
Proteomic Characterization of the Cellular Effects of AhR Activation by Microbial Tryptophan Catabolites in Endotoxin-Activated Human Macrophages

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
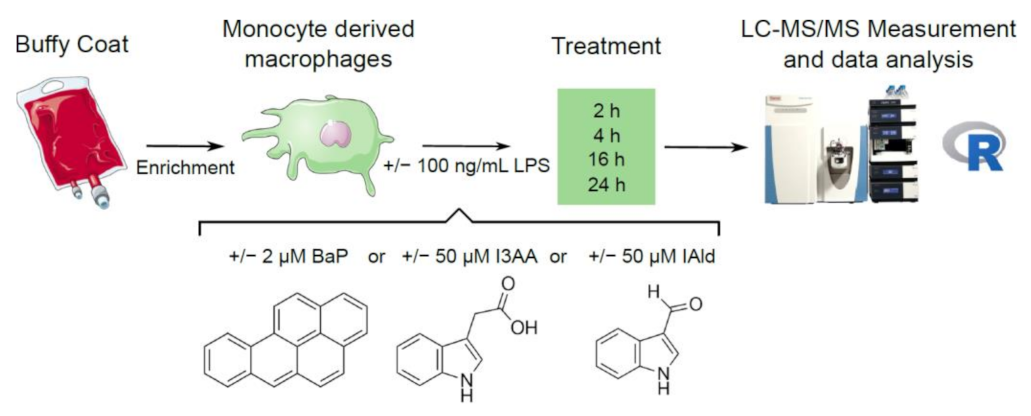
2.1. Cell Culture
2.2. Enzyme-Linked Immunosorbent Assay (ELISA)
2.3. Quantification of mRNA Expression Levels
2.4. Sample Preparation for LC-MS/MS-Based Proteomics
2.5. LC-MS/MS
2.6. Data Analysis
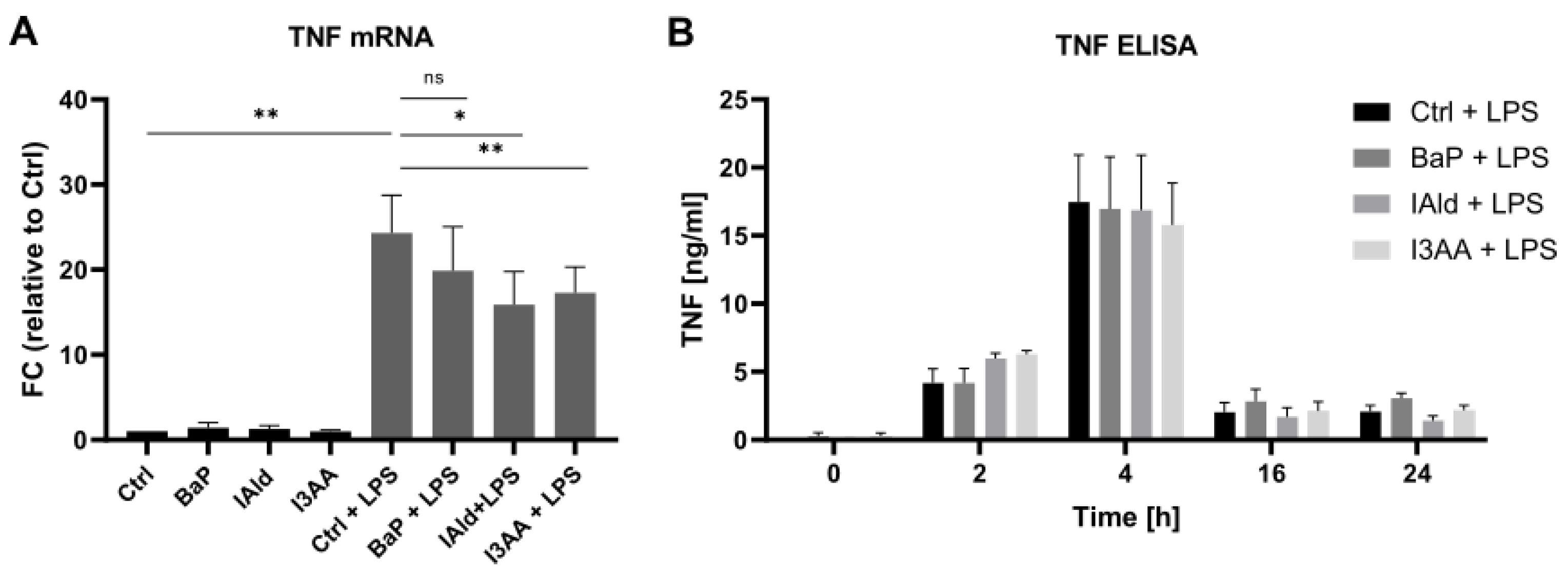
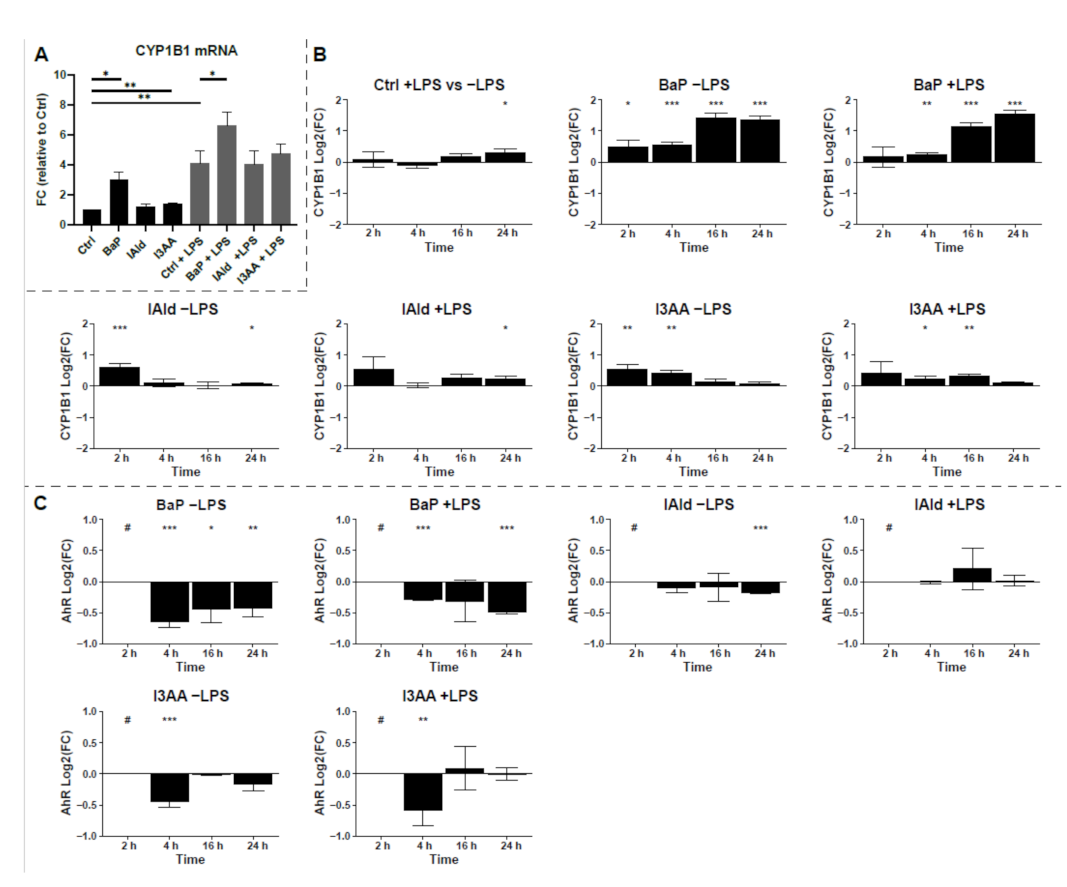
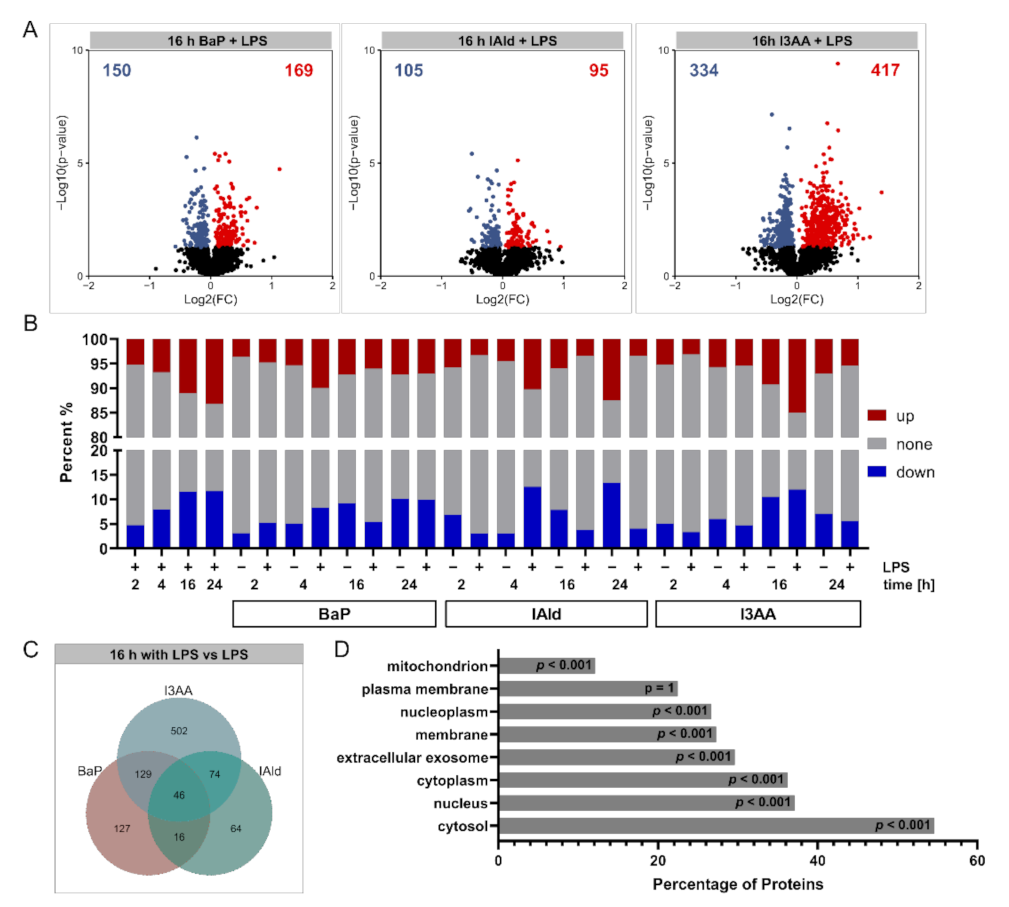
3. Results
4. Discussion
4.1. Anti-Inflammatory Modulation of Cell Metabolism in LPS-Activated Macrophages through AhR-Ligands
4.2. Potential Role of mTOR Signaling in AhR-Dependent Immunomodulation
4.3. Ligand-Specific Effects on Pathways of the Immune Response
4.4. Potential Role of AhR-Ligands in Gut Microbial Homeostasis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stockinger, B.; Shah, K.; Wincent, E. AHR in the intestinal microenvironment: Safeguarding barrier function. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 559–570. [Google Scholar] [CrossRef]
- Enan, E.; Matsumura, F. Identification of c-Src as the integral component of the cytosolic Ah receptor complex, transducing the signal of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) through the protein phosphorylation pathway. Biochem. Pharmacol. 1996, 52, 1599–1612. [Google Scholar] [CrossRef]
- Perdew, G.H. Association of the Ah receptor with the 90-kDa heat shock protein. J. Biol. Chem. 1988, 263, 13802–13805. [Google Scholar] [CrossRef]
- Carver, L.A.; Bradfield, C.A. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J. Biol. Chem. 1997, 272, 11452–11456. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.C.; Toran, E.J.; Rimerman, R.A.; Hjermstad, S.; Smithgall, T.E.; Smith, D.F. A pathway of multi-chaperone interactions common to diverse regulatory proteins: Estrogen receptor, Fes tyrosine kinase, heat shock transcription factor Hsf1, and the aryl hydrocarbon receptor. Cell Stress Chaperones 1996, 1, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Reyes, H.; Reisz-Porszasz, S.; Hankinson, O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 1992, 256, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.P.; Bradfield, C.A. