
Pathophysiology of Red Blood Cell Dysfunction in Diabetes and Its Complications

 ,
, 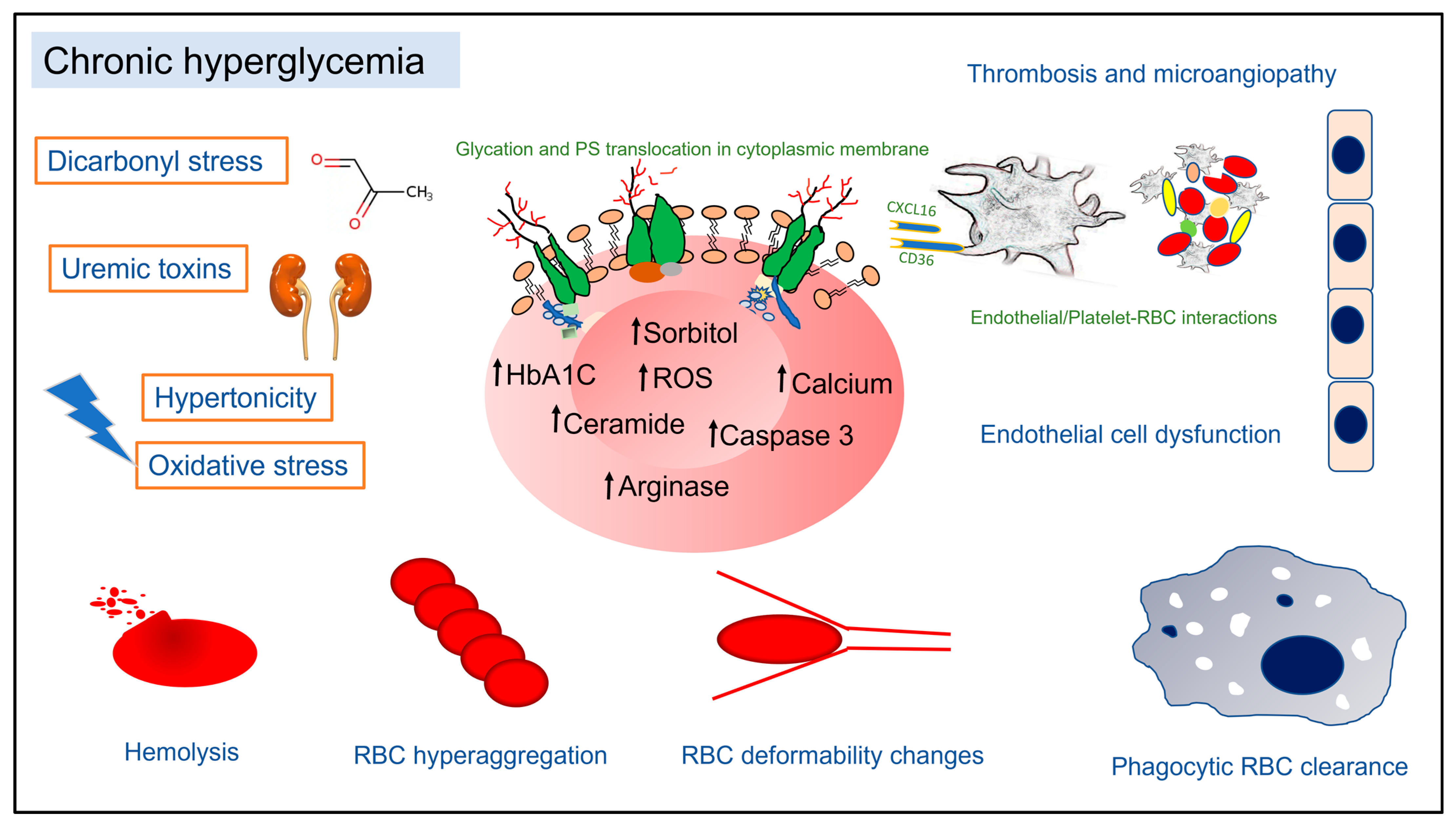{kind=link}
Abstract
:1. Introduction
2. Epidemiology and Subtypes of Anemia in Diabetes
3. Multifactorial Mechanisms of RBC Dysfunction and Anemia in Diabetes
3.1. Nephropathy and Reduced Erythropoiesis in Diabetes-Associated Anemia
3.2. Iron Deficiency in Diabetes-Associated Anemia
3.3. Role of Inflammation in Diabetes-Associated Anemia
3.4. Testosterone Deficiency as a Potential Link between Anemia and Diabetes
4. Multifactorial Mechanisms Explaining Shortened RBC Lifespan in Diabetes
4.1. Regulation of RBC Lifespan in Physiologic and Pathologic Conditions
4.2. Putative Intracellular Pathways of Cell Death Signaling in RBCs
4.3. Mechanisms of Reduced Lifespan of RBCs in Diabetes
4.4. Influence of Concurrent Systemic Conditions on RBC Survival in Diabetes
4.5. Impact of In Vitro Glycation on RBC Damage
4.6. Dicarbonyl Stress and Increased Endogenous Toxin Levels in Diabetes
5. Procoagulant RBC Phenotype and Microangiopathy in Diabetes
5.1. Implications of RBC Dysfunction for Thrombotic Risk in Diabetes
5.2. Increased RBC–Platelet Interactions in Diabetes
5.3. RBCs Promote Vascular Endothelial Cell Dysfunction in Diabetes
6. RBC Biochemical and Biophysical Alterations in Diabetes
6.1. Metabolic and Lipid Alterations of RBCs in Diabetes
6.2. Alterations in RBC Membrane Properties in Diabetes
6.3. Significance of Hemorheological Changes on Microangiopathic Complications in Diabetes
6.4. Enhanced Hemolysis and RBC Hyperaggregability in Diabetes
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kharroubi, A.T.; Darwish, H.M. Diabetes mellitus: The epidemic of the century. World J. Diabetes 2015, 6, 850–867. [Google Scholar] [CrossRef]
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, G.; Norhammar, A.; Gyberg, V.; Mellbin, L.; Ryden, L. Is Coronary Artery Disease Inevitable in Type 2 Diabetes? From a Glucocentric to a Holistic View on Patient Management. Diabetes Care 2020, 43, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Economic costs of diabetes in the U.S. in 2012. Diabetes Care 2013, 36, 1033–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punthakee, Z.; Goldenberg, R.; Katz, P. Definition, Classification and Diagnosis of Diabetes, Prediabetes and Metabolic Syndrome. Can. J. Diabetes 2018, 42 (Suppl. S1), S10–S15. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.L.; Kirkman, M.S.; Laffel, L.M.; Peters, A.L. Type 1 diabetes through the life span: A position statement of the American Diabetes Association. Diabetes Care 2014, 37, 2034–2054. [Google Scholar] [CrossRef]
- Pelletier, C.; Dai, S.; Roberts, K.C.; Bienek, A.; Onysko, J.; Pelletier, L. Report summary Diabetes in Canada: Facts and figures from a public health perspective. Chronic Dis. Inj. Can. 2012, 33, 53–54. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Ebrahim, H.; Fiseha, T.; Ebrahim, Y.; Bisetegn, H. Comparison of hematological parameters between type 2 diabetes mellitus patients and healthy controls at Dessie comprehensive specialized hospital, Northeast Ethiopia: Comparative cross-sectional study. PLoS ONE 2022, 17, e0272145. [Google Scholar]
- Adane, T.; Getaneh, Z.; Asrie, F. Red Blood Cell Parameters and Their Correlation with Renal Function Tests Among Diabetes Mellitus Patients: A Comparative Cross-Sectional Study. Diabetes Metab. Syndr. Obes. 2020, 13, 3937–3946. [Google Scholar] [CrossRef]
- Frater, J.L. Red Blood Cell Distribution Width as a Biomarker in Type 2 Diabetes Mellitus: Technical Notes [Letter]. Diabetes Metab. Syndr. Obes. 2023, 16, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Na, W.; Lee, S.B.; Ahn, C.W.; Moon, J.S.; Won, K.C.; Shin, S. Potential Diagnostic Hemorheological Indexes for Chronic Kidney Disease in Patients with Type 2 Diabetes. Front. Physiol. 2019, 10, 1062. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, P.; Yan, Z.; Liu, Z.; Ma, Q.; Zhang, Z.; Wang, Y.; Su, Y. The Relationship between Erythrocytes and Diabetes Mellitus. J. Diabetes Res. 2021, 2021, 6656062. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Fouque, D.; Soulage, C.O. Metabolic Abnormalities in Diabetes and Kidney Disease: Role of Uremic Toxins. Curr. Diab. Rep. 2018, 18, 97. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, A.; Cortese-Krott, M.M.; Kelm, M.; Li, N.; Pernow, J. Novel perspectives on redox signaling in red blood cells and platelets in cardiovascular disease. Free Radic. Biol. Med. 2021, 168, 95–109. [Google Scholar] [CrossRef]
