
Effects of Microencapsulated Ferulic Acid or Its Prodrug Methyl Ferulate on Neuroinflammation Induced by Muramyl Dipeptide

 , , ,
, , ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
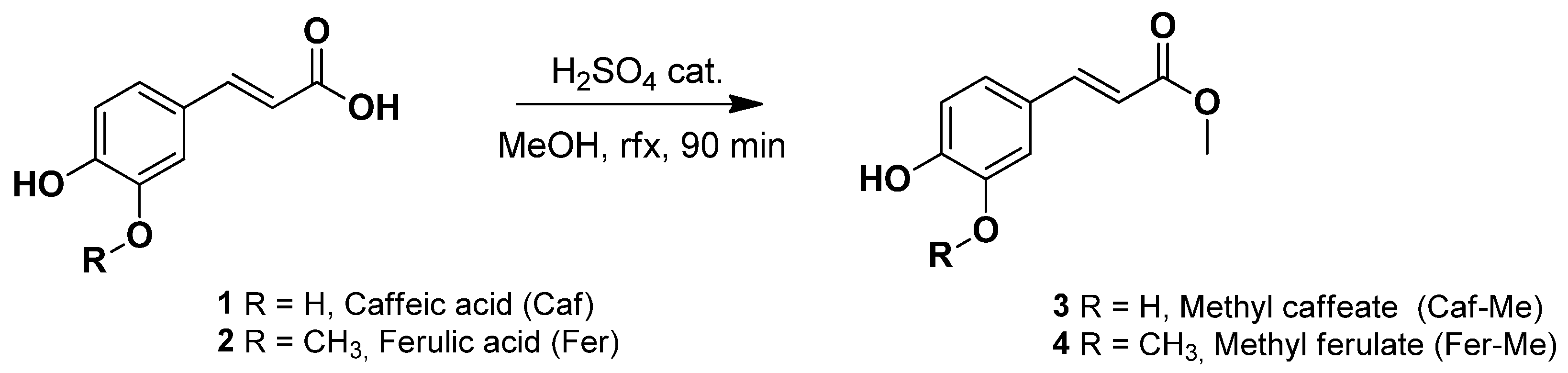
2.2. General Procedure for the Synthesis of Methyl Caffeate (3) and Methyl Ferulate (4) via Fischer Esterification
2.3. Antioxidant Activity
2.4. HPLC Analysis
2.5. Ferulic Acid and Methyl Ferulate Stock Solutions
2.6. Kinetic Analysis in Tris-HCl
2.7. Kinetic Analysis in Whole Blood
2.8. Preparation of Rat Liver Homogenates
2.9. Preparation of Rat Brain Homogenates
2.10. Kinetic Analysis in Rat Brain or Rat Liver Homogenates
2.11. Kinetic Calculations
2.12. PC12 Cell Culture and Treatment
2.13. Enzyme-Linked Immunosorbent Assay (ELISA)
2.14. Preparation of Ferulic Acid or Methyl Ferulate Loaded Microparticles
2.15. Microparticle Characterization
2.16. Ferulic Acid or Methyl Ferulate Content in the SLMs
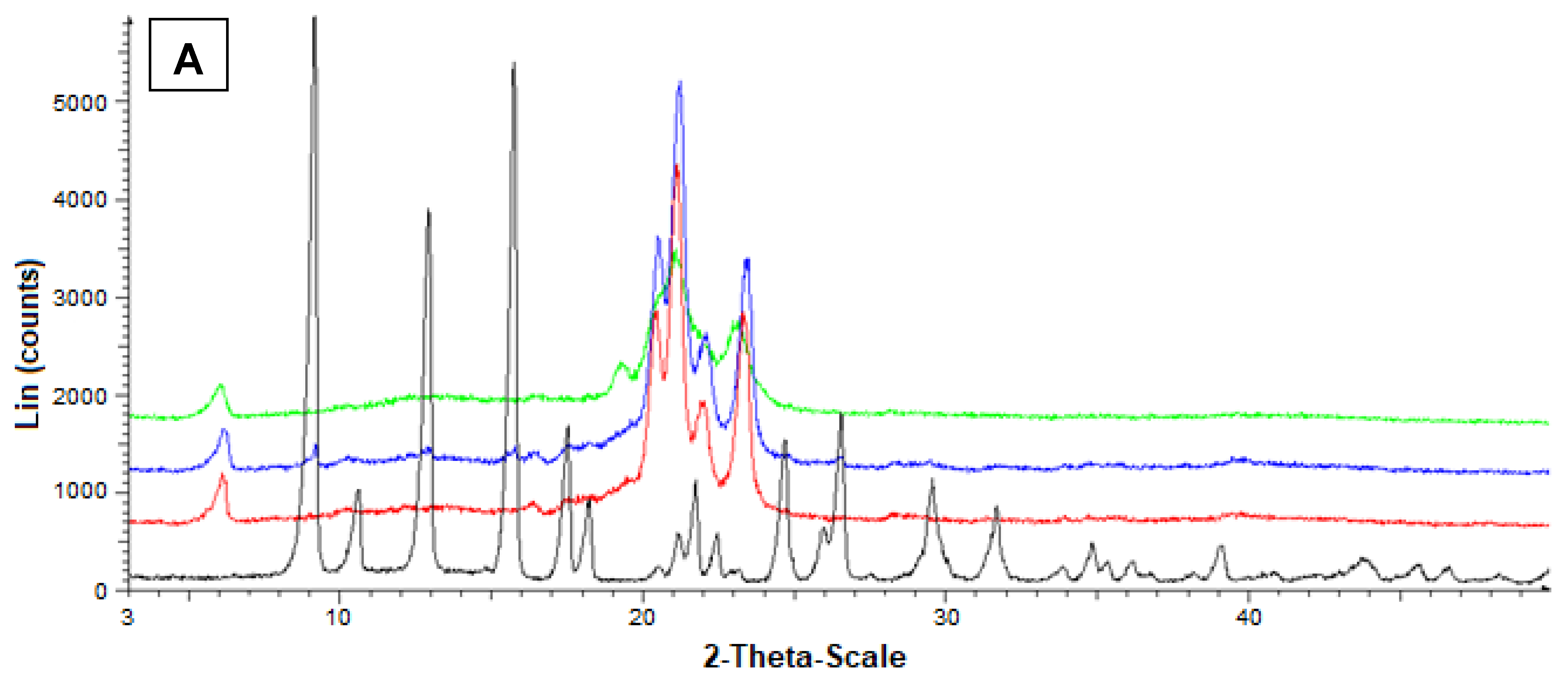
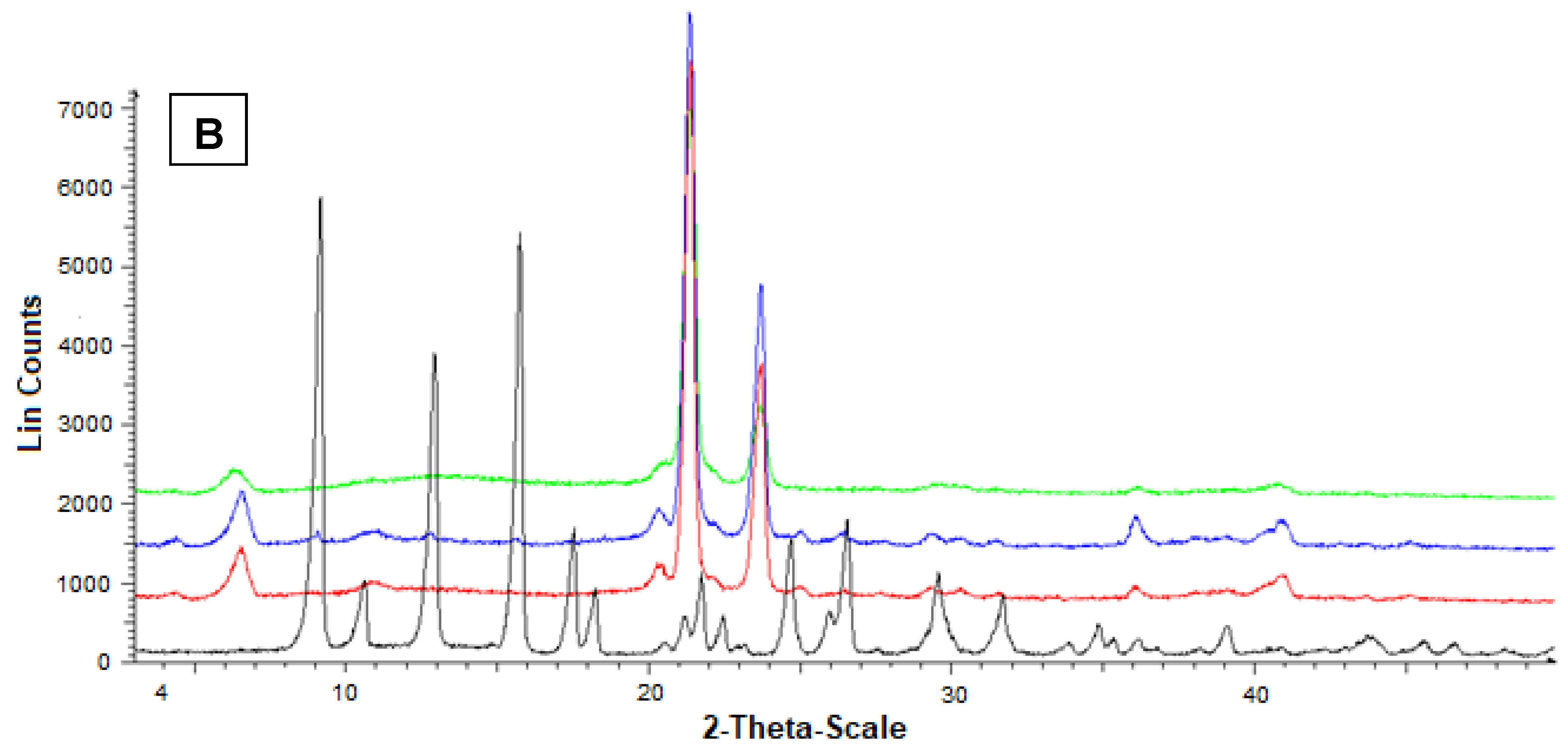
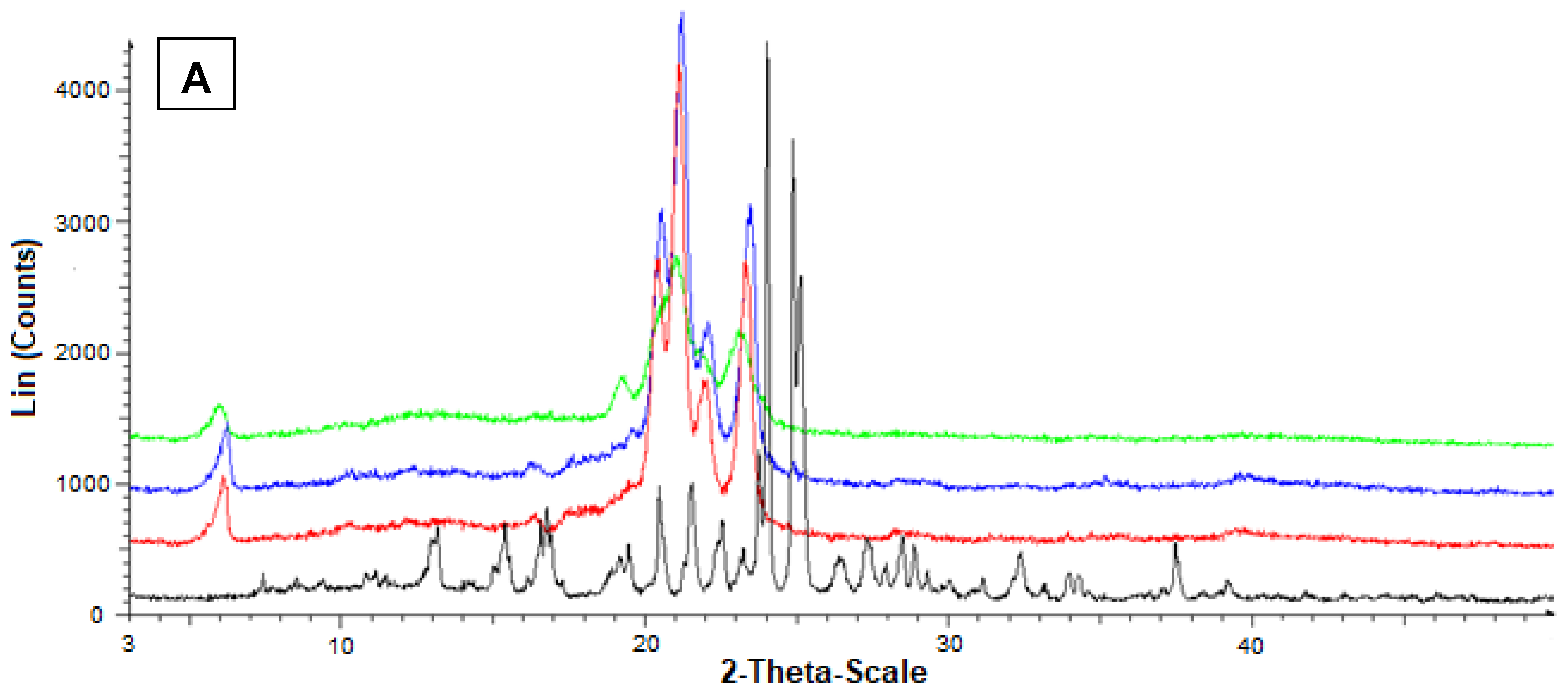
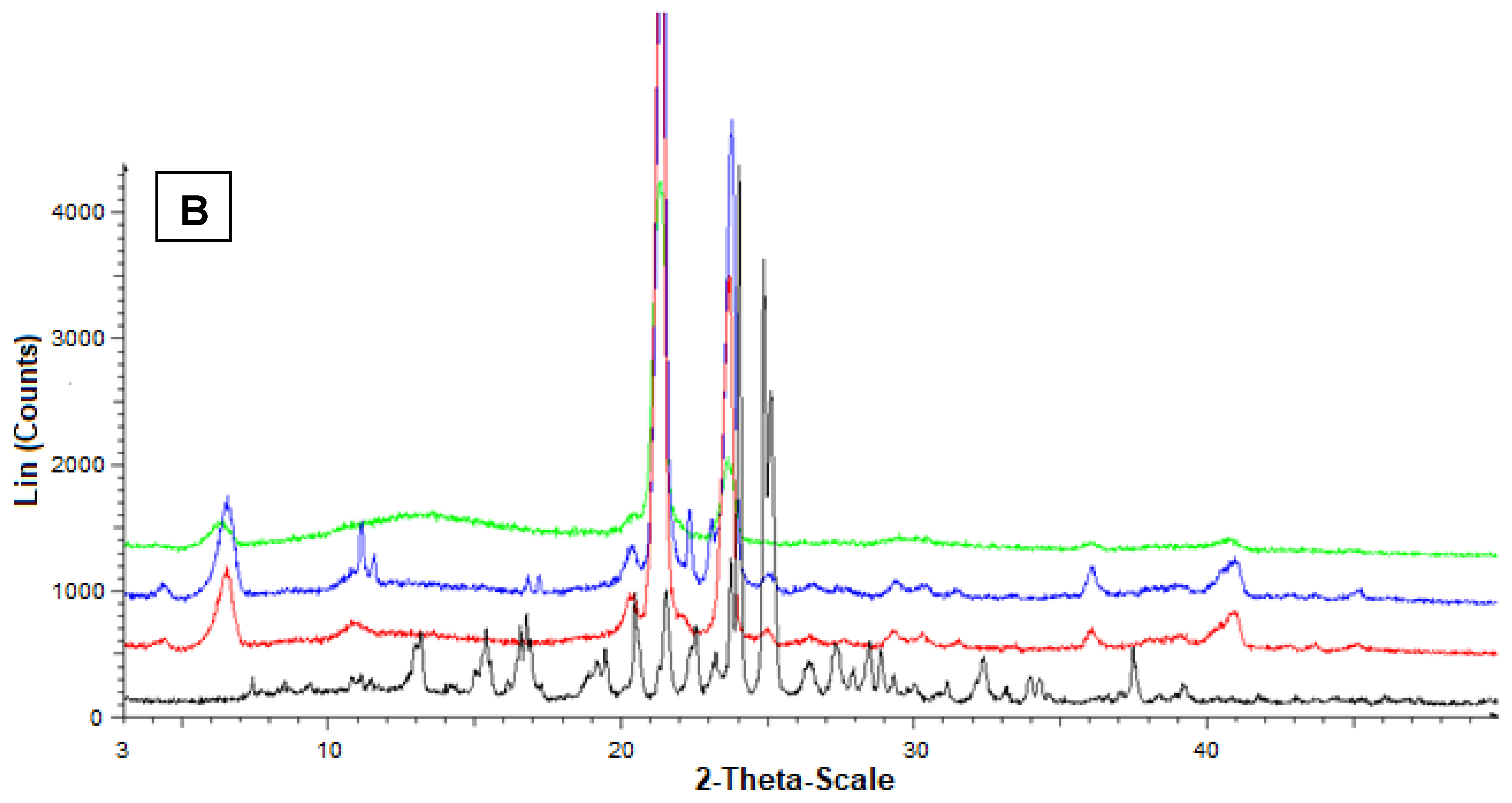
2.17. Powder X-ray Diffraction Analysis
2.18. Differential Scanning Calorimetry
2.19. Kinetic Analysis in Phosphate Buffer Saline
2.20. In Vitro Dissolution and Release Studies from SLMs
3. Results and Discussion
3.1. Synthesis of Methyl Ferulate and Methyl Caffeate
3.2. Antioxidant Activity
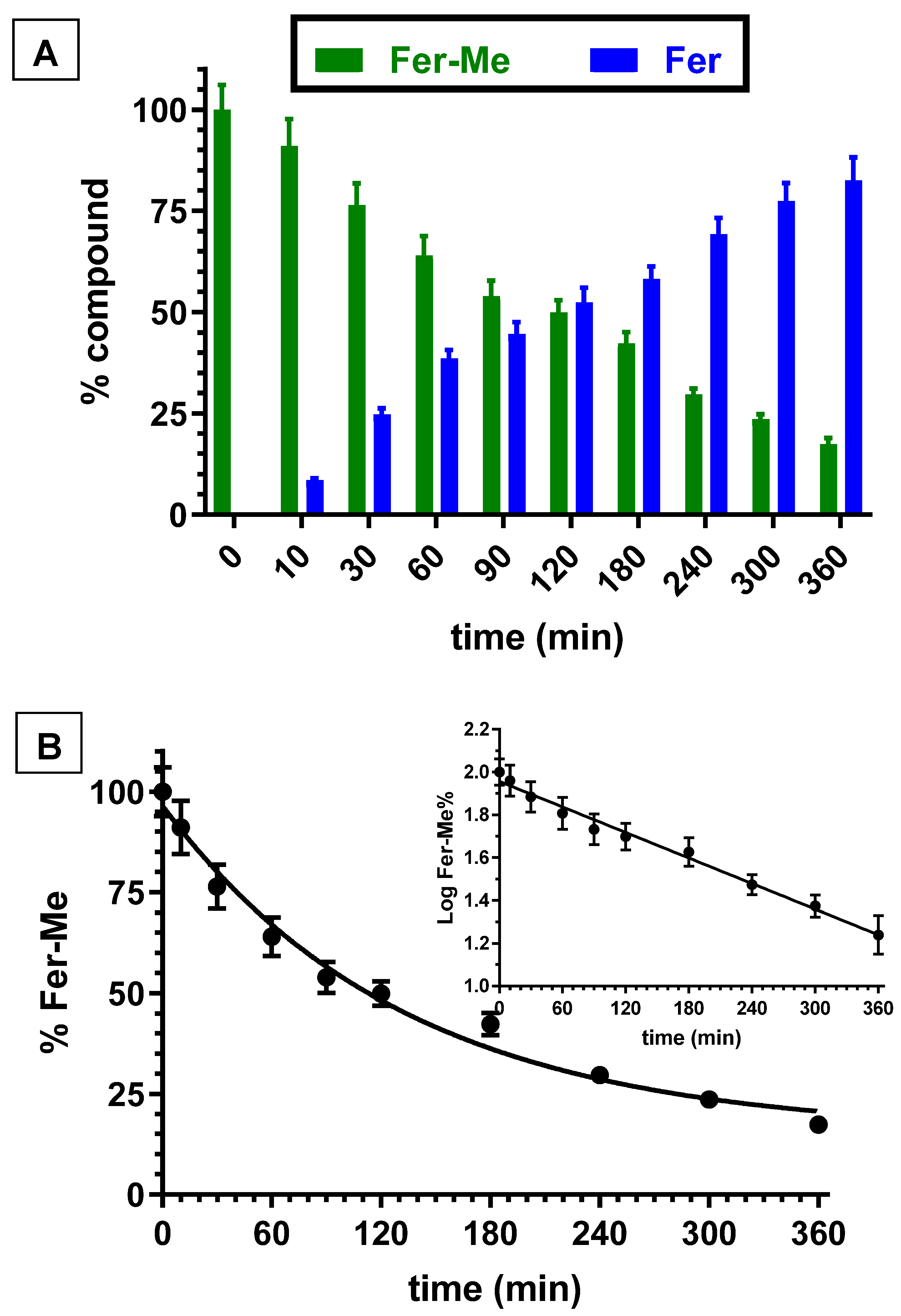
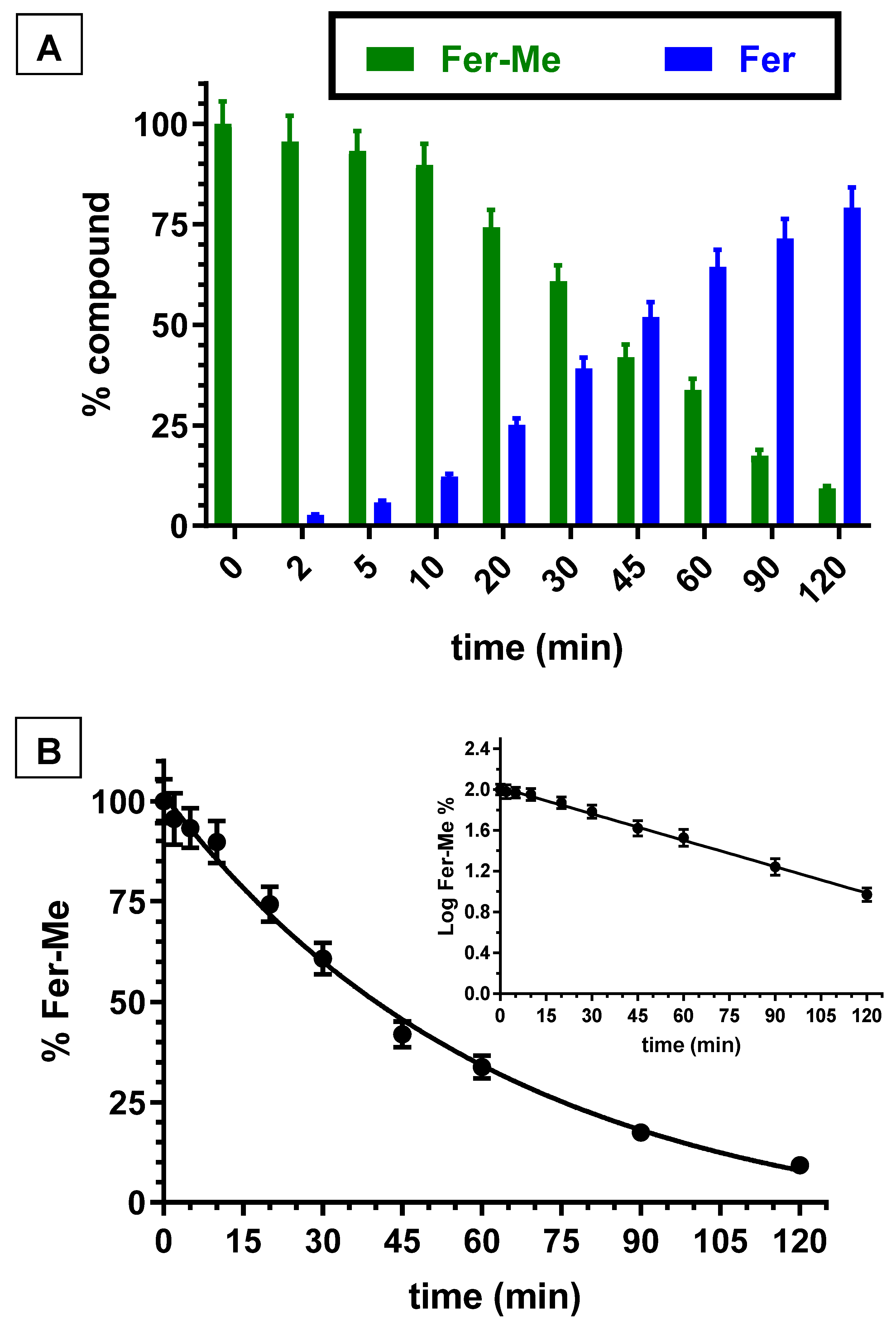
3.3. Hydrolysis Studies of Methyl Ferulate
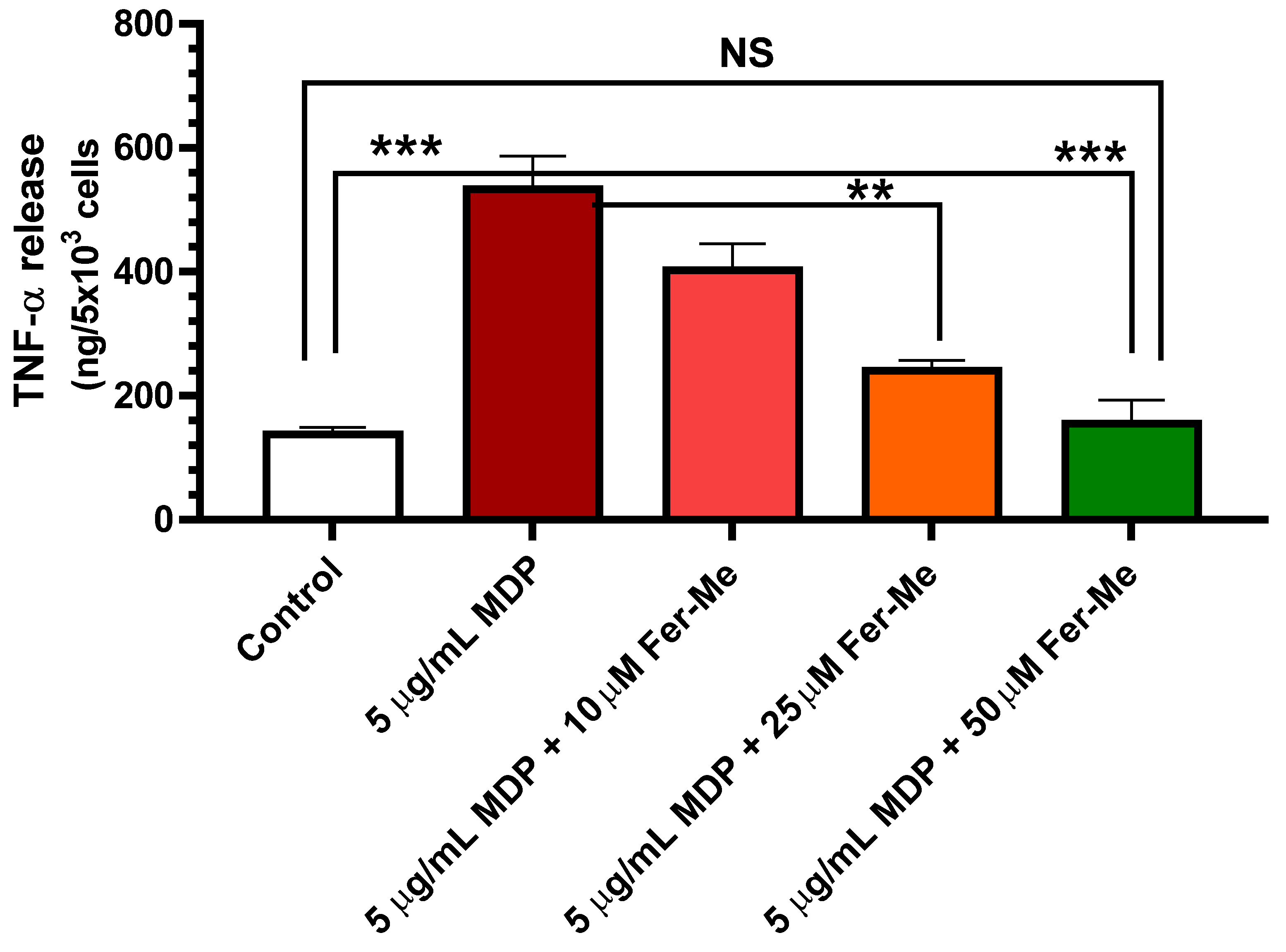
3.4. Fer-Me Counteracts the MDP-Evoked Release of Pro-Inflammatory Cytokine TNF-α in PC12 Cells
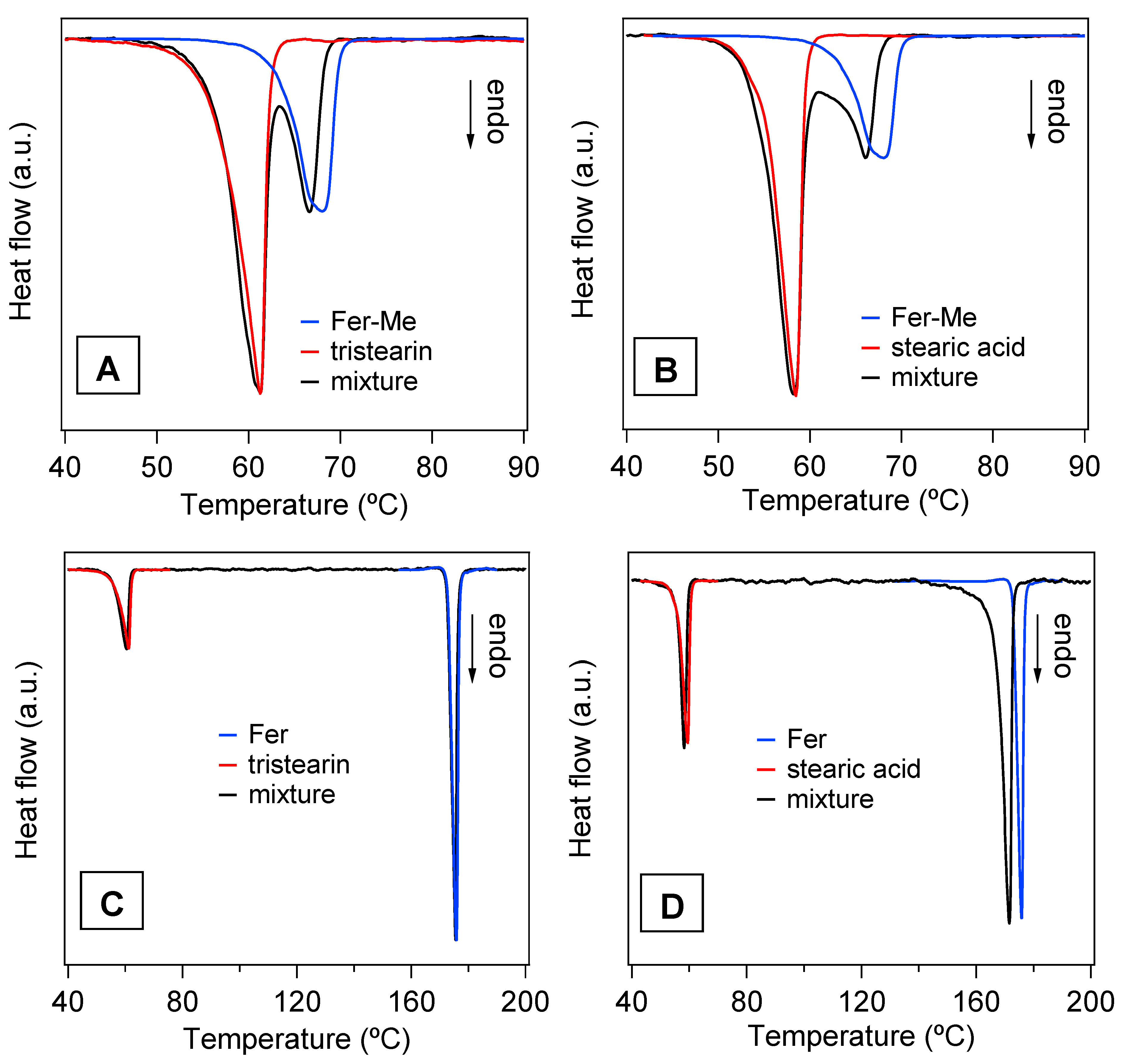
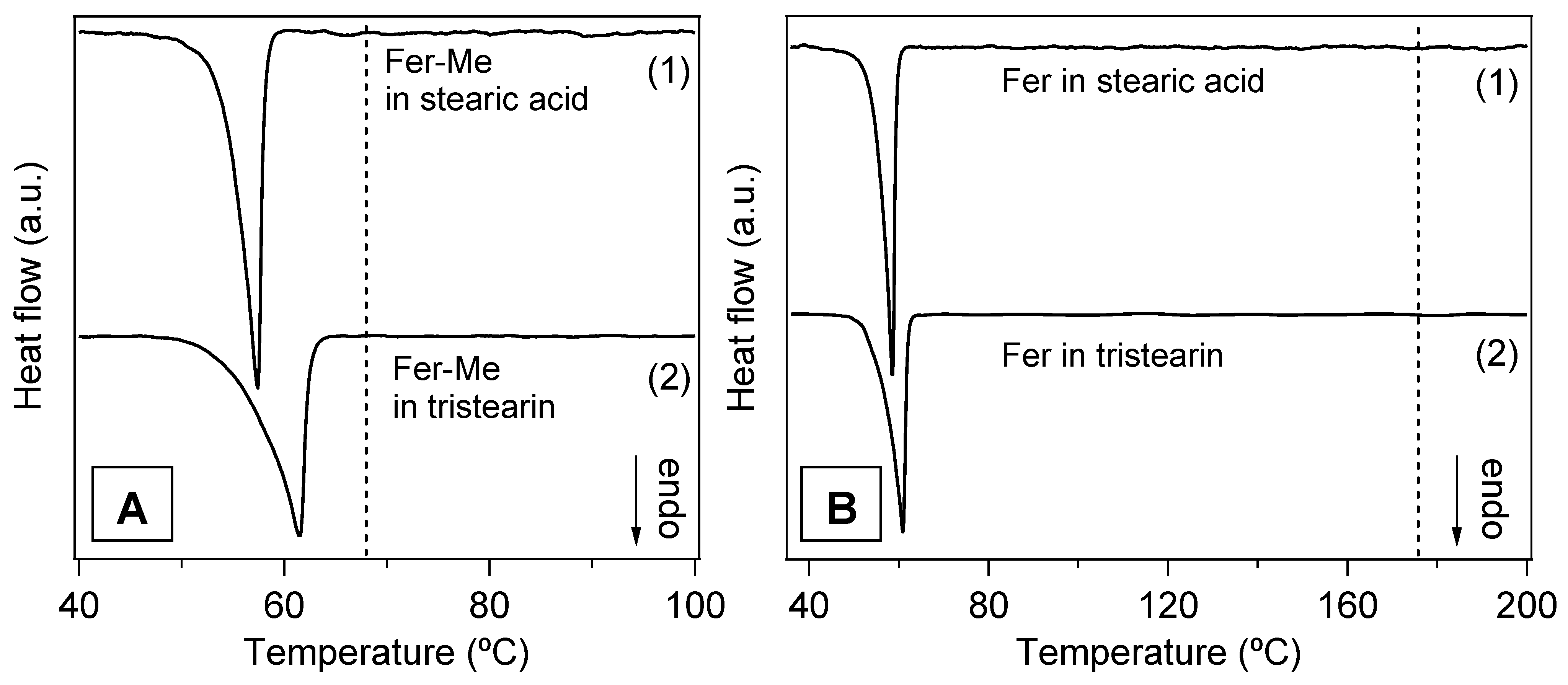
3.5. Preparation and Characterization of the SLMs
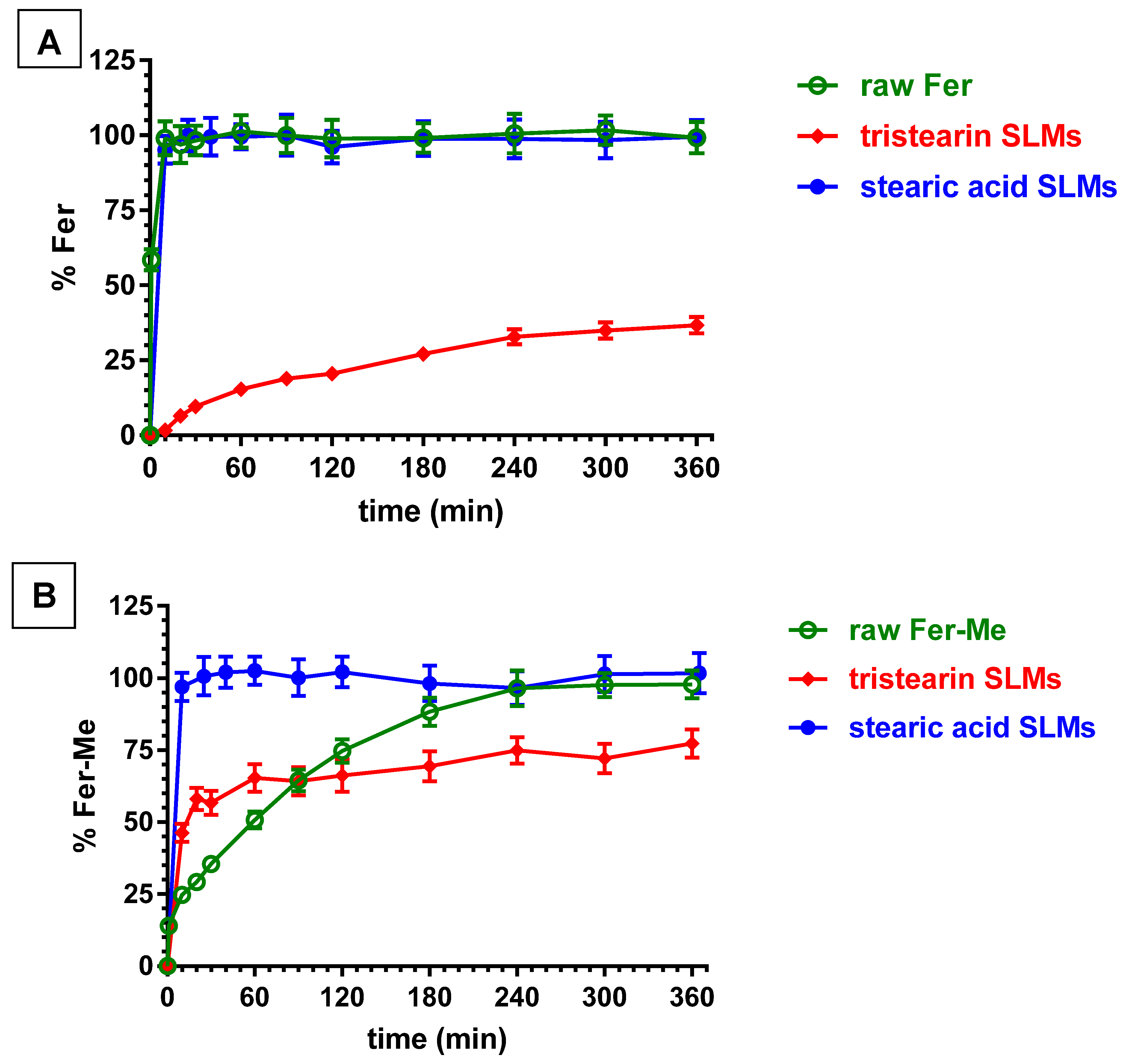
3.6. In Vitro Ferulic Acid or Methyl Ferulate Dissolution and Release from SLMs
4. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marcuzzi, A.; Secchiero, P.; Crovella, S.; Zauli, G. TRAIL administration down-modulated the acute systemic inflammatory response induced in a mouse model by muramyldipeptide or lipopolysaccharide. Cytokine 2012, 60, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Galeazzi, M.; Gasbarrini, G.; Ghirardello, A.; Grandemange, S.; Hoffman, H.M.; Manna, R.; Podswiadek, M.; Punzi, L.; Sebastiani, G.D.; Touitou, I.; et al. Autoinflammatory syndromes. Clin. Exp. Rheumatol. 2006, 24, S79–S85. [Google Scholar] [PubMed]