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 2008, 21, 102–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiserman, M.J.; Rickert, W.S. Carcinogens in tobacco smoke: Benzo[a]pyrene from Canadian cigarettes and cigarette tobacco. Am. J. Public Health 1992, 82, 1023–1026. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.K.; Prasad, S.; Patel, D.K.; Khan, A.H.; Tripathi, M.; Shukla, Y. Identification of polycyclic aromatic hydrocarbons in unleaded petrol and diesel exhaust emission. Environ. Monit. Assess. 2010, 168, 173–178. [Google Scholar] [CrossRef]
- Jaguin, M.; Fardel, O.; Lecureur, V. Exposure to diesel exhaust particle extracts (DEPe) impairs some polarization markers and functions of human macrophages through activation of AhR and Nrf2. PLoS ONE 2015, 10, e0116560. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Vazquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, U.H.; Lee, S.O.; Sridharan, G.; Lee, K.; Davidson, L.A.; Jayaraman, A.; Chapkin, R.S.; Alaniz, R.; Safe, S. Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol. Pharm. 2014, 85, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Heath-Pagliuso, S.; Rogers, W.J.; Tullis, K.; Seidel, S.D.; Cenijn, P.H.; Brouwer, A.; Denison, M.S. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry 1998, 37, 11508–11515. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, F.; Hao, F.; Murray, I.A.; Smith, P.B.; Koo, I.; Tindall, A.M.; Kris-Etherton, P.M.; Gowda, K.; Amin, S.G.; Patterson, A.D.; et al. Intestinal microbiota-derived tryptophan metabolites are predictive of Ah receptor activity. Gut Microbes 2020, 12, 1–24. [Google Scholar] [CrossRef]
- Natividad, J.M.; Agus, A.; Planchais, J.; Lamas, B.; Jarry, A.C.; Martin, R.; Michel, M.L.; Chong-Nguyen, C.; Roussel, R.; Straube, M.; et al. Impaired Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is a Key Factor in Metabolic Syndrome. Cell Metab. 2018, 28, 737–749.e734. [Google Scholar] [CrossRef] [Green Version]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharm. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [Green Version]
- Cervantes-Barragan, L.; Colonna, M. AHR signaling in the development and function of intestinal immune cells and beyond. Semin. Immunopathol. 2018, 40, 371–377. [Google Scholar] [CrossRef]
