
The Sources and Potential Hosts Identification of Antibiotic Resistance Genes in the Yellow River, Revealed by Metagenomic Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Quantification of MST Indicators
2.3. 16S Amplicon Sequencing
2.4. Metagenomic Analysis
2.5. Statistical Analysis
3. Results
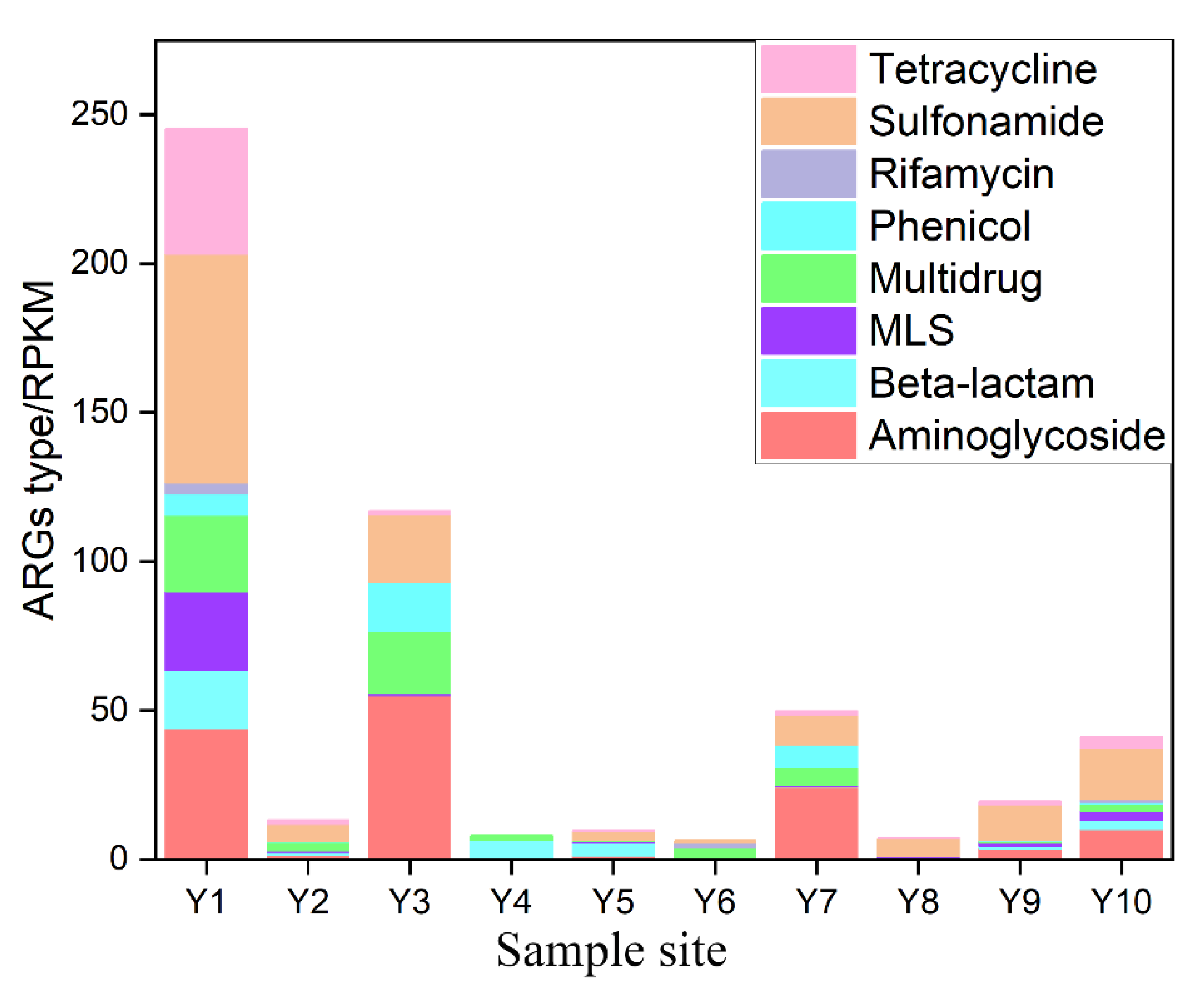
