
Evidence for Multiple Origins of De Novo Formed Vascular Smooth Muscle Cells in Pulmonary Hypertension: Challenging the Dominant Model of Pre-Existing Smooth Muscle Expansion

 ,
, 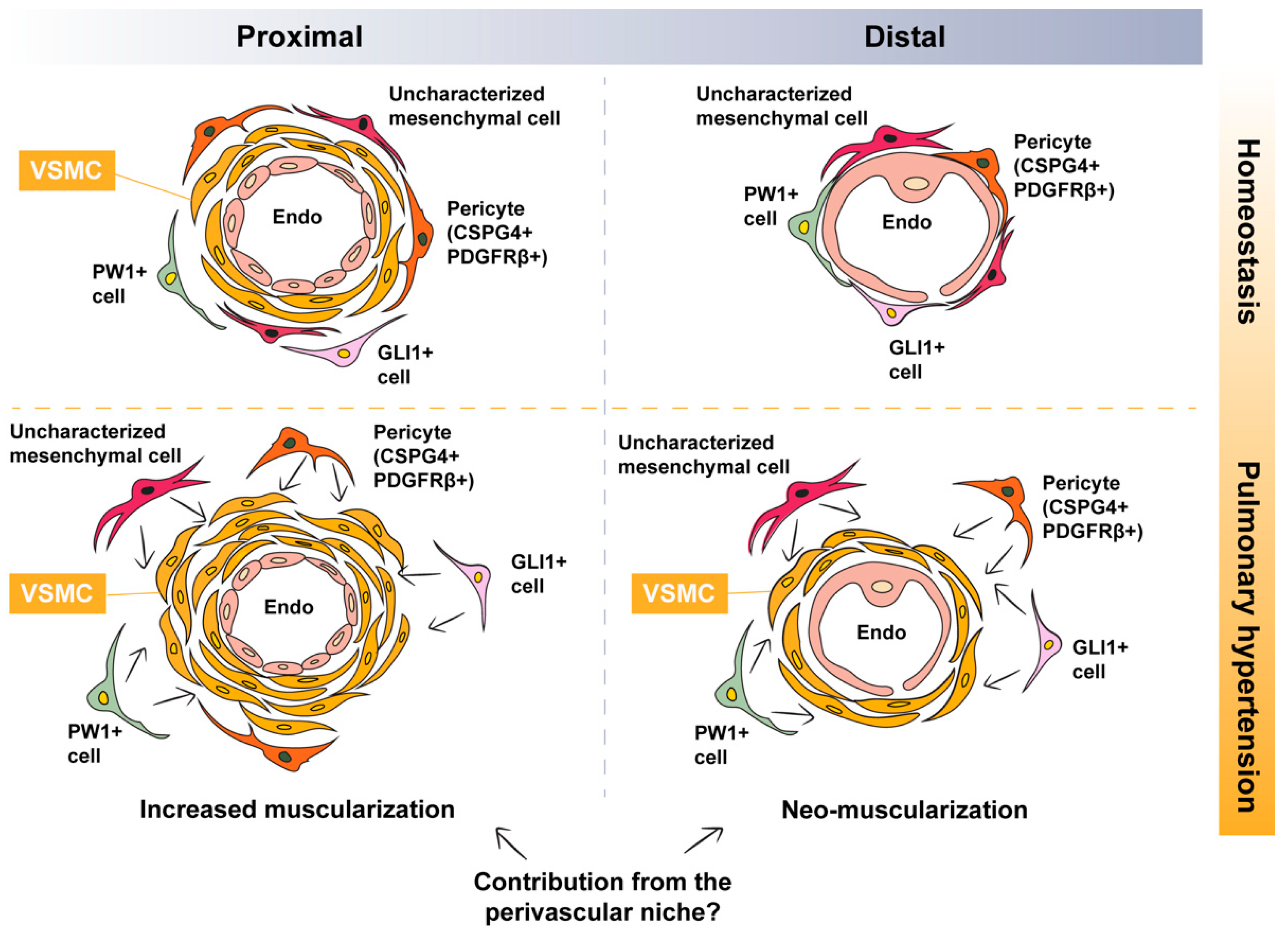{kind=link}
Abstract
:1. Introduction
2. Resident Vascular Smooth Muscle Cells
3. Endothelial-to-Mesenchymal Transition (EndMT)
4. Argument for the Contribution of Perivascular Mesenchymal (Progenitor) Cells
4.1. Are Pericytes a Source of PH-Associated VSMCs?
4.2. Are Mesenchymal Stem Cells Also Involved in Vascular Remodeling?
5. Insights from Other Non-Hypoxia-Driven PH Models
5.1. The Monocrotaline Model
5.2. The Sugen/Hypoxia Model
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the european society of cardiology (ESC) and the european respiratory society (ERS): Endorsed by: Association for european paediatric and congenital cardiology (AEPC), International society for heart and lung transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [Green Version]
- Schermuly, R.T.; Ghofrani, H.A.; Wilkins, M.R.; Grimminger, F. Mechanisms of disease: Pulmonary arterial hypertension. Nat. Rev. Cardiol. 2011, 8, 443–455. [Google Scholar] [CrossRef]
- Thenappan, T.; Chan, S.Y.; Weir, E.K. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1322–H1331. [Google Scholar] [CrossRef] [PubMed]
- Marsh, L.M.; Jandl, K.; Grünig, G.; Foris, V.; Bashir, M.; Ghanim, B.; Klepetko, W.; Olschewski, H.; Olschewski, A.; Kwapiszewska, G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1701214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolls, M.R.; Voelkel, N.F. The Roles of Immunity in the prevention and evolution of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2017, 195, 1292–1299. [Google Scholar] [CrossRef] [Green Version]
- Moiseenko, A.; Vazquez-Armendariz, A.I.; Kheirollahi, V.; Chu, X.; Tata, A.; Rivetti, S.; Günther, S.; Lebrigand, K.; Herold, S.; Braun, T.; et al. Identification of a repair-supportive mesenchymal cell population during airway epithelial regeneration. Cell Rep. 2020, 33, 108549. [Google Scholar] [CrossRef] [PubMed]
- Volckaert, T.; Dill, E.; Campbell, A.; Tiozzo, C.; Majka, S.; Bellusci, S.; De Langhe, S. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Investig. 2011, 121, 4409–4419. [Google Scholar] [CrossRef] [Green Version]
- El Agha, E.; Herold, S.; Al Alam, D.; Quantius, J.; MacKenzie, B.; Carraro, G.; Moiseenko, A.; Chao, C.-M.; Minoo, P.; Seeger, W.; et al. Fgf10-positive cells represent a progenitor cell population during lung development and postnatally. Dev. Camb. Engl. 2014, 141, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Agha, E.; Kheirollahi, V.; Moiseenko, A.; Seeger, W.; Bellusci, S. Ex Vivo Analysis of the contribution of FGF10+ cells to airway smooth muscle cell formation during early lung development. Dev. Dyn. 2017, 246, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Crnkovic, S.; Marsh, L.M.; El Agha, E.; Voswinckel, R.; Ghanim, B.; Klepetko, W.; Stacher-Priehse, E.; Olschewski, H.; Bloch, W.; Bellusci, S.; et al. Resident cell lineages are preserved in pulmonary vascular remodeling. J. Pathol. 2018, 244, 485–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, A.Q.; Lighthouse, J.K.; Greif, D.M. Recapitulation of developing artery muscularization in pulmonary hypertension. Cell Rep. 2014, 6, 809–817. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, A.Q.; Misra, A.; Rosas, I.O.; Adams, R.H.; Greif, D.M. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci. Transl. Med. 2015, 7, 308ra159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntokou, A.; Dave, J.M.; Kauffman, A.C.; Sauler, M.; Ryu, C.; Hwa, J.; Herzog, E.L.; Singh, I.; Saltzman, W.M.; Greif, D.M. Macrophage-derived PDGF-B induces muscularization in murine and human pulmonary hypertension. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Steffes, L.C.; Froistad, A.A.; Andruska, A.; Boehm, M.; McGlynn, M.; Zhang, F.; Zhang, W.; Hou, D.; Tian, X.; Miquerol, L.; et al. A notch3-marked subpopulation of vascular smooth muscle cells is the cell of origin for occlusive pulmonary vascular lesions. Circulation 2020, 142, 1545–1561. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, E.; Sutton, A.B.; Allen, T.D.; Schor, A.M. Transforming growth factor beta 1 promotes the differentiation of endothelial cells into smooth muscle-like cells In Vitro. J. Cell Sci. 1992, 103, 521–529. [Google Scholar] [CrossRef]
- Frid, M.G.; Kale, V.A.; Stenmark, K.R. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: In Vitro analysis. Circ. Res. 2002, 90, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopper, R.K.; Moonen, J.-R.A.J.; Diebold, I.; Cao, A.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.; Wang, L.; Rabinovitch, M. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation 2016, 133, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Nishimura, T.; Shi, L.; Sessions, D.; Thrasher, A.; Trudell, J.R.; Berry, G.J.; Pearl, R.G.; Kao, P.N. Endothelial fate mapping in mice with pulmonary hypertension. Circulation 2014, 129, 692–703. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Carrier, E.J.; Talati, M.H.; Rathinasabapathy, A.; Chen, X.; Nishimura, R.; Tada, Y.; Tatsumi, K.; West, J. Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L118–L126. [Google Scholar] [CrossRef]
- Sakao, S.; Hao, H.; Tanabe, N.; Kasahara, Y.; Kurosu, K.; Tatsumi, K. Endothelial-like cells in chronic thromboembolic pulmonary hypertension: Crosstalk with myofibroblast-like cells. Respir. Res. 2011, 12, 109. [Google Scholar] [CrossRef] [Green Version]
- Greif, D.M.; Kumar, M.; Lighthouse, J.K.; Hum, J.; An, A.; Ding, L.; Red-Horse, K.; Espinoza, F.H.; Olson, L.; Offermanns, S.; et al. Radial construction of an arterial wall. Dev. Cell 2012, 23, 482–493. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Flores, L.; Gutiérrez, R.; Madrid, J.F.; Varela, H.; Valladares, F.; Acosta, E.; Martín-Vasallo, P.; Díaz-Flores, L. Pericytes. morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol. Histopathol. 2009, 24, 909–969. [Google Scholar] [CrossRef]
- El Agha, E.; Kramann, R.; Schneider, R.K.; Li, X.; Seeger, W.; Humphreys, B.D.; Bellusci, S. Mesenchymal Stem cells in fibrotic disease. Cell Stem Cell 2017, 21, 166–177. [Google Scholar] [CrossRef]
- Mills, S.J.; Cowin, A.J.; Kaur, P. Pericytes, mesenchymal stem cells and the wound healing process. Cells 2013, 2, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricard, N.; Tu, L.; Hiress, M.L.; Huertas, A.; Phan, C.; Thuillet, R.; Sattler, C.; Fadel, E.; Seferian, A.; Montani, D.; et al. Increased pericyte coverage mediated by endothelial-derived fibroblast growth factor-2 and interleukin-6 is a source of smooth muscle-like cells in pulmonary hypertension. Circulation 2014, 129, 1586–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordenave, J.; Tu, L.; Berrebeh, N.; Thuillet, R.; Cumont, A.; Le Vely, B.; Fadel, E.; Nadaud, S.; Savale, L.; Humbert, M.; et al. Lineage tracing reveals the dynamic contribution of pericytes to the blood vessel remodeling in pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 766–782. [Google Scholar] [CrossRef] [PubMed]