- Lee, S.H.; Starkey, P.M.; Gordon, S. Quantitative analysis of total macrophage content in adult mouse tissues. Immunochemical studies with monoclonal antibody F4/80. J. Exp. Med. 1985, 161, 475–489. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.D.; Smythies, L.E.; Shen, R.; Greenwell-Wild, T.; Gliozzi, M.; Wahl, S.M. Intestinal macrophages and response to microbial encroachment. Mucosal. Immunol. 2011, 4, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, A.; Naka, T.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J. Exp. Med. 2009, 206, 2027–2035. [Google Scholar] [CrossRef] [Green Version]
- Sekine, H.; Mimura, J.; Oshima, M.; Okawa, H.; Kanno, J.; Igarashi, K.; Gonzalez, F.J.; Ikuta, T.; Kawajiri, K.; Fujii-Kuriyama, Y. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol. Cell. Biol. 2009, 29, 6391–6400. [Google Scholar] [CrossRef] [Green Version]
- Fueldner, C.; Kohlschmidt, J.; Riemschneider, S.; Schulze, F.; Zoldan, K.; Esser, C.; Hauschildt, S.; Lehmann, J. Benzo(a)pyrene attenuates the pattern-recognition-receptor induced proinflammatory phenotype of murine macrophages by inducing IL-10 expression in an aryl hydrocarbon receptor-dependent manner. Toxicology 2018, 409, 80–90. [Google Scholar] [CrossRef]
- Grosskopf, H.; Walter, K.; Karkossa, I.; von Bergen, M.; Schubert, K. Non-Genomic AhR-Signaling Modulates the Immune Response in Endotoxin-Activated Macrophages After Activation by the Environmental Stressor BaP. Front. Immunol. 2021, 12, 620270. [Google Scholar] [CrossRef]
- Schubert, K.; Collins, L.E.; Green, P.; Nagase, H.; Troeberg, L. LRP1 Controls TNF Release via the TIMP-3/ADAM17 Axis in Endotoxin-Activated Macrophages. J. Immunol. 2019, 202, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bannuscher, A.; Karkossa, I.; Buhs, S.; Nollau, P.; Kettler, K.; Balas, M.; Dinischiotu, A.; Hellack, B.; Wiemann, M.; Luch, A.; et al. A multi-omics approach reveals mechanisms of nanomaterial toxicity and structure-activity relationships in alveolar macrophages. Nanotoxicology 2020, 14, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Moggridge, S.; Muller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Wang, Z.; Karkossa, I.; Grosskopf, H.; Rolle-Kampczyk, U.; Hackermuller, J.; von Bergen, M.; Schubert, K. Comparison of quantitation methods in proteomics to define relevant toxicological information on AhR activation of HepG2 cells by BaP. Toxicology 2021, 448, 152652. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Karkossa, I.; Bannuscher, A.; Hellack, B.; Bahl, A.; Buhs, S.; Nollau, P.; Luch, A.; Schubert, K.; von Bergen, M.; Haase, A. An in-depth multi-omics analysis in RLE-6TN rat alveolar epithelial cells allows for nanomaterial categorization. Part. Fibre Toxicol. 