3.1. ARG Profile in the Sediments of the Yellow River
3.2. Distribution Pattern of ARG Subtypes in the Yellow River
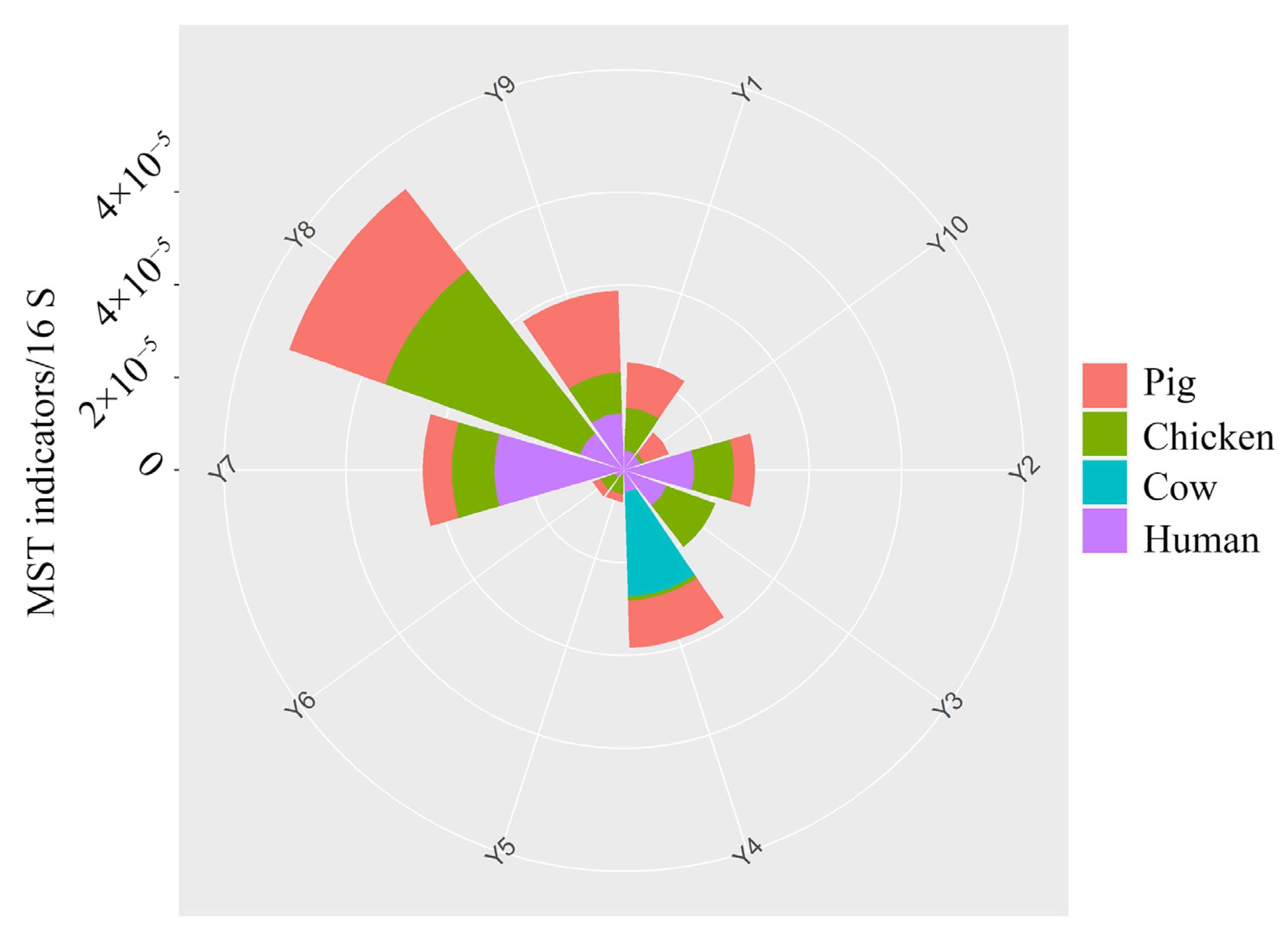
3.3. Microbial Source Tracking Genes Content and Their Relationship with ARGs
3.4. Characteristics of the Bacteria Communities
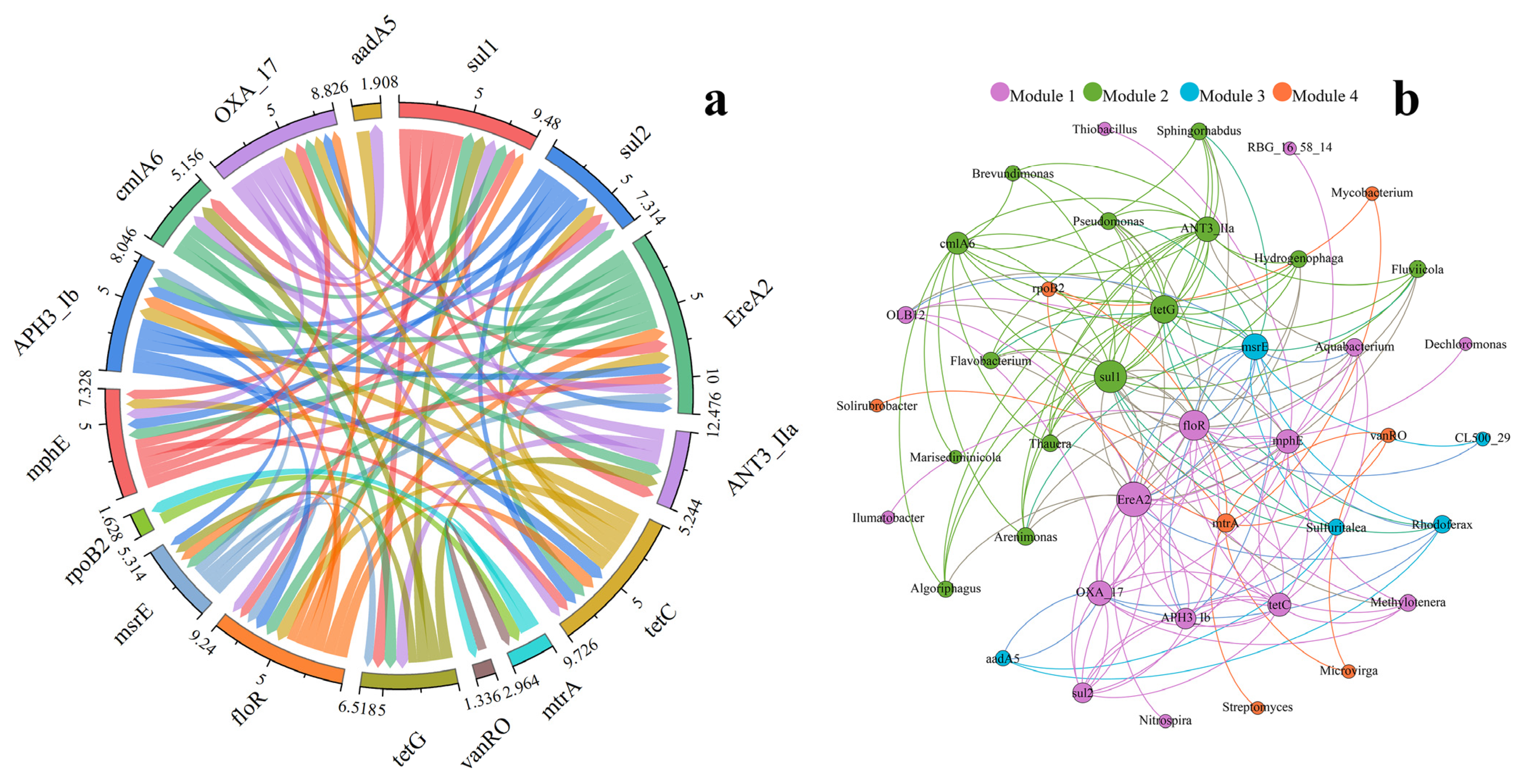
3.5. Relationship between ARGs and Bacterial Communities
4. Conclusions
- Sul1 and sul2 are the dominant ARGs in the Yellow River, which could be attributed to the huge usage of sulfonamides, horizontal gene transfer, and wide bacteria host of these two genes. Furthermore, the emergence of carbapenem-resistant genes indicates that the use of carbapenem antibiotics should be further restricted.