- Turpin, C.; Catan, A.; Guerin-Dubourg, A.; Debussche, X.; Bravo, S.B.; Alvarez, E.; Van Den Elsen, J.; Meilhac, O.; Rondeau, P.; Bourdon, E. Enhanced oxidative stress and damage in glycated erythrocytes. PLoS ONE 2020, 15, e0235335. [Google Scholar] [CrossRef]
- Buys, A.V.; Van Rooy, M.J.; Soma, P.; Van Papendorp, D.; Lipinski, B.; Pretorius, E. Changes in red blood cell membrane structure in type 2 diabetes: A scanning electron and atomic force microscopy study. Cardiovasc. Diabetol. 2013, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Szabo, E.; Kulin, A.; Koranyi, L.; Literati-Nagy, B.; Cserepes, J.; Somogyi, A.; Sarkadi, B.; Varady, G. Alterations in erythrocyte membrane transporter expression levels in type 2 diabetic patients. Sci. Rep. 2021, 11, 2765. [Google Scholar] [CrossRef]
- Radosinska, J.; Vrbjar, N. The role of red blood cell deformability and Na, K-ATPase function in selected risk factors of cardiovascular diseases in humans: Focus on hypertension, diabetes mellitus and hypercholesterolemia. Physiol. Res. 2016, 65 (Suppl. S1), S43–S54. [Google Scholar] [CrossRef]
- Spieker, C.; Fischer, S.; Zierden, E.; Schluter, H.; Tepel, M.; Zidek, W. Cellular Ca2+ ATPase activity in diabetes mellitus. Horm. Metab. Res. 1994, 26, 544–547. [Google Scholar] [CrossRef]
- Mazzanti, L.; Faloia, E.; Rabini, R.A.; Staffolani, R.; Kantar, A.; Fiorini, R.; Swoboda, B.; De Pirro, R.; Bertoli, E. Diabetes mellitus induces red blood cell plasma membrane alterations possibly affecting the aging process. Clin. Biochem. 1992, 25, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Winocour, P.; Farrington, K. Erythropoietic stress and anemia in diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 204–210. [Google Scholar] [CrossRef]
- Loyola-Leyva, A.; Loyola-Rodriguez, J.P.; Atzori, M.; Gonzalez, F.J. Morphological changes in erythrocytes of people with type 2 diabetes mellitus evaluated with atomic force microscopy: A brief review. Micron 2018, 105, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Sahay, M.; Kalra, S.; Badani, R.; Bantwal, G.; Bhoraskar, A.; Das, A.K.; Dhorepatil, B.; Ghosh, S.; Jeloka, T.; Khandelwal, D.; et al. Diabetes and Anemia: International Diabetes Federation (IDF)—Southeast Asian Region (SEAR) position statement. Diabetes Metab. Syndr. 2017, 11 (Suppl. S2), S685–S695. [Google Scholar] [CrossRef] [PubMed]
- AlDallal, S.M.; Jena, N. Prevalence of Anemia in Type 2 Diabetic Patients. J. Hematol. 2018, 7, 57–61. [Google Scholar] [CrossRef]
- Jorgensen, J.M.; Crespo-Bellido, M.; Dewey, K.G. Variation in hemoglobin across the life cycle and between males and females. Ann. N. Y. Acad. Sci. 2019, 1450, 105–125. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, J.; Fontela, P.C.; Winkelmann, E.R.; Zimmermann, C.E.; Sandri, Y.P.; Mallet, E.K.; Frizzo, M.N. Anemia in Patients with Type 2 Diabetes Mellitus. Anemia 2015, 2015, 354737. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.A.; Oddy, M.J.; Richards, T. Presence and characterisation of anaemia in diabetic foot ulceration. Anemia 2014, 2014, 104214. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.C.; MacIsaac, R.J.; Tsalamandris, C.; Power, D.; Jerums, G. Unrecognized anemia in patients with diabetes: A cross-sectional survey. Diabetes Care 2003, 26, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- Stevens, P.E.; O’Donoghue, D.J.; Lameire, N.R. Anaemia in patients with diabetes: Unrecognised, undetected and untreated? Curr. Med. Res. Opin. 2003, 19, 395–401. [Google Scholar] [CrossRef]
- Thomas, M.C.; Tsalamandris, C.; MacIsaac, R.J.; Jerums, G. The epidemiology of hemoglobin levels in patients with type 2 diabetes. Am. J. Kidney Dis. 2006, 48, 537–545. [Google Scholar] [CrossRef]
- Thomas, M.C.; MacIsaac, R.J.; Tsalamandris, C.; Molyneaux, L.; Goubina, I.; Fulcher, G.; Yue, D.; Jerums, G. The burden of anaemia in type 2 diabetes and the role of nephropathy: A cross-sectional audit. Nephrol. Dial. Transpl. 2004, 19, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Keane, W.F.; Lyle, P.A. Recent advances in management of type 2 diabetes and nephropathy: Lessons from the RENAAL study. Am. J. Kidney Dis. 2003, 41 (Suppl. S1), S22–S25. [Google Scholar] [CrossRef]
- Feteh, V.F.; Choukem, S.P.; Kengne, A.P.; Nebongo, D.N.; Ngowe-Ngowe, M. Anemia in type 2 diabetic patients and correlation with kidney function in a tertiary care sub-Saharan African hospital: A cross-sectional study. BMC Nephrol. 2016, 17, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erez, D.; Shefler, C.; Roitman, E.; Levy, S.; Dovrish, Z.; Ellis, M.; Twito, O. Anemia in Patients with Diabetes and Prediabetes with Normal Kidney Function: Prevalence and Clinical Outcomes. Endocr. Pract. 2022, 28, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Shams, N.; Osmani, M.H. Newly diagnosed anemia in admitted diabetics, frequency, etiology and associated factors. J. Coll. Physicians Surg. Pak. 2015, 25, 242–246. [Google Scholar] [PubMed]