- Doty, K.R.; Guillot-Sestier, M.V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Crispino, M.; Trinchese, G.; Penna, E.; Cimmino, F.; Catapano, A.; Villano, I.; Perrone-Capano, C.; Mollica, M.P. Interplay between peripheral and central inflammation in obesity-promoted disorders: The impact on synaptic mitochondrial functions. Int. J. Mol. Sci. 2020, 21, 5964. [Google Scholar] [CrossRef]
- Santa-Cecília, F.V.; Ferreira, D.W.; Guimaraes, R.M.; Cecilio, N.T.; Fonseca, M.M.; Lopes, A.H.; Davoli-Ferreira, M.; Kusuda, R.; Souza, G.R.; Nachbur, U.; et al. The NOD2 signaling in peripheral macrophages contributes to neuropathic pain development. Pain 2019, 160, 102–116. [Google Scholar] [CrossRef]
- Barnabei, L.; Laplantine, E.; Mbongo, W.; Rieux-Laucat, F.; Weil, R. NF-κB: At the borders of auto-immunity and inflammation. Front. Immunol. 2021, 12, 716469. [Google Scholar] [CrossRef]
- Lampiasi, N.; Montana, G. An in vitro inflammation model to study the Nrf2 and NF-κB crosstalk in presence of ferulic acid as modulator. Immunobiology 2018, 223, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Rui, Y.X.; Guo, S.D.; Luan, F.; Liu, R.; Zeng, N. Ferulic acid: A review of its pharmacology, pharmacokinetics and derivatives. Life Sci. 2021, 284, 119921. [Google Scholar] [CrossRef]
- Stompor-Gorący, M.; Machaczka, M. Recent advances in biological activity, new formulations and prodrugs of ferulic acid. Int. J. Mol. Sci. 2021, 22, 12889. [Google Scholar] [CrossRef]
- Tarnawski, M.; Depta, K.; Grejciun, D.; Szelepin, B. HPLC determination of phenolic acids and antioxidant activity in concentrated peat extract--a natural immunomodulator. J. Pharm. Biomed. Anal. 2006, 41, 182–188. [Google Scholar] [CrossRef]
- Zduńska, K.; Dana, A.; Kolodziejczak, A.; Rotsztejn, H. Antioxidant properties of ferulic acid and its possible application. Skin Pharmacol. Physiol. 2018, 31, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Basak, P.; Dutta, S.; Chowdhury, S.; Sil, P.C. New insights into the ameliorative effects of ferulic acid in pathophysiological conditions. Food Chem. Toxicol. 2017, 103, 41–55. [Google Scholar] [CrossRef]
- Thapliyal, S.; Singh, T.; Handu, S.; Bisht, M.; Kumari, P.; Arya, P.; Srivastava, P.; Gandham, R. A review on potential footprints of ferulic acid for treatment of neurological disorders. Neurochem. Res. 2021, 46, 1043–1057. [Google Scholar] [CrossRef]
