Smoking Related Diseases: The Central Role of Monoamine Oxidase

Abstract
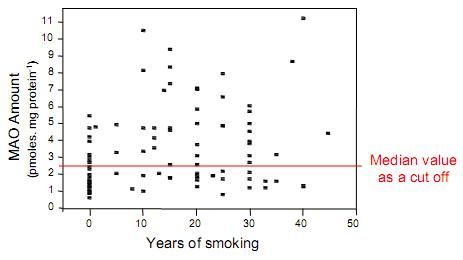
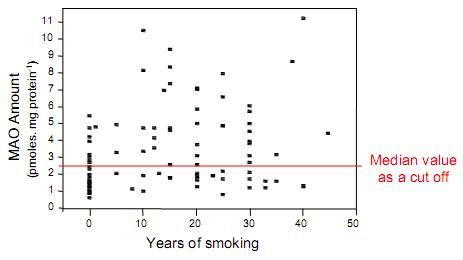
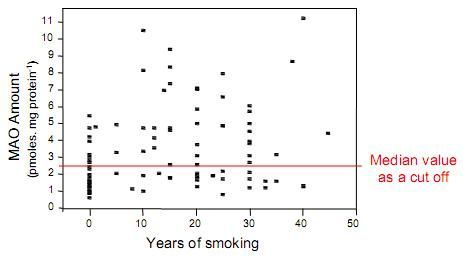
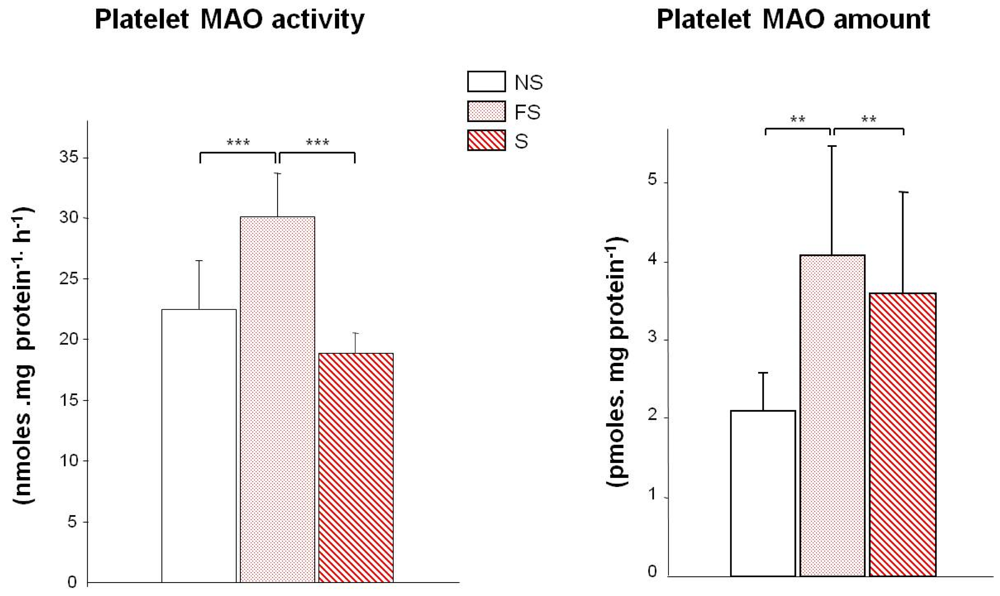
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Smoking Is a Prevalent Risk Factor of Morbidity and Mortality
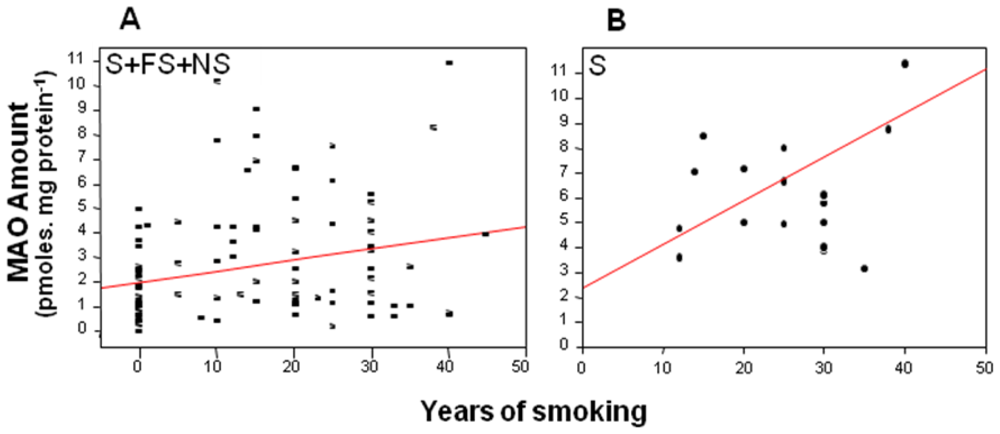
2. MAO and Smoking
3. Bioamines Catabolism in Smokers
3.1. 5-HT and 5-HIAA
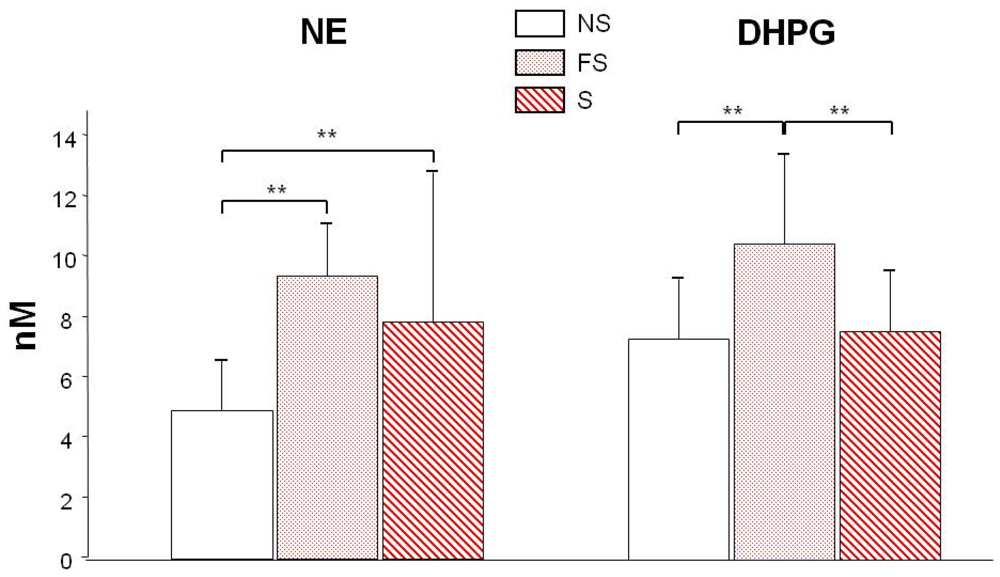
3.2. NE and DHPG
4. Is MAO a Risk Marker?
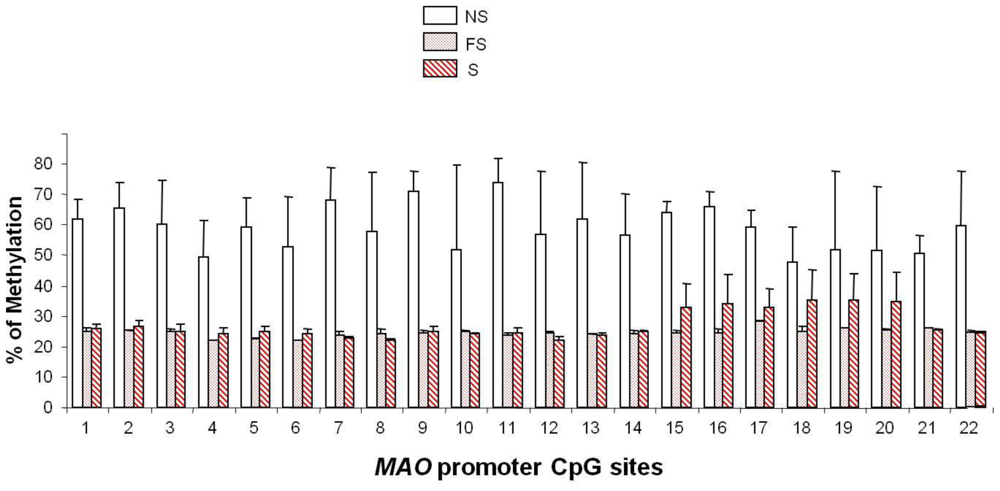
5. Smoking-induced Epigenetic Modification of MAO
6. MAO in Smokers: From Cardiovascular Diseases to Mood Disorders
7. Conclusions
References
- Ambrose, JA; Barua, RS. The pathophysiology of cigarette smoking and cardiovascular disease. An Update. J. Am. Coll. Cardiol 2004, 43, 1731–1737. [Google Scholar]
- Changeux, J-P. Nicotine addiction and nicotinic receptors: Lessons from genetically modified mice. Nature 2010, 11, 389–401. [Google Scholar]
- Benowitz, NL. Nicotine addiction. N. Engl. J. Med 2010, 362, 2295–2303. [Google Scholar]
- Rose, JE. Nicotine and nonnicotine factors in cigarette addiction. Psychopharmacology 2006, 184, 274–285. [Google Scholar]
- Lewis, A; Miller, JH; Lea, RA. Monoamine oxidase and tobacco dependence. Neurotoxicology 2007, 28, 182–195. [Google Scholar]
- Essman, WB. Serotonin and monoamine oxidase in mouse skin: Effects of cigarette smoke exposure. J. Med 1977, 8, 95–101. [Google Scholar]
- Oreland, L; Fowler, CJ; Schalling, D. Low platelet monoamine oxidase activity in cigarette smokers. Life Sci 1981, 29, 2511–2518. [Google Scholar]
- Berlin, I; Said, S; Spreux-Varoquaux, O; Olivares, R; Launay, J-M; Puech, AJ. Monoamine oxidase A and B activities in heavy smokers. Biol. Psychiat 1995, 38, 756–761. [Google Scholar]
- Fowler, JS; Volkow, ND; Wang, G-J; Pappas, N; Logan, J; Shea, C; Alexoff, D; MacGregor, R; Schyler, D; Zezulkova, I; Wolf, AP. Brain monoamine oxidase A inhibition in cigarette smokers. Proc. Natl. Acad. Sci. USA 1996, 93, 14065–14069. [Google Scholar]
- Fowler, JS; Volkow, ND; Wang, G-J; Pappas, N; Logan, J; MacGregor, R; Alexoff, D; Shea, C; Schyler, D; Wolf, AP; Warner, D; Zezulkova, I; Cilento, R. Inhibition of monoamine oxidase B in the brains of smokers. Nature 1996, 379, 733–736. [Google Scholar]