2019, 16, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathan, M.; Keerthikumar, S.; Chisanga, D.; Alessandro, R.; Ang, C.S.; Askenase, P.; Batagov, A.O.; Benito-Martin, A.; Camussi, G.; Clayton, A.; et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J. Extracell. Vesicles 2017, 6, 1321455. [Google Scholar] [CrossRef] [Green Version]
- Fonseka, P.; Pathan, M.; Chitti, S.V.; Kang, T.; Mathivanan, S. FunRich enables enrichment analysis of OMICs datasets. J. Mol. Biol. 2021, 433, 166747. [Google Scholar] [CrossRef]
- Ma, Q.; Baldwin, K.T. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR. J. Biol. Chem. 2000, 275, 8432–8438. [Google Scholar] [CrossRef] [Green Version]
- Davarinos, N.A.; Pollenz, R.S. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J. Biol. Chem. 1999, 274, 28708–28715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raught, B.; Peiretti, F.; Gingras, A.C.; Livingstone, M.; Shahbazian, D.; Mayeur, G.L.; Polakiewicz, R.D.; Sonenberg, N.; Hershey, J.W. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 2004, 23, 1761–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef]
- Ehrlich, A.M.; Pacheco, A.R.; Henrick, B.M.; Taft, D.; Xu, G.; Huda, M.N.; Mishchuk, D.; Goodson, M.L.; Slupsky, C.; Barile, D.; et al. Indole-3-lactic acid associated with Bifidobacterium-dominated microbiota significantly decreases inflammation in intestinal epithelial cells. BMC Microbiol. 2020, 20, 357. [Google Scholar] [CrossRef]
- Baron, J.M.; Zwadlo-Klarwasser, G.; Jugert, F.; Hamann, W.; Rübben, A.; Mukhtar, H.; Merk, H.F. Cytochrome P450 1B1: A major P450 isoenzyme in human blood monocytes and macrophage subsets. Biochem. Pharmacol. 1998, 56, 1105–1110. [Google Scholar] [CrossRef]
- Kim, J.H.; Stansbury, K.H.; Walker, N.J.; Trush, M.A.; Strickland, P.T.; Sutter, T.R. Metabolism of benzo[a]pyrene and benzo[a]pyrene-7,8-diol by human cytochrome P450 1B1. Carcinogenesis 1998, 19, 1847–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, R.J.; Deitrich, R.A. Isolation and Characterization of Human Liver Aldehyde Dehydrogenase. J. Biol. Chem. 1968, 243, 6402–6408. [Google Scholar] [CrossRef]