- Li, Y.; Liu, C.; Zhang, Y.; Mi, S.; Wang, N. Pharmacokinetics of ferulic acid and potential interactions with Honghua and clopidogrel in rats. J. Ethnopharmacol. 2011, 137, 562–567. [Google Scholar] [CrossRef]
- Zhao, Z.; Egashira, Y.; Sanada, H. Digestion and absorption of ferulic acid sugar esters in rat gastrointestinal tract. J. Agric. Food Chem. 2003, 51, 5534–5539. [Google Scholar] [CrossRef]
- Zafra-Gómez, A.; Luzón-Toro, B.; Jiménez-Diaz, I.; Ballesteros, O.; Navalón, A. Quantification of phenolic antioxidants in rat cerebrospinal fluid by GC-MS after oral administration of compounds. J. Pharm. Biomed. Anal. 2010, 53, 103–108. [Google Scholar] [CrossRef]
- Liu, C.S.; Chen, L.; Hu, Y.N.; Dai, J.L.; Ma, B.; Tang, Q.F.; Tan, X.M. Self-microemulsifying drug delivery system for improved oral delivery and hypnotic efficacy of ferulic acid. Int. J. Nanomed. 2020, 15, 2059–2070. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, W.; Zhang, Y.; Wang, Q.; Qin, C.; Du, S.; Huang, L.; Ye, F.; Chen, L.; Zheng, T. Pharmacokinetics, bioavailability, and tissue distribution study of angoroside c and its metabolite ferulic acid in rat using UPLC-MS/MS. Front. Pharmacol. 2018, 9, 1186. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Lee, S.Y.; Wu, X.; Tyler, J.Y.; Wang, H.; Ouyang, Z.; Park, K.; Xu, X.M.; Cheng, J.X. Neuroprotective ferulic acid (FA)-glycol chitosan (GC) nanoparticles for functional restoration of traumatically injured spinal cord. Biomaterials 2014, 35, 2355–2364. [Google Scholar] [CrossRef]
- Hassanzadeh, P.; Arbabi, E.; Atyabi, F.; Dinarvand, R. Ferulic acid-loaded nanostructured lipid carriers: A promising nanoformulation against the ischemic neural injuries. Life Sci. 2018, 193, 64–76. [Google Scholar] [CrossRef]
- Saini, S.; Sharma, T.; Jain, A.; Kaur, H.; Katare, O.P.; Singh, B. Systematically designed chitosan-coated solid lipid nanoparticles of ferulic acid for effective management of Alzheimer’s disease: A preclinical evidence. Colloids Surf. B Biointerfaces 2021, 205, 111838. [Google Scholar] [CrossRef] [PubMed]