- Berlin, I; Anthenelli, RM. Monoamine oxidases and tobacco smoking. Int. J. Neuropsychopharmacol 2001, 4, 33–42. [Google Scholar]
- Pietri, M; Schneider, B; Mouillet-Richard, S; Ermonval, M; Mutel, V; Launay, JM; Kellerman, O. Reactive oxygen species-dependent TNF-alpha converting enzyme activation through stimulation of 5-HT2B and alpha1D autoreceptors in neuronal cells. FASEB J 2005, 19, 1078–1087. [Google Scholar]
- Eddahibi, S; Guignabert, C; Barlier-Mur, AM; Dawachter, L; Fadel, E; Dartevelle, P; Humbert, M; Simonneau, G; Hanoun, N; Saurini, F; Hamon, M; Adnot, S. Cross-talk between endothelial and smoothmuscle cells in pulmonary hypertension: critical role for serotonin-induced smooth muscle hyperplasia. Circulation 2006, 113, 1857–1864. [Google Scholar]
- Liu, Y; Li, M; Warburton, RR; Hill, NS; Fanburg, BL. The 5-HT transpoter transactivates the PDGF-{beta} receptor in pulmonary artery smooth muscle cells. FASEB J 2007, 21, 2725–2734. [Google Scholar]
- Toninello, A; Pietrangeli, P; De Marchi, U; Salvi, M; Mondovi, B. Amine oxidases in apoptosis and cancer. Biochim. Biophys. Acta 2006, 1765, 1–13. [Google Scholar]
- Lan, NC; Heinzmann, C; Gal, A; Klisak, I; Orth, U; Lai, E; Grimsby, J; Sparkes, RS; Mohandas, T; Shih, JC. Human monoamine oxidase A and B genes map to Xp11.23 and are deleted in a patient with Norrie disease. Genomics 1989, 4, 552–559. [Google Scholar]
- Grimsby, J; Chen, K; Wang, L-J; Lan, NC; Shih, JC. Human monoamine oxidase A and B genes exhibit identical exon-intron organization. Proc. Natl. Acad. Sci. USA 1991, 88, 3637–3641. [Google Scholar]
- Pinsonneault, JK; Papp, AC; Sadée, W. Allelic mRNA expression of X-linked monoamine oxidase a (MAOA) in human brain: dissection of epigenetic and genetic factors. Hum. Mol. Genet 2006, 15, 2636–2649. [Google Scholar]
- Shumay, E; Fowler, JS. Identification and characterization of putative methylation targets in the MAOA locus using bioinformatics approaches. Epigenetics 2010, 16, 325–342. [Google Scholar]
- Costa-Mallen, P; Costa, LG; Checkoway, H. Genotype combinations for monoamine oxidase-B intron 13 polymorphism and dopamine D2 receptor TaqIB polymorphism are associated with ever-smoking status among men. Neurosci. Lett 2005, 385, 158–162. [Google Scholar]