- Antwi-Bafour, S.; Hammond, S.; Adjei, J.K.; Kyeremeh, R.; Martin-Odoom, A.; Ekem, I. A case-control study of prevalence of anemia among patients with type 2 diabetes. J. Med. Case Rep. 2016, 10, 110. [Google Scholar] [CrossRef] [Green Version]
- Rajab, A.M.; Rahman, S.; Rajab, T.M.; Haider, K.H. Morphology and Chromic Status of Red Blood Cells Are Significantly Influenced by Gestational Diabetes. J. Hematol. 2018, 7, 140–148. [Google Scholar] [CrossRef]
- Hosseini, M.S.; Rostami, Z.; Saadat, A.; Saadatmand, S.M.; Naeimi, E. Anemia and microvascular complications in patients with type 2 diabetes mellitus. Nephrourol. Mon. 2014, 6, e19976. [Google Scholar] [CrossRef] [Green Version]
- Pinero-Pilona, A.; Litonjua, P.; Devaraj, S.; Aviles-Santa, L.; Raskin, P. Anemia associated with new-onset diabetes: Improvement with blood glucose control. Endocr. Pract. 2002, 8, 276–281. [Google Scholar] [CrossRef]
- NeamTu, M.C.; CraiToiu, S.; Avramescu, E.T.; Margina, D.M.; Bacanoiu, M.V.; Turneanu, D.; Danciulescu Miulescu, R. The prevalence of the red cell morphology changes in patients with type 2 diabetes mellitus. Rom. J. Morphol. Embryol. 2015, 56, 183–189. [Google Scholar]
- Nunes, J.M.; Pretorius, E. Red blood cell membrane cholesterol in type 2 diabetes mellitus. Thromb. Res. 2019, 178, 91–98. [Google Scholar] [CrossRef]
- Pretorius, E.; Bester, J.; Vermeulen, N.; Alummoottil, S.; Soma, P.; Buys, A.V.; Kell, D.B. Poorly controlled type 2 diabetes is accompanied by significant morphological and ultrastructural changes in both erythrocytes and in thrombin-generated fibrin: Implications for diagnostics. Cardiovasc. Diabetol. 2015, 14, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George-Gay, B.; Parker, K. Understanding the complete blood count with differential. J. Perianesth Nurs. 2003, 18, 96–117. [Google Scholar] [CrossRef]
- Kurt, Y.G.; Cayci, T.; Aydin, F.N.; Agilli, M. Is red cell distribution width a useful biomarker for risk assessment of diabetes mellitus? J. Intern. Med. 2014, 276, 537. [Google Scholar] [CrossRef] [Green Version]
- Engstrom, G.; Smith, J.G.; Persson, M.; Nilsson, P.M.; Melander, O.; Hedblad, B. Response to letter to the editor ‘Is red cell distribution width a biomarker in risk assessment of diabetes mellitus?’. J. Intern. Med. 2014, 276, 538. [Google Scholar] [CrossRef] [Green Version]
- Engstrom, G.; Smith, J.G.; Persson, M.; Nilsson, P.M.; Melander, O.; Hedblad, B. Red cell distribution width, haemoglobin A1c and incidence of diabetes mellitus. J. Intern. Med. 2014, 276, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.F.; Yang, Y.; Chen, X.; Zhu, X.; Hu, C.; Han, Y.; Zhao, L.; Liu, F.; Sun, L. Red cell distribution width as a significant indicator of medication and prognosis in type 2 diabetic patients. Sci. Rep. 2017, 7, 2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nada, A.M. Red cell distribution width in type 2 diabetic patients. Diabetes Metab. Syndr. Obes. 2015, 8, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhang, Y.; Li, C.; He, L. Association between red blood cell distribution and renal function in patients with untreated type 2 diabetes mellitus. Ren. Fail. 2015, 37, 659–663. [Google Scholar] [CrossRef]
- Tsuboi, S.; Miyauchi, K.; Kasai, T.; Ogita, M.; Dohi, T.; Miyazaki, T.; Yokoyama, T.; Kojima, T.; Yokoyama, K.; Kurata, T.; et al. Impact of red blood cell distribution width on long-term mortality in diabetic patients after percutaneous coronary intervention. Circ. J. 2013, 77, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaslov, K.; Kruljac, I.; Mirosevic, G.; Gacina, P.; Kolonic, S.O.; Vrkljan, M. The prognostic value of red blood cell characteristics on diabetic retinopathy development and progression in type 2 diabetes mellitus. Clin. Hemorheol. Microcirc. 2019, 71, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Magri, C.J.; Fava, S. Red blood cell distribution width and diabetes-associated complications. Diabetes Metab. Syndr. 2014, 8, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Inomata, S.; Itoh, M.; Imai, H.; Sato, T. Serum levels of erythropoietin as a novel marker reflecting the severity of diabetic nephropathy. Nephron 1997, 75, 426–430. [Google Scholar] [CrossRef]