- Puris, E.; Gynther, M.; Huttunen, J.; Auriola, S.; Huttunen, K.M. L-type amino acid transporter 1 utilizing prodrugs of ferulic acid revealed structural features supporting the design of prodrugs for brain delivery. Eur. J. Pharm. Sci. 2019, 129, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Jaspart, S.; Piel, G.; Delattre, L.; Evrard, B. Solid lipid microparticles: Formulation, preparation, characterization, drug release and applications. Expert Opin. Drug Deliv. 2005, 2, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Trotta, M.; Cavalli, R.; Carlotti, M.E.; Battaglia, L.; Debernardi, F. Solid lipid micro-particles carrying insulin formed by solvent-in-water emulsion–diffusion technique. Int. J. Pharm. 2005, 288, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Jaspart, S.; Bertholet, P.; Piel, G.; Dogné, J.G.; Delattre, L.; Evrard, B. Solid lipid microparticles as a sustained release system for pulmunary drug delivery. Eur. J. Pharm. Biopharm. 2007, 65, 47–56. [Google Scholar] [CrossRef]
- Tursilli, R.; Piel, G.; Delattre, L.; Scalia, S. Solid lipid microparticles containing the sunscreen agent, octyl-dimethylaminobenzoate: Effect of the vehicle. Eur. J. Pharm. Biopharm. 2007, 66, 483–487. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Cacciari, B.; Mezzena, M.; Strada, M.; Scalia, S. Solid lipid microparticles for the stability enhancement of a dopamine prodrug. J. Pharm. Sci. 2010, 99, 4730–4737. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Ferraro, L.; Perrone, D.; Leo, E.; Iannuccelli, V.; Pavan, B.; Paganetto, G.; Beggiato, S.; Scalia, S. Brain uptake of a Zidovudine prodrug after nasal administration of solid lipid microparticles. Mol. Pharm. 2014, 11, 1550–1561. [Google Scholar] [CrossRef]
- De Oliveira Junior, E.R.; Truzzi, E.; Ferraro, L.; Fogagnolo, M.; Pavan, B.; Beggiato, S.; Rustichelli, C.; Maretti, E.; Lima, E.M.; Leo, E.; et al. Nasal administration of nanoencapsulated geraniol/ursodeoxycholic acid conjugate: Towards a new approach for the management of Parkinson’s disease. J. Control. Release 2020, 321, 540–552. [Google Scholar] [CrossRef]
- Fukumoto, L.R.; Mazza, G. Assessing antioxidant and prooxidant activities of phenolic compounds. J. Agric. Food Chem. 2000, 48, 3597–3604. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Perez-Castillo, Y.; Lima, T.C.; Ferreira, A.R.; Silva, C.R.; Campos, R.S.; Neto, J.B.A.; Magalhães, H.I.F.; Cavalcanti, B.C.; Júnior, H.V.N.; de Sousa, D.P. Bioactivity and molecular docking studies of derivatives from cinnamic and benzoic acids. Biomed Res. Int. 2020, 2020, 6345429. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.H.N.; Andrade, A.C.M.; Nóbrega, D.F.; de Castro, R.D.; Pessôa, H.L.F.; Rani, N.; de Sousa, D.P. Antimicrobial activity of 4-chlorocinnamic acid derivatives. Biomed Res. Int. 2019, 2019, 3941242. [Google Scholar] [CrossRef]