- Passman, J.N.; Dong, X.R.; Wu, S.-P.; Maguire, C.T.; Hogan, K.A.; Bautch, V.L.; Majesky, M.W. A sonic hedgehog signaling domain in the arterial adventitia supports resident sca1+ smooth muscle progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9349–9354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.-W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.K.; Mullally, A.; Dugourd, A.; Peisker, F.; Hoogenboezem, R.; Strien, P.M.H.V.; Bindels, E.M.; Heckl, D.; Büsche, G.; Fleck, D.; et al. Gli1+ mesenchymal stromal cells are a key driver of bone marrow fibrosis and an important cellular therapeutic target. Cell Stem Cell 2017, 20, 785–800.e8. [Google Scholar] [CrossRef] [Green Version]
- Kramann, R.; Goettsch, C.; Wongboonsin, J.; Iwata, H.; Schneider, R.K.; Kuppe, C.; Kaesler, N.; Chang-Panesso, M.; Machado, F.G.; Gratwohl, S.; et al. Adventitial MSC-like cells are progenitors of vascular smooth muscle cells and drive vascular calcification in chronic kidney disease. Cell Stem Cell 2016, 19, 628–642. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Li, M.; Li, S.; Xing, Y.; Yang, C.-Y.; Li, A.; Borok, Z.; De Langhe, S.; Minoo, P. Progenitors of secondary crest myofibroblasts are developmentally committed in early lung mesoderm. Stem Cells Dayt. Ohio 2015, 33, 999–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moiseenko, A.; Kheirollahi, V.; Chao, C.-M.; Ahmadvand, N.; Quantius, J.; Wilhelm, J.; Herold, S.; Ahlbrecht, K.; Morty, R.E.; Rizvanov, A.A.; et al. Origin and characterization of alpha smooth muscle actin-positive cells during murine lung development. Stem Cells 2017, 35, 1566–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierick, F.; Héry, T.; Hoareau-Coudert, B.; Mougenot, N.; Monceau, V.; Claude, C.; Crisan, M.; Besson, V.; Dorfmüller, P.; Marodon, G.; et al. Resident PW1+ progenitor cells participate in vascular remodeling during pulmonary arterial hypertension. Circ. Res. 2016, 118, 822–833. [Google Scholar] [CrossRef] [Green Version]
- Pannérec, A.; Formicola, L.; Besson, V.; Marazzi, G.; Sassoon, D.A. Defining skeletal muscle resident progenitors and their cell fate potentials. Dev. Camb. Engl. 2013, 140, 2879–2891. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Arroyo, J.G.; Farkas, L.; Alhussaini, A.A.; Farkas, D.; Kraskauskas, D.; Voelkel, N.F.; Bogaard, H.J. The monocrotaline model of pulmonary hypertension in perspective. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L363–L369. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Kay, J.M.; Heath, D. Hypertensive pulmonary vascular disease in rats after prolonged feeding with crotalaria spectabilis seeds. J. Pathol. 1970, 102, 97–106. [Google Scholar] [CrossRef]
- Lee, J.; Reich, R.; Xu, F.; Sehgal, P.B. Golgi, trafficking, and mitosis dysfunctions in pulmonary arterial endothelial cells exposed to monocrotaline pyrrole and no scavenging. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L715–L728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Lee, J.; Sehgal, P.B. Depletion of the atpase nsf from golgi membranes with hypo-s-nitrosylation of vasorelevant proteins in endothelial cells exposed to monocrotaline pyrrole. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1943–H1955. [Google Scholar] [CrossRef]