- Catrina, S.B.; Okamoto, K.; Pereira, T.; Brismar, K.; Poellinger, L. Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes 2004, 53, 3226–3232. [Google Scholar] [CrossRef] [Green Version]
- Zou, A.P.; Cowley, A.W., Jr. Reactive oxygen species and molecular regulation of renal oxygenation. Acta Physiol. Scand. 2003, 179, 233–241. [Google Scholar] [CrossRef]
- Bosman, D.R.; Winkler, A.S.; Marsden, J.T.; Macdougall, I.C.; Watkins, P.J. Anemia with erythropoietin deficiency occurs early in diabetic nephropathy. Diabetes Care 2001, 24, 495–499. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.C. The high prevalence of anemia in diabetes is linked to functional erythropoietin deficiency. Semin. Nephrol. 2006, 26, 275–282. [Google Scholar] [CrossRef]
- Thomas, M.C.; Tsalamandris, C.; Macisaac, R.; Jerums, G. Functional erythropoietin deficiency in patients with Type 2 diabetes and anaemia. Diabet. Med. 2006, 23, 502–509. [Google Scholar] [CrossRef]
- Thomas, M.C.; Cooper, M.E.; Tsalamandris, C.; MacIsaac, R.; Jerums, G. Anemia with impaired erythropoietin response in diabetic patients. Arch. Intern. Med. 2005, 165, 466–469. [Google Scholar] [CrossRef]
- Thomas, M.C. Anemia in diabetes: Marker or mediator of microvascular disease? Nat. Clin. Pract. Nephrol. 2007, 3, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Lorber, D.; Reddan, D. Clinical characteristics of chronic kidney disease patients with and without diabetes: A subanalysis of the PAERI study. Clin. Nephrol. 2006, 66, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, E.; Nishizawa, Y.; Okuno, S.; Matsumoto, N.; Emoto, M.; Inaba, M.; Kawagishi, T.; Kim, C.W.; Morii, H. Diabetes mellitus increases the severity of anemia in non-dialyzed patients with renal failure. J. Nephrol. 1998, 11, 83–86. [Google Scholar] [PubMed]
- Bissinger, R.; Nemkov, T.; D’Alessandro, A.; Grau, M.; Dietz, T.; Bohnert, B.N.; Essigke, D.; Worn, M.; Schaefer, L.; Xiao, M.; et al. Proteinuric chronic kidney disease is associated with altered red blood cell lifespan, deformability and metabolism. Kidney Int. 2021, 100, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Drueke, T.B.; Massy, Z.A. Role of proteinuria in the anemia of chronic kidney disease. Kidney Int. 2021, 100, 1160–1162. [Google Scholar] [CrossRef]
- Mahr, N.; Neyer, U.; Prischl, F.; Kramar, R.; Mayer, G.; Kronenberg, F.; Lhotta, K. Proteinuria and hemoglobin levels in patients with primary glomerular disease. Am. J. Kidney Dis. 2005, 46, 424–431. [Google Scholar] [CrossRef]
- Fadini, G.P. Is bone marrow another target of diabetic complications? Eur. J. Clin. Investig. 2011, 41, 457–463. [Google Scholar] [CrossRef]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yu, J.; Wang, T.; Shen, Y.; Lin, D.; Xu, X.; Wang, Y. Identification of megakaryocytes as a target of advanced glycation end products in diabetic complications in bone marrow. Acta Diabetol. 2018, 55, 419–427. [Google Scholar] [CrossRef]
- Kim, A.; Nemeth, E. New insights into iron regulation and erythropoiesis. Curr. Opin. Hematol. 2015, 22, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Kautz, L.; Nemeth, E. Molecular liaisons between erythropoiesis and iron metabolism. Blood 2014, 124, 479–482. [Google Scholar] [CrossRef]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar] [PubMed]
- Ioannou, G.N.; Spector, J.; Scott, K.; Rockey, D.C. Prospective evaluation of a clinical guideline for the diagnosis and management of iron deficiency anemia. Am. J. Med. 2002, 113, 281–287. [Google Scholar] [CrossRef]
- Guo, W.; Zhou, Q.; Jia, Y.; Xu, J. Increased Levels of Glycated Hemoglobin A1c and Iron Deficiency Anemia: A Review. Med. Sci. Monit. 2019, 25, 8371–8378. [Google Scholar] [CrossRef] [PubMed]
- Christy, A.L.; Manjrekar, P.A.; Babu, R.P.; Hegde, A.; Rukmini, M.S. Influence of iron deficiency anemia on hemoglobin A1c levels in diabetic individuals with controlled plasma glucose levels. Iran. Biomed. J. 2014, 18, 88–93. [Google Scholar]
- Thomas, M.C.; MacIsaac, R.J.; Tsalamandris, C.; Jerums, G. Elevated iron indices in patients with diabetes. Diabet. Med. 2004, 21, 798–802. [Google Scholar] [CrossRef]
- Fujimoto, S.; Kawakami, N.; Ohara, A. Nonenzymatic glycation of transferrin: Decrease of iron-binding capacity and increase of oxygen radical production. Biol. Pharm. Bull. 1995, 18, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Soliman, A.T.; De Sanctis, V.; Yassin, M.; Soliman, N. Iron deficiency anemia and glucose metabolism. Acta Biomed. 2017, 88, 112–118. [Google Scholar] [PubMed]
- Macdougall, I.C.; Cooper, A.C. Erythropoietin resistance: The role of inflammation and pro-inflammatory cytokines. Nephrol. Dial. Transpl. 2002, 17 (Suppl. S11), 39–43. [Google Scholar] [CrossRef] [Green Version]
