction of pro-oxidant molecules. | Yubero-Serrano et al. (2015) [136] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salazar, J.; Navarro, C.; Ortega, Á.; Nava, M.; Morillo, D.; Torres, W.; Hernández, M.; Cabrera, M.; Angarita, L.; Ortiz, R.; et al. Advanced Glycation End Products: New Clinical and Molecular Perspectives. Int. J. Environ. Res. Public Health 2021, 18, 7236. https://doi.org/10.3390/ijerph18147236
Salazar J, Navarro C, Ortega Á, Nava M, Morillo D, Torres W, Hernández M, Cabrera M, Angarita L, Ortiz R, et al. Advanced Glycation End Products: New Clinical and Molecular Perspectives. International Journal of Environmental Research and Public Health. 2021; 18(14):7236. https://doi.org/10.3390/ijerph18147236
Chicago/Turabian StyleSalazar, Juan, Carla Navarro, Ángel Ortega, Manuel Nava, Daniela Morillo, Wheeler Torres, Marlon Hernández, Mayela Cabrera, Lissé Angarita, Rina Ortiz, and et al. 2021. "Advanced Glycation End Products: New Clinical and Molecular Perspectives" International Journal of Environmental Research and Public Health 18, no. 14: 7236. https://doi.org/10.3390/ijerph18147236
APA StyleSalazar, J., Navarro, C., Ortega, Á., Nava, M., Morillo, D., Torres, W., Hernández, M., Cabrera, M., Angarita, L., Ortiz, R., Chacín, M., D’Marco, L., & Bermúdez, V. (2021). Advanced Glycation End Products: New Clinical and Molecular Perspectives. International Journal of Environmental Research and Public Health, 18(14), 7236. https://doi.org/10.3390/ijerph18147236