- Chen, L.; Liang, Y.; Song, T.; Anjum, K.; Wang, W.; Yu, S.; Huang, H.; Lian, X.Y.; Zhang, Z. Synthesis and bioactivity of tripolinolate A from Tripolium vulgare and its analogs. Bioorg. Med. Chem. Lett. 2015, 25, 2629–2633. [Google Scholar] [CrossRef]
- Prevost, M.S.; Delarue-Cochin, S.; Marteaux, J.; Colas, C.; Van Renterghem, C.; Blondel, A.; Malliavin, T.; Corringer, P.J.; Joseph, D. Identification of cinnamic acid derivatives as novel antagonists of the prokaryotic proton-gated ion channel GLIC. J. Med. Chem. 2013, 56, 4619–4630. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, Y.G.; Zheng, X.L.; Dang, Y.L.; Zhu, C.M.; Zhang, R.R.; Fu, Y.Y.; Zhou, T.Y.; Li, J.H. Lipophilic ferulic acid derivatives protect PC12 cells against oxidative damage via modulating β-amyloid aggregation and activating Nrf2 enzymes. Food Funct. 2020, 11, 4707–4718. [Google Scholar] [CrossRef] [PubMed]
- Jobsis, P.D.; Rothstein, E.C.; Balaban, R.S. Limited utility of acetoxymethyl (AM)-based intracellular delivery systems, in vivo: Interference by extracellular esterases. J. Microsc. 2007, 226, 74–81. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Paganetto, G.; Pavan, B.; Fogagnolo, M.; Medici, A.; Beggiato, S.; Perrone, D. Zidovudine and ursodeoxycholic acid conjugation: Design of a new prodrug potentially able to bypass the active efflux transport systems of the central nervous system. Mol. Pharm. 2012, 9, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Pavan, B.; Simoni, D.; Baruchello, R.; Rondanin, R.; Mischiati, C.; Feriotto, G.; Ferraro, L.; Hsu, L.C.; Lee, R.M.; et al. A novel hybrid drug between two potent anti-tubulin agents as a potential prolonged anticancer approach. Eur. J. Pharm. Sci. 2016, 91, 50–63. [Google Scholar] [CrossRef]
- Wang, H.; Xu, Y.S.; Wang, M.L.; Cheng, C.; Bian, R.; Yuan, H.; Wang, Y.; Guo, T.; Zhu, L.L.; Zhou, H. Protective effect of naringin against the LPS-induced apoptosis of PC12 cells: Implications for the treatment of neurodegenerative disorders. Int. J. Mol. Med. 2017, 39, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Hong, Q.; Tan, H.L.; Xiao, C.R.; Gao, Y. Ferulic acid prevents LPS-induced