- Lang, E.; Bissinger, R.; Qadri, S.M.; Lang, F. Suicidal death of erythrocytes in cancer and its chemotherapy: A potential target in the treatment of tumor-associated anemia. Int. J. Cancer 2017, 141, 1522–1528. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.; Goodnough, L.T. Anemia of chronic disease. N. Engl. J. Med. 2005, 352, 1011–1023. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Fung, E.; Parikh, S.G.; Valore, E.V.; Gabayan, V.; Nemeth, E.; Ganz, T. A mouse model of anemia of inflammation: Complex pathogenesis with partial dependence on hepcidin. Blood 2014, 123, 1129–1136. [Google Scholar] [CrossRef]
- Dulmovits, B.M.; Tang, Y.; Papoin, J.; He, M.; Li, J.; Yang, H.; Addorisio, M.E.; Kennedy, L.; Khan, M.; Brindley, E.; et al. HMGB1-mediated restriction of EPO signaling contributes to anemia of inflammation. Blood 2022, 139, 3181–3193. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, J.D.; Kang, J.Y.; Yoo, T.K.; Park, Y.W. Testosterone deficiency and the risk of anemia: A propensity score-matched analysis. Am. J. Hum. Biol. 2022, 34, e23751. [Google Scholar] [CrossRef]
- Valancy, D.; Blachman-Braun, R.; Kuchakulla, M.; Nackeeran, S.; Ramasamy, R. Association between low testosterone and anaemia: Analysis of the National Health and Nutrition Examination Survey. Andrologia 2021, 53, e14210. [Google Scholar] [CrossRef]
- Ferrucci, L.; Maggio, M.; Bandinelli, S.; Basaria, S.; Lauretani, F.; Ble, A.; Valenti, G.; Ershler, W.B.; Guralnik, J.M.; Longo, D.L. Low testosterone levels and the risk of anemia in older men and women. Arch. Intern. Med. 2006, 166, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malgor, L.A.; Valsecia, M.; Verges, E.; De Markowsky, E.E. Blockade of the in vitro effects of testosterone and erythropoietin on Cfu-E and Bfu-E proliferation by pretreatment of the donor rats with cyproterone and flutamide. Acta Physiol. Pharmacol. Ther. Latinoam 1998, 48, 99–105. [Google Scholar] [PubMed]
- Kanias, T.; Sinchar, D.; Osei-Hwedieh, D.; Baust, J.J.; Jordan, A.; Zimring, J.C.; Waterman, H.R.; de Wolski, K.S.; Acker, J.P.; Gladwin, M.T. Testosterone-dependent sex differences in red blood cell hemolysis in storage, stress, and disease. Transfusion 2016, 56, 2571–2583. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, M.; Panagiotopolous, S.; Sharpe, K.; MacIsaac, R.J.; Clarke, S.; Zajac, J.D.; Jerums, G.; Thomas, M.C. Low testosterone and anaemia in men with type 2 diabetes. Clin. Endocrinol. 2009, 70, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Benedik, P.S.; Hamlin, S.K. The physiologic role of erythrocytes in oxygen delivery and implications for blood storage. Crit. Care Nurs. Clin. N. Am. 2014, 26, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Bosman, G.J.; Willekens, F.L.; Werre, J.M. Erythrocyte aging: A more than superficial resemblance to apoptosis? Cell Physiol. Biochem. 2005, 16, 1–8. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, A.; Zimring, J.C.; Busch, M. Chronological storage age and metabolic age of stored red blood cells: Are they the same? Transfusion 2019, 59, 1620–1623. [Google Scholar] [CrossRef]
- Antonelou, M.H.; Kriebardis, A.G.; Papassideri, I.S. Aging and death signalling in mature red cells: From basic science to transfusion practice. Blood Transfus. 2010, 8 (Suppl. S3), s39–s47. [Google Scholar]
- Yoshida, T.; Prudent, M.; D’Alessandro, A. Red blood cell storage lesion: Causes and potential clinical consequences. Blood Transfus. 2019, 17, 27–52. [Google Scholar] [PubMed]
- Dreischer, P.; Duszenko, M.; Stein, J.; Wieder, T. Eryptosis: Programmed Death of Nucleus-Free, Iron-Filled Blood Cells. Cells 2022, 11, 503. [Google Scholar] [CrossRef]
- Brun, J.F.; Varlet-Marie, E.; Myzia, J.; Raynaud de Mauverger, E.; Pretorius, E. Metabolic Influences Modulating Erythrocyte Deformability and Eryptosis. Metabolites 2021, 12, 4. [Google Scholar] [CrossRef]
- Restivo, I.; Attanzio, A.; Tesoriere, L.; Allegra, M. Suicidal Erythrocyte Death in Metabolic Syndrome. Antioxidants 2021, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Repsold, L.; Joubert, A.M. Eryptosis: An Erythrocyte’s Suicidal Type of Cell Death. Biomed. Res. Int. 2018, 2018, 9405617. [Google Scholar] [CrossRef] [PubMed]
- Alghareeb, S.A.; Alfhili, M.A.; Fatima, S. Molecular Mechanisms and Pathophysiological Significance of Eryptosis. Int. J. Mol. Sci. 2023, 24, 5079. [Google Scholar] [CrossRef]
- Pretorius, E. Erythrocyte deformability and eryptosis during inflammation, and impaired blood rheology. Clin. Hemorheol. Microcirc. 2018, 69, 545–550. [Google Scholar] [CrossRef]