- Tuder, R.M.; Marecki, J.C.; Richter, A.; Fijalkowska, I.; Flores, S. Pathology of pulmonary hypertension. Clin. Chest Med. 2007, 28, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Frid, M.G.; Graham, B.B.; Tuder, R.M. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc. Res. 2018, 114, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Meyrick, B.; Reid, L. Hypoxia and incorporation of 3h-thymidine by cells of the rat pulmonary arteries and alveolar wall. Am. J. Pathol. 1979, 96, 51–70. [Google Scholar] [PubMed]
- Meyrick, B.; Reid, L. Development of pulmonary arterial changes in rats fed crotalaria spectabilis. Am. J. Pathol. 1979, 94, 37–51. [Google Scholar] [PubMed]
- Meyrick, B.; Hislop, A.; Reid, L. Pulmonary arteries of the normal rat: The thick walled oblique muscle segment. J. Anat. 1978, 125, 209–221. [Google Scholar]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/akt signal transduction pathway. requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [Green Version]
- Alon, T.; Hemo, I.; Itin, A.; Pe’er, J.; Stone, J.; Keshet, E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat. Med. 1995, 1, 1024–1028. [Google Scholar] [CrossRef]
- Fong, T.A.; Shawver, L.K.; Sun, L.; Tang, C.; App, H.; Powell, T.J.; Kim, Y.H.; Schreck, R.; Wang, X.; Risau, W.; et al. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) That inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999, 59, 99–106. [Google Scholar]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef]
- Taraseviciene-Stewart, L.; Kasahara, Y.; Alger, L.; Hirth, P.; Mc Mahon, G.; Waltenberger, J.; Voelkel, N.F.; Tuder, R.M. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Toba, M.; Alzoubi, A.; Ito, M.; Fagan, K.A.; Cool, C.D.; Voelkel, N.F.; McMurtry, I.F.; Oka, M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 2010, 121, 2747–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, D.L.; Frid, M.G.; Kunrath, C.L.; Karoor, V.; Anwar, A.; Wagner, B.D.; Strassheim, D.; Stenmark, K.R. Sustained hypoxia promotes the development of a pulmonary artery-specific chronic inflammatory microenvironment. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L238–L250. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, X.; Ahmadvand, N.; Zhang, J.-S.; Seeger, W.; Bellusci, S.; El Agha, E. Evidence for Multiple Origins of De Novo Formed Vascular Smooth Muscle Cells in Pulmonary Hypertension: Challenging the Dominant Model of Pre-Existing Smooth Muscle Expansion. Int. J. Environ. Res. Public Health 2021, 18, 8584. https://doi.org/10.3390/ijerph18168584
Chu X, Ahmadvand N, Zhang J-S, Seeger W, Bellusci S, El Agha E. Evidence for Multiple Origins of De Novo Formed Vascular Smooth Muscle Cells in Pulmonary Hypertension: Challenging the Dominant Model of Pre-Existing Smooth Muscle Expansion. International Journal of Environmental Research and Public Health. 2021; 18(16):8584. https://doi.org/10.3390/ijerph18168584
Chicago/Turabian StyleChu, Xuran, Negah Ahmadvand, Jin-San Zhang, Werner Seeger, Saverio Bellusci, and Elie El Agha. 2021. "Evidence for Multiple Origins of De Novo Formed Vascular Smooth Muscle Cells in Pulmonary Hypertension: Challenging the Dominant Model of Pre-Existing Smooth Muscle Expansion" International Journal of Environmental Research and Public Health 18, no. 16: 8584. https://doi.org/10.3390/ijerph18168584
APA StyleChu, X., Ahmadvand, N., Zhang, J. -S., Seeger, W., Bellusci, S., & El Agha, E. (2021). Evidence for Multiple Origins of De Novo Formed Vascular Smooth Muscle Cells in Pulmonary Hypertension: Challenging the Dominant Model of Pre-Existing Smooth Muscle Expansion. International Journal of Environmental Research and Public Health, 18(16), 8584. https://doi.org/10.3390/ijerph18168584