- Animal and human feces are found in the Yellow River, while the prohibition of antibiotics in the breeding industry could minimize the contribution of the breeding industry to ARGs.
- Numerous dominant bacteria are associated with denitrification, and the prevalence of some opportunistic pathogens (e.g., Corynebacterium and Pseudomonas) may pose an adverse health risk to humans.
- There exists a close relationship among ARGs. Furthermore, Pseudomonas is the potential host of sul1, tetG, and ANT(3′′)-IIa, which may pose a risk to human health.
5. Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gray, D.A.; Wenzel, M. Multitarget approaches against multiresistant superbugs. ACS Infect. Dis. 2020, 6, 1346–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, E.T.; Ojemaye, M.O.; Okoh, O.O.; Okoh, A.I. A critical review on the occurrence of resistomes in the environment and their removal from wastewater using apposite treatment technologies: Limitations, successes and future improvement. Environ. Pollut. 2020, 263, 113791. [Google Scholar] [CrossRef] [PubMed]
- Le, T.-H.; Ng, C.; Tran, N.H.; Chen, H.; Gin, K.Y.-H. Removal of antibiotic residues, antibiotic resistant bacteria and antibiotic resistance genes in municipal wastewater by membrane bioreactor systems. Water Res. 2018, 145, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Reddy, B.; Singh, V.K.; Singh, A.K.; Kesari, K.K.; Tripathi, P.; Kumaret, P.; Tripathi, V.; Simal-Gandara, J. Potential environmental and human health risks caused by antibiotic-resistant bacteria (ARB), antibiotic resistance genes (ARGs) and emerging contaminants (ECs) from municipal solid waste (MSW) landfill. Antibiotics 2021, 10, 374. [Google Scholar] [CrossRef]
- Wang, S.; Xue, N.; Li, W.; Zhang, D.; Pan, X.; Luo, Y. Selectively enrichment of antibiotics and ARGs by microplastics in river, estuary and marine waters. Sci. Total Environ. 2020, 708, 134594. [Google Scholar] [CrossRef]
- Zhang, K.; Li, K.; Xin, R.; Han, Y.; Guo, Z.; Zou, W.; Wei, W.; Cui, X.; Zhang, Z.; Zhang, Y.; et al. Antibiotic resistomes in water supply reservoirs sediments of central China: Main biotic drivers and distribution pattern. Environ. Sci. Pollut. Res. 2022, 29, 37712–37721. [Google Scholar] [CrossRef]
- Liang, X.; Guan, F.; Chen, B.; Luo, P.; Guo, C.; Wu, G.; Ye, Y.; Zhou, Q.; Feng, H. Spatial and seasonal variations of antibiotic resistance genes and antibiotics in the surface waters of Poyang Lake in China. Ecotoxicol. Environ. Saf. 2020, 196, 110543. [Google Scholar] [CrossRef]
- Zhang, K.; Xin, R.; Zhao, Z.; Ma, Y.; Zhang, Y.; Niu, Z. Antibiotic resistance genes in drinking water of China: Occurrence, distribution and influencing factors. Ecotoxicol. Environ. Saf. 2020, 188, 109837. [Google Scholar] [CrossRef]
- Gogry, F.A.; Siddiqui, M.T.; Haq, Q.M.R. Emergence of mcr-1 conferred colistin resistance among bacterial isolates from urban sewage water in India. Environ. Sci. Pollut. Res. 2020, 26, 33715–33717. [Google Scholar] [CrossRef]
- Gogry, F.A.; Siddiqui, M.T.; Sultan, I.; Husain, F.M.; Al-Kheraif, A.A.; Ali, A.; Haq, Q.M.R. Colistin Interaction and Surface Changes Associated with mcr-1 Conferred Plasmid Mediated Resistance in E. coli and veronii Strains. Pharmaceutics 2022, 14, 295. [Google Scholar] [CrossRef]
- Gogry, F.A.; Siddiqui, M.T.; Sultan, I.; Haq, Q.M.R. Current update on intrinsic and acquired colistin resistance mechanisms in bacteria. Front. Med. 2021, 8, 677720. [Google Scholar] [CrossRef]