- Lepri, S.; Ceccarelli, M.; Milani, N.; Tortorella, S.; Cucco, A.; Valeri, A.; Goracci, L.; Brink, A.; Cruciani, G. Structure-metabolism relationships in human-AOX: Chemical insights from a large database of aza-aromatic and amide compounds. Proc. Natl. Acad. Sci. USA 2017, 114, E3178–E3187. [Google Scholar] [CrossRef] [Green Version]
- Langan, D.; Perkins, D.J.; Vogel, S.N.; Moudgil, K.D. Microbiota-Derived Metabolites, Indole-3-aldehyde and Indole-3-acetic Acid, Differentially Modulate Innate Cytokines and Stromal Remodeling Processes Associated with Autoimmune Arthritis. Int. J. Mol. Sci. 2021, 22, 2017. [Google Scholar] [CrossRef]
- Bouchon, A.; Dietrich, J.; Colonna, M. Cutting edge: Inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 2000, 164, 4991–4995. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, A.; Facchetti, F.; Weigand, M.A.; Colonna, M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001, 410, 1103–1107. [Google Scholar] [CrossRef]
- Malmgaard, L. Induction and regulation of IFNs during viral infections. J. Interferon Cytokine Res. 2004, 24, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Zhao, Q.; Yang, T.; Ding, W.; Zhao, Y. Cellular metabolism and macrophage functional polarization. Int. Rev. Immunol. 2015, 34, 82–100. [Google Scholar] [CrossRef]
- Noël, W.; Raes, G.; Hassanzadeh Ghassabeh, G.; De Baetselier, P.; Beschin, A. Alternatively activated macrophages during parasite infections. Trends Parasitol. 2004, 20, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, S.; Vuckovic, I.; Jeon, R.; Lerman, A.; Folmes, C.D.; Dzeja, P.P.; Herrmann, J. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 2018, 28, 463–475.e464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funk, J.L.; Feingold, K.R.; Moser, A.H.; Grunfeld, C. Lipopolysaccharide stimulation of RAW 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis 1993, 98, 67–82. [Google Scholar] [CrossRef]
- Feingold, K.R.; Shigenaga, J.K.; Kazemi, M.R.; McDonald, C.M.; Patzek, S.M.; Cross, A.S.; Moser, A.; Grunfeld, C. Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 2012, 92, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malandrino, M.I.; Fucho, R.; Weber, M.; Calderon-Dominguez, M.; Mir, J.F.; Valcarcel, L.; Escote, X.; Gomez-Serrano, M.; Peral, B.; Salvado, L.; et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E756–E769. [Google Scholar] [CrossRef] [Green Version]