up-regulation of PDE4B and stimulates the cAMP/CREB signaling pathway in PC12 cells. Acta Pharmacol. Sin. 2016, 37, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Laman, J.D.; Bert, A.; Power, C.; Dziarski, R. Bacterial peptidoglycan as a driver of chronic brain inflammation. Trends Mol. Med. 2020, 26, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.K.; Kesharwani, R.; Kumar, V. Etodolac loaded solid lipid nanoparticle based topical gel for enhanced skin delivery. Biocatal. Agric. Biotechnol. 2020, 29, 101810. [Google Scholar] [CrossRef]
- Boettinger, W.J.; Kattner, U.R.; Moon, K.-W.; Perepezko, J.H. DTA and heat-flux DSC measurements of alloy melting and freezing. In Methods for Phase Diagram Determination, 1st ed.; Zhao, J.C., Ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2007; pp. 151–221. [Google Scholar] [CrossRef]
- Jenning, V.; Thünemann, A.F.; Gohla, S.H. Characterisation of a novel solid lipid nanoparticle carrier system based on binary mixtures of liquid and solid lipids. Int. J. Pharm. 2000, 199, 167–177. [Google Scholar] [CrossRef]
- Kovačević, A.B.; Müller, R.H.; Savić, S.D.; Vuleta, G.M.; Keck, C.M. Solid lipid nanoparticles (SLN) stabilized with polyhydroxy surfactants: Preparation, characterization and physical stability investigation. Colloids Surf. A Physicochem. Eng. Asp. 2014, 444, 15–25. [Google Scholar] [CrossRef]
- Stosch, R.; Bauerecker, S.; Cammenga, H.K. The fusion behaviour of mixed crystals. A comparison between experimental and calculated calorimetric curves. Z. Phys. Chem. 1996, 194, 231–241. [Google Scholar] [CrossRef]
- Ekowati, J.; Pratama, R.P.; Nofianti, K.A.; Diyah, N.W. The temperature effect on ultrasonic-assisted of synthesis methyl ferulate and its antiplatelet assay. ALCHEMY J. Penelit. Kim. 2019, 15, 272–286. [Google Scholar] [CrossRef]
- Lerdkanchanaporn, S.; Dollimore, D. An investigation of the evaporation of stearic acid using a simultaneous TG-DTA unit. Thermochim. Acta 1998, 324, 15–23. [Google Scholar] [CrossRef]
- Ravikumar, P.; Katariya, M.; Patil, S.; Tatke, P.; Pillai, R. Skin delivery of resveratrol encapsulated lipidic formulation for melanoma chemoprevention. J. Microencapsul. 2019, 36, 535–551. [Google Scholar] [CrossRef]
- Chiu, M.; Prenner, E. Differential scanning calorimetry: An invaluable tool for a detailed thermodynamic characterization of macromolecules and their interactions. J. Pharm. Bioallied Sci. 2011, 3, 39–59. [Google Scholar] [CrossRef]