- Bissinger, R.; Bhuyan, A.A.M.; Qadri, S.M.; Lang, F. Oxidative stress, eryptosis and anemia: A pivotal mechanistic nexus in systemic diseases. FEBS J. 2019, 286, 826–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qadri, S.M.; Bissinger, R.; Solh, Z.; Oldenborg, P.A. Eryptosis in health and disease: A paradigm shift towards understanding the (patho)physiological implications of programmed cell death of erythrocytes. Blood Rev. 2017, 31, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; du Plooy, J.N.; Bester, J. A Comprehensive Review on Eryptosis. Cell Physiol. Biochem. 2016, 39, 1977–2000. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Bissinger, R.; Gulbins, E.; Lang, F. Ceramide in the regulation of eryptosis, the suicidal erythrocyte death. Apoptosis 2015, 20, 758–767. [Google Scholar] [CrossRef]
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly—Suicidal erythrocyte death. Int. J. Biochem. Cell Biol. 2012, 44, 1236–1243. [Google Scholar] [CrossRef]
- Lang, F.; Qadri, S.M. Mechanisms and significance of eryptosis, the suicidal death of erythrocytes. Blood Purif. 2012, 33, 125–130. [Google Scholar] [CrossRef]
- Bissinger, R.; Petkova-Kirova, P.; Mykhailova, O.; Oldenborg, P.A.; Novikova, E.; Donkor, D.A.; Dietz, T.; Bhuyan, A.A.M.; Sheffield, W.P.; Grau, M.; et al. Thrombospondin-1/CD47 signaling modulates transmembrane cation conductance, survival, and deformability of human red blood cells. Cell Commun. Signal. 2020, 18, 155. [Google Scholar] [CrossRef]
- Kempe-Teufel, D.S.; Bissinger, R.; Qadri, S.M.; Wagner, R.; Peter, A.; Lang, F. Cellular markers of eryptosis are altered in type 2 diabetes. Clin. Chem. Lab. Med. 2018, 56, e177–e180. [Google Scholar] [CrossRef]
- Muhlberger, T.; Balach, M.M.; Bisig, C.G.; Santander, V.S.; Monesterolo, N.E.; Casale, C.H.; Campetelli, A.N. Inhibition of flippase-like activity by tubulin regulates phosphatidylserine exposure in erythrocytes from hypertensive and diabetic patients. J. Biochem. 2021, 169, 731–745. [Google Scholar] [CrossRef]
- Maellaro, E.; Leoncini, S.; Moretti, D.; Del Bello, B.; Tanganelli, I.; De Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2013, 50, 489–495. [Google Scholar] [CrossRef]
- Savu, O.; Bradescu, O.M.; Serafinceanu, C.; Iosif, L.; Tirgoviste, C.I.; Stoian, I. Erythrocyte caspase-3 and antioxidant defense is activated in red blood cells and plasma of type 2 diabetes patients at first clinical onset. Redox Rep. 2013, 18, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Moin, A.S.M.; Nandakumar, M.; Al-Qaissi, A.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Potential Biomarkers to Predict Acute Ischemic Stroke in Type 2 Diabetes. Front. Mol. Biosci. 2021, 8, 744459. [Google Scholar] [CrossRef]
- Choi, K.Y.; Kim, D.B.; Kim, M.J.; Kwon, B.J.; Chang, S.Y.; Jang, S.W.; Cho, E.J.; Rho, T.H.; Kim, J.H. Higher plasma thrombospondin-1 levels in patients with coronary artery disease and diabetes mellitus. Korean Circ. J. 2012, 42, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, J.Y.; Lim, K.M.; Bae, O.N.; Chung, S.M.; Lee, S.W.; Joo, K.M.; Lee, S.D.; Chung, J.H. Procoagulant and prothrombotic activation of human erythrocytes by phosphatidic acid. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H347–H355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eligini, S.; Porro, B.; Werba, J.P.; Capra, N.; Genovese, S.; Greco, A.; Cavalca, V.; Banfi, C. Oxidative Stress and Arginine/Nitric Oxide Pathway in Red Blood Cells Derived from Patients with Prediabetes. Biomedicines 2022, 10, 1407. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Zentella, M.L.; Sanchez-Sevilla, L.; Suarez-Cuenca, J.A.; Olguin-Martinez, M.; Alatriste-Contreras, M.G.; Garcia-Garcia, N.; Orozco, L.; Hernandez-Munoz, R. The role of oxidant stress and gender in the erythrocyte arginine metabolism and ammonia management in patients with type 2 diabetes. PLoS ONE 2019, 14, e0219481. [Google Scholar] [CrossRef] [Green Version]
- Khorasani, V.; Jeddi, S.; Yaghmaei, P.; Tohidi, M.; Ghasemi, A. Effect of long-term sodium nitrate administration on diabetes-induced anemia and glucose homeostasis in obese type 2 diabetic male rats. Nitric. Oxide 2019, 86, 21–30. [Google Scholar] [CrossRef]
- Su, Y.; Chen, J.; Dong, Z.; Zhang, Y.; Ma, R.; Kou, J.; Wang, F.; Shi, J. Procoagulant Activity of Blood and Endothelial Cells via Phosphatidylserine Exposure and Microparticle Delivery in Patients with Diabetic Retinopathy. Cell Physiol. Biochem. 2018, 45, 2411–2420. [Google Scholar] [CrossRef]