- Wang, J.; Chu, L.; Wojnárovits, L.; Takács, E. Occurrence and fate of antibiotics, antibiotic resistant genes (ARGs) and antibiotic resistant bacteria (ARB) in municipal wastewater treatment plant: An overview. Sci. Total Environ. 2020, 744, 140997. [Google Scholar] [CrossRef] [PubMed]
- Komijani, M.; Shamabadi, N.S.; Shahin, K.; Eghbalpour, F.; Tahsili, M.R.; Bahram, M. Heavy metal pollution promotes antibiotic resistance potential in the aquatic environment. Environ. Pollut. 2021, 274, 116569. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Xu, Z.; Fan, L. Response of heavy metal and antibiotic resistance genes and related microorganisms to different heavy metals in activated sludge. J. Environ. Manag. 2021, 300, 113754. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, Z.; Mu, Q.; Wu, X.; Zhang, J.; Mao, D.; Luo, Y.; Alvarez, P. Ionic liquid enriches the antibiotic resistome, especially efflux pump genes, before significantly affecting microbial community structure. Environ. Sci. Technol. 2020, 54, 4305–4315. [Google Scholar] [CrossRef]
- Zhou, C.; Pan, Y.; Ge, S.; Coulon, F.; Yang, Z. Rapid methods for antimicrobial resistance diagnosis in contaminated soils for effective remediation strategy. TrAC Trends Anal. Chem. 2021, 137, 116203. [Google Scholar] [CrossRef]
- Qin, K.; Wei, L.; Li, J.; Lai, B.; Zhu, F.; Yu, H.; Zhaob, Q.; Wang, K. A review of ARGs in WWTPs: Sources, stressors and elimination. Chin. Chem. Lett. 2020, 31, 2603–2613. [Google Scholar] [CrossRef]
- Karkman, A.; Pärnänen, K.; Larsson, D. Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nat. Commun. 2019, 10, 80. [Google Scholar] [CrossRef]
- Hou, L.; Wang, H.; Chen, Q.; Su, J.-Q.; Gad, M.; Li, J.; Mulla, S.I.; Yu, C.P.; Hu, A. Fecal pollution mediates the dominance of stochastic assembly of antibiotic resistome in an urban lagoon (Yundang lagoon), China. J. Hazard. Mater. 2021, 417, 126083. [Google Scholar] [CrossRef]
- Unno, T.; Staley, C.; Brown, C.M.; Han, D.; Sadowsky, M.J.; Hur, H.-G. Fecal pollution: New trends and challenges in microbial source tracking using next-generation sequencing. Environ. Microbiol. 2018, 20, 3132–3140. [Google Scholar] [CrossRef] [Green Version]
- Schill, W.B.; Mathes, M.V. Real-time PCR detection and quantification of nine potential sources of fecal contamination by analysis of mitochondrial cytochrome b targets. Environ. Sci. Technol. 2008, 42, 5229–5234. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhou, S.; Han, X.; Zhang, L.; Ding, S.; Li, Y.; Zhang, D.; Zarin, K. Occurrence, distribution, and source track of antibiotics and antibiotic resistance genes in the main rivers of Chongqing city, southwest China. J. Hazard. Mater. 2020, 389, 122110. [Google Scholar] [CrossRef] [PubMed]
- Stange, C.; Tiehm, A. Occurrence of antibiotic resistance genes and microbial source tracking markers in the water of a karst spring in Germany. Sci. Total Environ. 2020, 742, 140529. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Hao, J.; Tao, L. Concentrations, spatial distribution, and pollution assessment of heavy metals in surficial sediments from upstream of Yellow River, China. Environ. Sci. Pollut. Res. 2021, 28, 2904–2913. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Liu, Q.; Ru, X.; Xi, N.; Sun, J. Occurrence and distribution of priority pharmaceuticals in the Yellow River and the Huai River in Henan, China. Environ. Sci. Pollut. Res. 2020, 27, 16816–16826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, K.; Xin, R.; Xu, Z.; Zhang, Z.; He, S.; Zhao, Z.; Tong, M.; Cui, Y. Distribution pattern and ecological risk assessment of heavy metals in Henan section of the Yellow River. Water Supply 2022, 22, 6195–6204. [Google Scholar] [CrossRef]