- Wiesbeck, GA; Wodarz, N; Weijers, HG; Dursteler-McFarland, KM; Wurts, FM; Walter, M; Boening, J. A functional polymorphism in the promoter region of the monoamine oxidase A gene is associated with the cigarette smoking quantity in alcohol-dependent heavy smokers. Neuropsychobiology 2006, 53, 181–185. [Google Scholar]
- Valette, H; Bottlaender, M; Dollé, F; Coulon, C; Ottaviani, M; Syrota, A. Acute inhibition of cardiac monoamine oxidase A after tobacco smoke inhalation : validation study of [11C]befloxatone in rats followed by a positrion emission tomography application in baboons. J. Pharmacol. Exp. Ther 2005, 314, 431–436. [Google Scholar]
- Cesura, AM; Galva, MD; Imhof, R; Da Prada, M. Binding of [3H]Ro 16–6491, a reversible inhibitor of monoamine oxidase type B, to human brain mitochondria and platelet membranes. J. Neurochem 1987, 48, 170–176. [Google Scholar]
- Launay, J-M; Del Pino, M; Chironi, G; Callebert, J; Peoc’h, K; Mégnien, J-L; Mallet, J; Simon, A; Rendu, F. Smoking induces long-lasting effects through a monoamineOxidase epigenetic regulation. PLoS ONE 2009, 4, e7959. [Google Scholar]
- Vikenes, K; Farstad, M; Nordrehaug, JE. Serotonin is associated with coronary artery disease and cardiac events. Circulation 1999, 100, 483–489. [Google Scholar]
- Lindemann, S; Krämer, B; Seizer, P; Gawaz, M. Platelets, inflammation and atherosclerosis. J. Thromb. Haemost 2007, 5, 203–211. [Google Scholar]
- Seizer, P; Gawaz, M; May, AE. Platelet-monocytes interactions—A dangerous liaison linking thrombosis, inflammation and atherosclerosis. Curr. Med. Chem 2008, 15, 1976–1980. [Google Scholar]
- Weyrich, AS; Prescott, SM; Zimmerman, GA. Platelets, endothelial cells, inflammatory chemokines, and restenosis. Complex signalling in the vascular play book. Circulation 2002, 106, 1433–1435. [Google Scholar]
- Schober, A; Manka, D; von Hundelshausen, P; Huo, Y; Hanrath, P; Sarembock, IJ; Ley, K; Weber, C. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation 2002, 106, 1523–1529. [Google Scholar]
- Harding, SA; Sarma, J; Joseph, DH; Cruden, NL; Din, JN; Twomey, PJ; Fox, KAA; Newby, DE. Upregulation of the CD40/CD40 ligand dyad and platelet-monocyte aggregation in cigarette smokers. Circulation 2004, 109, 1926–1929. [Google Scholar]
- Launay, JM; Hervé, P; Peoc’h, K; Tournois, C; Callebert, J; Nebigil, CG; Etienne, N; Drouet, L; Humbert, M; Simonneau, G; Maroteaux, L. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nature Med 2002, 10, 1129–1135. [Google Scholar]