- Meyer, J.A.; Subasinghe, W.; Sima, A.A.; Keltner, Z.; Reid, G.E.; Daleke, D.; Spence, D.M. Zinc-activated C-peptide resistance to the type 2 diabetic erythrocyte is associated with hyperglycemia-induced phosphatidylserine externalization and reversed by metformin. Mol. Biosyst. 2009, 5, 1157–1162. [Google Scholar] [CrossRef]
- Manodori, A.B.; Kuypers, F.A. Altered red cell turnover in diabetic mice. J. Lab. Clin. Med. 2002, 140, 161–165. [Google Scholar] [CrossRef]
- Firat, U.; Kaya, S.; Cim, A.; Buyukbayram, H.; Gokalp, O.; Dal, M.S.; Tamer, M.N. Increased caspase-3 immunoreactivity of erythrocytes in STZ diabetic rats. Exp. Diabetes Res. 2012, 2012, 316384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sola, E.; Vaya, A.; Martinez, M.; Moscardo, A.; Corella, D.; Santaolaria, M.L.; Espana, F.; Hernandez-Mijares, A. Erythrocyte membrane phosphatidylserine exposure in obesity. Obesity 2009, 17, 318–322. [Google Scholar] [CrossRef]
- Wiewiora, M.; Piecuch, J.; Sedek, L.; Mazur, B.; Sosada, K. The effects of obesity on CD47 expression in erythrocytes. Cytom. B Clin. Cytom. 2017, 92, 485–491. [Google Scholar] [CrossRef]
- Unruh, D.; Srinivasan, R.; Benson, T.; Haigh, S.; Coyle, D.; Batra, N.; Keil, R.; Sturm, R.; Blanco, V.; Palascak, M.; et al. Red Blood Cell Dysfunction Induced by High-Fat Diet: Potential Implications for Obesity-Related Atherosclerosis. Circulation 2015, 132, 1898–1908. [Google Scholar] [CrossRef] [Green Version]
- Cilla, A.; Lopez-Garcia, G.; Collado-Diaz, V.; Amparo Blanch-Ruiz, M.; Garcia-Llatas, G.; Barbera, R.; Martinez-Cuesta, M.A.; Real, J.T.; Alvarez, A.; Martinez-Hervas, S. Hypercholesterolemic patients have higher eryptosis and erythrocyte adhesion to human endothelium independently of statin therapy. Int. J. Clin. Pract. 2021, 75, e14771. [Google Scholar] [CrossRef] [PubMed]
- Pinzon-Diaz, C.E.; Calderon-Salinas, J.V.; Rosas-Flores, M.M.; Hernandez, G.; Lopez-Betancourt, A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in hypertensive and dyslipidemic patients. Mol. Cell Biochem. 2018, 440, 105–113. [Google Scholar] [CrossRef]
- Lang, F.; Bissinger, R.; Abed, M.; Artunc, F. Eryptosis—The Neglected Cause of Anemia in End Stage Renal Disease. Kidney Blood Press. Res. 2017, 42, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Gok, M.G.; Paydas, S.; Boral, B.; Onan, E.; Kaya, B. Evaluation of eryptosis in patients with chronic kidney disease. Int. Urol. Nephrol. 2022, 54, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Dias, G.F.; Grobe, N.; Rogg, S.; Jorg, D.J.; Pecoits-Filho, R.; Moreno-Amaral, A.N.; Kotanko, P. The Role of Eryptosis in the Pathogenesis of Renal Anemia: Insights From Basic Research and Mathematical Modeling. Front. Cell Dev. Biol. 2020, 8, 598148. [Google Scholar] [CrossRef]
- Meyring-Wosten, A.; Kuntsevich, V.; Campos, I.; Williams, S.; Ma, J.; Patel, S.; Ornillo, C.; Thijssen, S.; Kotanko, P. Erythrocyte Sodium Sensitivity and Eryptosis in Chronic Hemodialysis Patients. Kidney Blood Press. Res. 2017, 42, 314–326. [Google Scholar] [CrossRef]
- Di Pietro, N.; Giardinelli, A.; Sirolli, V.; Riganti, C.; Di Tomo, P.; Gazzano, E.; Di Silvestre, S.; Panknin, C.; Cortese-Krott, M.M.; Csonka, C.; et al. Nitric oxide synthetic pathway and cGMP levels are altered in red blood cells from end-stage renal disease patients. Mol. Cell Biochem. 2016, 417, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zheng, X.; Zhang, Y.; Li, X.; Chen, X.; Yin, Y.; Hu, J.; Li, J.; Guo, M.; Wang, X. What Should Be Responsible for Eryptosis in Chronic Kidney Disease? Kidney Blood Press. Res. 2022, 47, 375–390. [Google Scholar] [CrossRef]
- Bissinger, R.; Artunc, F.; Qadri, S.M.; Lang, F. Reduced Erythrocyte Survival in Uremic Patients Under Hemodialysis or Peritoneal Dialysis. Kidney Blood Press. Res. 2016, 41, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.H.; Cheng, L.T.; Wang, T. The role of erythrocytes phosphatidylserine exposure in anemia in peritoneal dialysis patients. Ren. Fail. 2006, 28, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Salinas, J.V.; Munoz-Reyes, E.G.; Guerrero-Romero, J.F.; Rodriguez-Moran, M.; Bracho-Riquelme, R.L.; Carrera-Gracia, M.A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in type 2 diabetic mellitus patients with chronic kidney disease. Mol. Cell Biochem. 2011, 357, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fang, K.; Peng, H.; He, L.; Wang, Y. The relationship between glycosylated hemoglobin level and red blood cell storage lesion in blood donors. Transfusion 2022, 62, 663–674. [Google Scholar] [CrossRef]