- Xi, N.; Guo, W.; Feng, J.; Liu, S.; Sun, J. Distribution, source, and ecological risk assessment of PAHs among size and density fractions in contaminated sediments from the Yellow River of Henan section. Environ. Forensics 2019, 20, 171–181. [Google Scholar] [CrossRef]
- Song, W.T.; Wang, Z.J. Occurrence and biological effects of endocrine disrupting chemicals in the Yellow River (Zhengzhou section). Bull. Environ. Contam. Toxicol. 2016, 97, 763–769. [Google Scholar] [CrossRef]
- Yu, Q.; Yang, J.; Su, W.; Li, T.; Feng, T.; Li, H. Heavy metals and microbiome are negligible drivers than mobile genetic elements in determining particle-attached and free-living resistomes in the Yellow River. J. Hazard. Mater. 2022, 424, 127564. [Google Scholar] [CrossRef]
- Wang, Z.-W.; Lei, T.-Z.; Yan, X.-Y.; Li, Y.-L.; He, X.-F.; Zhu, J.-L. Assessment and utilization of agricultural residue resources in Henan province, China. Bioresources 2012, 7, 3847–3861. [Google Scholar]
- Ren, Y.; Yu, G.; Shi, C.; Liu, L.; Guo, Q.; Han, C.; Zhang, D.; Zhang, L.; Liu, B.; Gao, H.; et al. Majorbio Cloud: A one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta 2022, 1, e12. [Google Scholar] [CrossRef]
- Baran, W.; Adamek, E.; Ziemiańska, J.; Sobczak, A. Effects of the presence of sulfonamides in the environment and their influence on human health. J. Hazard. Mater. 2011, 196, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sarmah, A.K.; Meyer, M.T.; Boxall, A.B. A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment. Chemosphere 2006, 65, 725–759. [Google Scholar] [CrossRef] [PubMed]
- Ling, W.; Fan, Y.; Fang, Y.; Sun, C.; Liu, G. Antibiotics pollution of livestock and poultry breeding in Beijing-Tianjin-Hebei region. J. Environ. Eng. Technol. 2018, 8, 390–397. [Google Scholar]
- Mutuku, C.; Gazdag, Z.; Melegh, S. Occurrence of antibiotics and bacterial resistance genes in wastewater: Resistance mechanisms and antimicrobial resistance control approaches. World J. Microbiol. Biotechnol. 2022, 38, 152. [Google Scholar] [CrossRef]
- Wang, L.; Li, H.; Dang, J.; Guo, H.; Zhu, Y.; Han, W. Occurrence, distribution, and partitioning of antibiotics in surface water and sediment in a typical tributary of Yellow River, China. Environ. Sci. Pollut. Res. 2021, 28, 28207–28221. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Li, H.; Zhu, Y.; Liu, R. Occurrence, source apportionment and source-specific risk assessment of antibiotics in a typical tributary of the Yellow River basin. J. Environ. Manage. 2022, 305, 114382. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Liu, R.; Li, L.; Cao, L.; Jiao, L.; Xia, X. Source-specific risk apportionment and critical risk source identification of antibiotic resistance in Fenhe River basin, China. Chemosphere 2022, 287, 131997. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gu, J.; Gao, H.; Qian, X.; Li, H. Abundances of clinically relevant antibiotic resistance genes and bacterial community diversity in the Weihe River, China. Int. J. Environ. Res. Public Health 2018, 15, 708. [Google Scholar] [CrossRef] [Green Version]