- Dennis, P.B.; Jaeschke, A.; Saitoh, M.; Fowler, B.; Kozma, S.C.; Thomas, G. Mammalian TOR: A homeostatic ATP sensor. Science 2001, 294, 1102–1105. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Rocher, C.; Singla, D.K. SMAD-PI3K-Akt-mTOR pathway mediates BMP-7 polarization of monocytes into M2 macrophages. PLoS ONE 2013, 8, e84009. [Google Scholar] [CrossRef] [Green Version]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katholnig, K.; Linke, M.; Pham, H.; Hengstschlager, M.; Weichhart, T. Immune responses of macrophages and dendritic cells regulated by mTOR signalling. Biochem. Soc. Trans. 2013, 41, 927–933. [Google Scholar] [CrossRef]
- Saric, A.; Hipolito, V.E.; Kay, J.G.; Canton, J.; Antonescu, C.N.; Botelho, R.J. mTOR controls lysosome tubulation and antigen presentation in macrophages and dendritic cells. Mol. Biol. Cell 2016, 27, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Antonia, R.J.; Baldwin, A.S. PI3K/Akt promotes feedforward mTORC2 activation through IKKalpha. Oncotarget 2016, 7, 21064–21075. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, Y.; Kado, S.Y.; Hoeper, C.; Harel, S.; Vogel, C.F.A. Role of NF-kB RelB in Aryl Hydrocarbon Receptor-Mediated Ligand Specific Effects. Int. J. Mol. Sci. 2019, 20, 2652. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, A.K.; Pennington, J.M.; Bisson, W.H.; Kolluri, S.K.; Kerkvliet, N.I. TCDD, FICZ, and Other High Affinity AhR Ligands Dose-Dependently Determine the Fate of CD4+ T Cell Differentiation. Toxicol. Sci. 2018, 161, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Jin, U.H.; Park, H.; Chapkin, R.S.; Jayaraman, A. Aryl Hydrocarbon Receptor (AHR) Ligands as Selective AHR Modulators (SAhRMs). Int. J. Mol. Sci. 2020, 21, 6654. [Google Scholar] [CrossRef]
- Kopec, A.K.; Burgoon, L.D.; Ibrahim-Aibo, D.; Burg, A.R.; Lee, A.W.; Tashiro, C.; Potter, D.; Sharratt, B.; Harkema, J.R.; Rowlands, J.C.; et al. Automated dose-response analysis and comparative toxicogenomic evaluation of the hepatic effects elicited by TCDD, TCDF, and PCB126 in C57BL/6 mice. Toxicol. Sci. 2010, 118, 286–297. [Google Scholar] [CrossRef]
- Ji, Y.; Yin, W.; Liang, Y.; Sun, L.; Yin, Y.; Zhang, W. Anti-Inflammatory and Anti-Oxidative Activity of Indole-3-Acetic Acid Involves Induction of HO-1 and Neutralization of Free Radicals in RAW264.7 Cells. Int. J. Mol. Sci. 2020, 21, 1579. [Google Scholar] [CrossRef] [Green Version]