- Osanlou, R.; Emtyazjoo, M.; Banaei, A.; Hesarinejad, M.A.; Ashrafi, F. Preparation of solid lipid nanoparticles and nanostructured lipid carriers containing zeaxanthin and evaluation of physicochemical properties. Colloids Surf. A Physicochem. Eng. Asp. 2022, 641, 128588. [Google Scholar] [CrossRef]
- Müller, R.H.; Radtke, M.; Wissing, S. Solid lipid nanoparticles (SLN) and nanostructured lipid carrier (NLC) in cosmetic and dermatological preparations. Adv. Drug Deliv. Rev. 2002, 54, S131–S155. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Mezzena, M.; Scatturin, A.; Scalia, S. Solid lipid microparticles for the stability enhancement of the polar drug N6-cyclopentyladenosine. Int. J. Pharm. 2008, 355, 81–86. [Google Scholar] [CrossRef]
- Salem, H.F. Sustained-release progesterone nanosuspension following intramuscular injection in ovariectomized rats. Int. J. Nanomed. 2010, 5, 943–954. [Google Scholar] [CrossRef]
- Umeyor, E.C.; Kenechukwu, F.C.; Ogbonna, J.D.; Chime, S.A.; Attama, A. Preparation of novel solid lipid microparticles loaded with gentamicin and its evaluation in vitro and in vivo. J. Microencapsul. 2012, 29, 296–307. [Google Scholar] [CrossRef]
- Elbrink, K.; Van Hees, S.; Chamanza, R.; Roelant, D.; Loomans, T.; Holm, R.; Kiekens, F. Application of solid lipid nanoparticles as a long-term drug delivery platform for intramuscular and subcutaneous administration: In vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2021, 163, 158–170. [Google Scholar] [CrossRef]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-to-brain drug delivery: An update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J. Control. Release 2018, 281, 139–177. [Google Scholar] [CrossRef]
- Botti, G.; Dalpiaz, A.; Pavan, B. Targeting systems to the brain obtained by merging prodrugs, nanoparticles, and nasal administration. Pharmaceutics 2021, 13, 1144. [Google Scholar] [CrossRef]
- Gavini, E.; Rassu, G.; Ferraro, L.; Beggiato, S.; Alhalaweh, A.; Velaga, S.; Marchetti, N.; Bandiera, P.; Giunchedi, P.; Dalpiaz, A. Influence of polymeric microcarriers on the in vivo intranasal uptake of an anti-migraine drug for brain targeting. Eur. J. Pharm. Biopharm. 2013, 83, 174–183. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Fogagnolo, M.; Ferraro, L.; Capuzzo, A.; Pavan, B.; Rassu, G.; Salis, A.; Giunchedi, P.; Gavini, E. Nasal chitosan microparticles target a zidovudine prodrug to brain HIV sanctuaries. Antivir. Res. 2015, 123, 146–157. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SLMs Based Lipid | Loaded Compound | Drug Loading (%) | Encapsulation Efficiency (%) |