- Auguet, O.; Pijuan, M.; Borrego, C.M.; Rodriguez-Mozaz, S.; Triadó-Margarit, X.; Della Giustina, S.V.; Gutierrez, O. Sewers as potential reservoirs of antibiotic resistance. Sci. Total Environ. 2017, 605, 1047–1054. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, X.; Liu, R.; Zhang, T.; Li, M.; Zhang, Z.; Qu, Z.; Yuan, Z.; Yu, H. Influence of reclaimed water discharge on the dissemination and relationships of sulfonamide, sulfonamide resistance genes along the Chaobai River, Beijing. Front. Environ. Sci. Eng. 2019, 13, 8. [Google Scholar] [CrossRef]
- Wei, Z.; Feng, K.; Li, S.; Zhang, Y.; Chen, H.; Yin, H.; Xu, M.; Deng, Y. Exploring abundance, diversity and variation of a widespread antibiotic resistance gene in wastewater treatment plants. Environ. Int. 2018, 117, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Sun, W.; Jin, D.; Yu, Q.; Yang, Y.; Zhang, Z.; Sun, P.; Ma, J. Effect of composting on the conjugative transmission of sulfonamide resistance and sulfonamide-resistant bacterial population. J. Clean. Prod. 2021, 285, 125483. [Google Scholar] [CrossRef]
- Ploy, M.-C.; Denis, F.; Courvalin, P.; Lambert, T. Molecular characterization of integrons in Acinetobacter baumannii: Description of a hybrid class 2 integron. Antimicrob. Agents Chemother. 2000, 44, 2684–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soufi, L.; Sáenz, Y.; Vinué, L.; Abbassi, M.S.; Ruiz, E.; Zarazaga, M.; Hassen, A.B.; Hammami, S.; Torres, C. Escherichia coli of poultry food origin as reservoir of sulphonamide resistance genes and integrons. Int. J. Food Microbiol. 2011, 144, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Suhadolnik, M.L.S.; Costa, P.S.; Paiva, M.C.; de Matos Salim, A.C.; Barbosa, F.A.R.; Lobo, F.P.; Nascimento, A.M.A. Spatiotemporal dynamics of the resistome and virulome of riverine microbiomes disturbed by a mining mud tsunami. Sci. Total Environ. 2022, 806, 150936. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Cerniglia, C.E. Influence of erythromycin A on the microbial populations in aquaculture sediment microcosms. Aquat. Toxicol. 2005, 73, 230–241. [Google Scholar] [CrossRef]
- Sheu, C.-C.; Chang, Y.-T.; Lin, S.-Y.; Chen, Y.-H.; Hsueh, P.-R. Infections caused by carbapenem-resistant Enterobacteriaceae: An update on therapeutic options. Front. Microbiol. 2019, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Moellering, R.C., Jr.; Eliopoulos, G.M.; Sentochnik, D.E. The carbapenems: New broad spectrum β-lactam antibiotics. J. Antimicrob. Chemother. 1989, 24 (Suppl. A), 1–7. [Google Scholar] [CrossRef]
- Zhang, K.; Xin, R.; Zhao, Z.; Li, W.; Wang, Y.; Wang, Q.; Niu, Z.; Zhang, Y. Mobile genetic elements are the Major driver of High antibiotic resistance genes abundance in the Upper reaches of huaihe River Basin. J. Hazard. Mater. 2020, 401, 123271. [Google Scholar] [CrossRef]
- Kiama, C.W.; Njire, M.M.; Kambura, A.K.; Mugweru, J.N.; Matiru, V.N.; Wafula, E.N.; Kagali, R.N.; Kuja, O.K. Prokaryotic diversity and composition within equatorial lakes Olbolosat and Oloiden in Kenya (Africa). Curr. Res. Microb. Sci. 2021, 2, 100066. [Google Scholar] [CrossRef] [PubMed]
- Kuang, B.; Xiao, R.; Wang, C.; Zhang, L.; Wei, Z.; Bai, J.; Zhang, K.; Campos, M.; Jorquera, M.A. Bacterial community assembly in surface sediments of a eutrophic shallow lake in northern China. Ecohydrol. Hydrobiol. 2022. [Google Scholar] [CrossRef]
- Smith, E.; Thavamani, P.; Ramadass, K.; Naidu, R.; Srivastava, P.; Megharaj, M. Remediation trials for hydrocarbon-contaminated soils in arid environments: Evaluation of bioslurry and biopiling techniques. Int. Biodeter Biodegr. 2015, 101, 56–65. [Google Scholar] [CrossRef]