- Sullivan, CC; Du, L; Chu, D; Cho, AJ; Kido, M; Wolf, PL; Jamieson, SW; Thistlethwaite, PA. Induction of pulmonary hypertension by an angiopoietin 1/TIE2/serotonin pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 12331–12336. [Google Scholar]
- Hervé, P; Drouet, L; Dosquet, C; Launay, JM; Rain, B; Simonneau, G; Caen, J; Duroux, P. Primary pulmonary hypertension in a patient with a familial platelet storage pool disease: Role of serotonin. Am. J. Med 1990, 89, 117–120. [Google Scholar]
- Nemecek, GM; Coughlin, SR; Handley, DA; Moskowitz, MA. Stimulation of aortic smooth muscle cell mitogenesis by serotonin. Proc. Natl. Acad. Sci. USA 1986, 83, 674–678. [Google Scholar]
- Crowley, ST; Dempsey, EC; Horwitz, KB. Platelet-induced vascular smooth muscle cell proliferation is modulated by the growth amplification factors serotonin and adenosine diphosphate. Circulation 1994, 90, 1908–1918. [Google Scholar]
- Ito, T; Ikeda, U; Shimpo, M; Yamamoto, K; Shimada, K. Serotonin increases interleukin-6 synthesis in human vascular smooth muscle cells. Circulation 2000, 102, 2522–2527. [Google Scholar]
- King, SM; McNamee, RA; Houng, AK; Patel, R; Brands, M; Reed, GL. Platelet dense granule secretion plays a critical role in thrombosis and subsequent vascular remodelling in atherosclerotic mice. Circulation 2009, 120, 785–791. [Google Scholar]
- Frishman, WH; Grewall, P. Serotonin and the heart. Ann. Med 2000, 32, 195–209. [Google Scholar]
- Anderson, HV; McNatt, J; Clubb, FJ; Herman, M; Maffrand, JP; DeClerck, F; Ahn, C; Buja, LM. Platelet inhibition reduces cyclic flow variations and neointimal proliferation in normal and hypercholesterolemic-atherosclerotic canine coronary arteries. Circulation 2001, 104, 2331–2337. [Google Scholar]
- Fusegawa, Y; Goto, S; Handa, S; Kawada, T; Ando, Y. Platelet spontaneous aggregation in platelet-rich plasma is increased in habitual smokers. Thromb. Res 1999, 93, 271–278. [Google Scholar]
- Rubenstein, D; Jesty, J; Bluestein, D. Differences between mainstream and sidestream cigarette smoke extracts and nicotine in the activation of platelets under static and flow conditions. Circulation 2004, 109, 78–83. [Google Scholar]