|---|---|---|---|
| Tristearin | Fer | 0.375 ± 0.004 | 14.9 ± 0.2 |
| Fer-Me | 0.719 ± 0.005 | 27.9 ± 0.2 | |
| Stearic Acid | Fer | 0.946 ± 0.015 | 37.8 ± 0.6 |
| Fer-Me | 1.507 ± 0.014 | 59.3 ± 0.6 |
| Compound | TO (°C) (±0.4 °C) | TP (°C) (±0.4 °C) |
|---|---|---|
| Stearic acid | 55.7 | 59.5 |
| Tristearin | 56.5 | 61.7 |
| Fer-Me | 62.8 | 68.0 |
| Fer | 172.8 | 175.8 |
| Stearic acid in stearic acid + Fer-Me | 53.5 | 57.4 |
| Stearic acid in stearic acid + Fer | 54.2 | 58.5 |
| Tristearin in tristearin + Fer-Me | 54.9 | 61.5 |
| Tristearin in tristearin + Fer | 54.5 | 60.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botti, G.; Bianchi, A.; Pavan, B.; Tedeschi, P.; Albanese, V.; Ferraro, L.; Spizzo, F.; Del Bianco, L.; Dalpiaz, A. Effects of Microencapsulated Ferulic Acid or Its Prodrug Methyl Ferulate on Neuroinflammation Induced by Muramyl Dipeptide. Int. J. Environ. Res. Public Health 2022, 19, 10609. https://doi.org/10.3390/ijerph191710609
Botti G, Bianchi A, Pavan B, Tedeschi P, Albanese V, Ferraro L, Spizzo F, Del Bianco L, Dalpiaz A. Effects of Microencapsulated Ferulic Acid or Its Prodrug Methyl Ferulate on Neuroinflammation Induced by Muramyl Dipeptide. International Journal of Environmental Research and Public Health. 2022; 19(17):10609. https://doi.org/10.3390/ijerph191710609
Chicago/Turabian StyleBotti, Giada, Anna Bianchi, Barbara Pavan, Paola Tedeschi, Valentina Albanese, Luca Ferraro, Federico Spizzo, Lucia Del Bianco, and Alessandro Dalpiaz. 2022. "Effects of Microencapsulated Ferulic Acid or Its Prodrug Methyl Ferulate on Neuroinflammation Induced by Muramyl Dipeptide" International Journal of Environmental Research and Public Health 19, no. 17: 10609. https://doi.org/10.3390/ijerph191710609
APA StyleBotti, G., Bianchi, A., Pavan, B., Tedeschi, P., Albanese, V., Ferraro, L., Spizzo, F., Del Bianco, L., & Dalpiaz, A. (2022). Effects of Microencapsulated Ferulic Acid or Its Prodrug Methyl Ferulate on Neuroinflammation Induced by Muramyl Dipeptide. International Journal of Environmental Research and Public Health, 19(17), 10609. https://doi.org/10.3390/ijerph191710609