- Lin, Y.; Ye, Y.; Hu, Y.; Shi, H. The variation in microbial community structure under different heavy metal contamination levels in paddy soils. Ecotoxicol. Environ. Saf. 2019, 180, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Hemmat-Jou, M.; Safari-Sinegani, A.; Mirzaie-Asl, A.; Tahmourespour, A.A. Analysis of microbial communities in heavy metals-contaminated soils using the metagenomic approach. Ecotoxicology 2018, 27, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Palansooriya, K.N.; Sang, M.K.; Igalavithana, A.D.; Zhang, M.; Hou, D.; Oleszczuk, P.; Sung, J.; Ok, Y.S. Biochar alters chemical and microbial properties of microplastic-contaminated soil. Environ. Res. 2022, 209, 112807. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Zhang, H.; Guo, Y.; Wang, Z.; Huang, T. Temporal and Spatial Patterns of Sediment Microbial Communities and Driving Environment Variables in a Shallow Temperate Mountain River. Microorganisms 2022, 10, 816. [Google Scholar] [CrossRef]
- Lee, J.; Mills, M.; Lee, S.; Mollenkopf, D.; Wittum, T.; Sullivan, M.P. Comparison of Environmental Microbiomes in an Antibiotic Resistance-Polluted Urban River Highlights Periphyton and Fish Gut Communities as Reservoirs of Concern. Sci. Total Environ. 2022, 158042. [Google Scholar]
- Zhang, B.; Wu, X.; Tai, X.; Sun, L.; Wu, M.; Zhang, W.; Chen, X.; Zhang, G.; Chen, T.; Liu, G.; et al. Variation in actinobacterial community composition and potential function in different soil ecosystems belonging to the arid Heihe River Basin of Northwest China. Front. Microbiol. 2019, 10, 2209. [Google Scholar] [CrossRef]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Klenk, H.-P.; Clément, C.; Ouhdouch, Y.; Wezel, G.P. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 25. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.-J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, K.; Wu, N.; Li, W.; Xu, W.; Zhang, Y.; Niu, Z. Estuarine sediments are key hotspots of intracellular and extracellular antibiotic resistance genes: A high-throughput analysis in Haihe Estuary in China. Environ. Int. 2020, 135, 105385. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Li, K.; Liu, Z.; Li, Q.; Li, W.; Chen, Q.; Xia, Y.; Hu, F.; Yang, F. The Sources and Potential Hosts Identification of Antibiotic Resistance Genes in the Yellow River, Revealed by Metagenomic Analysis. Int. J. Environ. Res. Public Health 2022, 19, 10420. https://doi.org/10.3390/ijerph191610420
Zhang K, Li K, Liu Z, Li Q, Li W, Chen Q, Xia Y, Hu F, Yang F. The Sources and Potential Hosts Identification of Antibiotic Resistance Genes in the Yellow River, Revealed by Metagenomic Analysis. International Journal of Environmental Research and Public Health. 2022; 19(16):10420. https://doi.org/10.3390/ijerph191610420
Chicago/Turabian StyleZhang, Kai, Kuangjia Li, Ziyi Liu, Qidi Li, Wenpeng Li, Qi Chen, Yangchun Xia, Feiyue Hu, and Fengxia Yang. 2022. "The Sources and Potential Hosts Identification of Antibiotic Resistance Genes in the Yellow River, Revealed by Metagenomic Analysis" International Journal of Environmental Research and Public Health 19, no. 16: 10420. https://doi.org/10.3390/ijerph191610420
APA StyleZhang, K., Li, K., Liu, Z., Li, Q., Li, W., Chen, Q., Xia, Y., Hu, F., & Yang, F. (2022). The Sources and Potential Hosts Identification of Antibiotic Resistance Genes in the Yellow River, Revealed by Metagenomic Analysis. International Journal of Environmental Research and Public Health, 19(16), 10420. https://doi.org/10.3390/ijerph191610420