- Wong, WK; Chen, K; Shih, JC. Decreased methylation and transcription repressor Sp3 up-regulated human monoamine oxidase (MAO) B expression during Caco-2 differentiation. J. Biol. Chem 2003, 278, 36227–36235. [Google Scholar]
- Caiafa, P; Guastafierro, T; Zampieri, M. Epigenetics: poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. FASEB J 2009, 23, 672–678. [Google Scholar]
- Krishnakumar, R; Kraus, WL. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar]
- Nordquist, N; Oreland, L. Serotonin, genetic variability, behaviour, and psychiatric disorders—A review. Ups. J. Med. Sci 2010, 115, 2–10. [Google Scholar]
- Li, YF; Langholz, B; Salam, MT; Gilliland, FD. Maternal and grandmaternal smoking patterns are associated with early childhood asthma. Chest 2005, 127, 1232–1241. [Google Scholar]
- Berlin, I; Heilbronner, C; Georgieu, S; Meier, C; Launay, JM; Spreux-Varoquaux, O. Reduced monoamine oxidase A activity in pregnant smokers and in their newborns. Biol. Psychiat 2009, 66, 728–733. [Google Scholar]
- Rappaport, SM; Smith, MT. Environment and disease risks. Science 2010, 330, 460–461. [Google Scholar]
- Relton, CL; Smith, GD. Epigenetic epidemiology of common complex disease: Prospects for prediction, prevention, and treatment. PLoS Med 2010, 7, e10000356. [Google Scholar]




© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rendu, F.; Peoc’h, K.; Berlin, I.; Thomas, D.; Launay, J.-M. Smoking Related Diseases: The Central Role of Monoamine Oxidase. Int. J. Environ. Res. Public Health 2011, 8, 136-147. https://doi.org/10.3390/ijerph8010136
Rendu F, Peoc’h K, Berlin I, Thomas D, Launay J-M. Smoking Related Diseases: The Central Role of Monoamine Oxidase. International Journal of Environmental Research and Public Health. 2011; 8(1):136-147. https://doi.org/10.3390/ijerph8010136
Chicago/Turabian StyleRendu, Francine, Katell Peoc’h, Ivan Berlin, Daniel Thomas, and Jean-Marie Launay. 2011. "Smoking Related Diseases: The Central Role of Monoamine Oxidase" International Journal of Environmental Research and Public Health 8, no. 1: 136-147. https://doi.org/10.3390/ijerph8010136
APA StyleRendu, F., Peoc’h, K., Berlin, I., Thomas, D., & Launay, J. -M. (2011). Smoking Related Diseases: The Central Role of Monoamine Oxidase. International Journal of Environmental Research and Public Health, 8(1), 136-147. https://doi.org/10.3390/ijerph8010136