- Sciullo, E.M.; Vogel, C.F.; Li, W.; Matsumura, F. Initial and extended inflammatory messages of the nongenomic signaling pathway of the TCDD-activated Ah receptor in U937 macrophages. Arch. Biochem. Biophys. 2008, 480, 143–155. [Google Scholar] [CrossRef]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Iacob, S.; Iacob, D.G.; Luminos, L.M. Intestinal Microbiota as a Host Defense Mechanism to Infectious Threats. Front. Microbiol. 2018, 9, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasaly, N.; de Vos, P.; Hermoso, M.A. Impact of Bacterial Metabolites on Gut Barrier Function and Host Immunity: A Focus on Bacterial Metabolism and Its Relevance for Intestinal Inflammation. Front. Immunol. 2021, 12, 658354. [Google Scholar] [CrossRef]
- Puccetti, M.; Pariano, M.; Borghi, M.; Barola, C.; Moretti, S.; Galarini, R.; Mosci, P.; Ricci, M.; Costantini, C.; Giovagnoli, S. Enteric formulated indole-3-carboxaldehyde targets the aryl hydrocarbon receptor for protection in a murine model of metabolic syndrome. Int. J. Pharm. 2021, 602, 120610. [Google Scholar] [CrossRef] [PubMed]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walter, K.; Grosskopf, H.; Karkossa, I.; von Bergen, M.; Schubert, K. Proteomic Characterization of the Cellular Effects of AhR Activation by Microbial Tryptophan Catabolites in Endotoxin-Activated Human Macrophages. Int. J. Environ. Res. Public Health 2021, 18, 10336. https://doi.org/10.3390/ijerph181910336
Walter K, Grosskopf H, Karkossa I, von Bergen M, Schubert K. Proteomic Characterization of the Cellular Effects of AhR Activation by Microbial Tryptophan Catabolites in Endotoxin-Activated Human Macrophages. International Journal of Environmental Research and Public Health. 2021; 18(19):10336. https://doi.org/10.3390/ijerph181910336
Chicago/Turabian StyleWalter, Katharina, Henning Grosskopf, Isabel Karkossa, Martin von Bergen, and Kristin Schubert. 2021. "Proteomic Characterization of the Cellular Effects of AhR Activation by Microbial Tryptophan Catabolites in Endotoxin-Activated Human Macrophages" International Journal of Environmental Research and Public Health 18, no. 19: 10336. https://doi.org/10.3390/ijerph181910336
APA StyleWalter, K., Grosskopf, H., Karkossa, I., von Bergen, M., & Schubert, K. (2021). Proteomic Characterization of the Cellular Effects of AhR Activation by Microbial Tryptophan Catabolites in Endotoxin-Activated Human Macrophages. International Journal of Environmental Research and Public Health, 18(19), 10336. https://doi.org/10.3390/ijerph181910336