- Jagadish, S.; Hemshekhar, M.; NaveenKumar, S.K.; Sharath Kumar, K.S.; Sundaram, M.S.; Basappa; Girish, K.S.; Rangappa, K.S. Novel oxolane derivative DMTD mitigates high glucose-induced erythrocyte apoptosis by regulating oxidative stress. Toxicol. Appl. Pharmacol. 2017, 334, 167–179. [Google Scholar] [CrossRef]
- Sompong, W.; Cheng, H.; Adisakwattana, S. Protective Effects of Ferulic Acid on High Glucose-Induced Protein Glycation, Lipid Peroxidation, and Membrane Ion Pump Activity in Human Erythrocytes. PLoS ONE 2015, 10, e0129495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.K.; Palmer, M.; Chen, Y. Effect of vitamin E and N-acetylcysteine on phosphatidylserine externalization and induction of coagulation by high-glucose-treated human erythrocytes. Metabolism 1999, 48, 957–959. [Google Scholar] [CrossRef]
- Kucherenko, Y.V.; Bhavsar, S.K.; Grischenko, V.I.; Fischer, U.R.; Huber, S.M.; Lang, F. Increased cation conductance in human erythrocytes artificially aged by glycation. J. Membr. Biol. 2010, 235, 177–189. [Google Scholar] [CrossRef]
- Lai, S.W.T.; Lopez Gonzalez, E.J.; Zoukari, T.; Ki, P.; Shuck, S.C. Methylglyoxal and Its Adducts: Induction, Repair, and Association with Disease. Chem. Res. Toxicol. 2022, 35, 1720–1746. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef] [PubMed]
- Maessen, D.E.; Stehouwer, C.D.; Schalkwijk, C.G. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. 2015, 128, 839–861. [Google Scholar] [CrossRef]
- Sachdeva, R.; Schlotterer, A.; Schumacher, D.; Matka, C.; Mathar, I.; Dietrich, N.; Medert, R.; Kriebs, U.; Lin, J.; Nawroth, P.; et al. TRPC proteins contribute to development of diabetic retinopathy and regulate glyoxalase 1 activity and methylglyoxal accumulation. Mol. Metab. 2018, 9, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, N.M.J.; Scheijen, J.; Jorsal, A.; Parving, H.H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D.A.; Schalkwijk, C.G. Higher Plasma Methylglyoxal Levels Are Associated with Incident Cardiovascular Disease in Individuals with Type 1 Diabetes: A 12-Year Follow-up Study. Diabetes 2017, 66, 2278–2283. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.J. Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem. J. 1988, 254, 751–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishibashi, Y.; Matsui, T.; Nakamura, N.; Sotokawauchi, A.; Higashimoto, Y.; Yamagishi, S.I. Methylglyoxal-derived hydroimidazolone-1 evokes inflammatory reactions in endothelial cells via an interaction with receptor for advanced glycation end products. Diab. Vasc. Dis. Res. 2017, 14, 450–453. [Google Scholar] [CrossRef]
- Prestes, A.S.; Dos Santos, M.M.; Ecker, A.; Zanini, D.; Schetinger, M.R.; Rosemberg, D.B.; da Rocha, J.B.; Barbosa, N.V. Evaluation of methylglyoxal toxicity in human erythrocytes, leukocytes and platelets. Toxicol. Mech. Methods 2017, 27, 307–317. [Google Scholar] [CrossRef]
- Nicolay, J.P.; Schneider, J.; Niemoeller, O.M.; Artunc, F.; Portero-Otin, M.; Haik, G., Jr.; Thornalley, P.J.; Schleicher, E.; Wieder, T.; Lang, F. Stimulation of suicidal erythrocyte death by methylglyoxal. Cell Physiol. Biochem. 2006, 18, 223–232. [Google Scholar] [CrossRef]
- Delveaux, J.; Turpin, C.; Veeren, B.; Diotel, N.; Bravo, S.B.; Begue, F.; Alvarez, E.; Meilhac, O.; Bourdon, E.; Rondeau, P. Antirhea borbonica Aqueous Extract Protects Albumin and Erythrocytes from Glycoxidative Damages. Antioxidants 2020, 9, 415. [Google Scholar] [CrossRef]
- Virzi, G.M.; Mattiotti, M.; Clementi, A.; Milan Manani, S.; Battaglia, G.G.; Ronco, C.; Zanella, M. In Vitro Induction of Eryptosis by Uremic Toxins and Inflammation Mediators in Healthy Red Blood Cells. J. Clin. Med. 2022, 11, 5329. [Google Scholar] [CrossRef]
- Lu, C.L.; Zheng, C.M.; Lu, K.C.; Liao, M.T.; Wu, K.L.; Ma, M.C. Indoxyl-Sulfate-Induced Redox Imbalance in Chronic Kidney Disease. Antioxidants 2021, 10, 936. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Langer, H.; Abed, M.; Voelkl, J.; Lang, F. The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press. Res. 2013, 37, 158–167. [Google Scholar] [CrossRef]
- Voelkl, J.; Alzoubi, K.; Mamar, A.K.; Ahmed, M.S.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by increased extracellular phosphate concentrations. Kidney Blood Press. Res. 2013, 38, 42–51. [Google Scholar] [CrossRef]
- Byrnes, J.R.; Wolberg, A.S. Red blood cells in thrombosis. Blood 2017, 130, 1795–1799. [Google Scholar] [CrossRef]
- Byrnes, J.R.; Duval, C.; Wang, Y.; Hansen, C.E.; Ahn, B.; Mooberry, M.J.; Clark, M.A.; Johnsen, J.M.; Lord, S.T.; Lam, W.A.; et al. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin alpha-chain crosslinking. Blood 2015, 126, 1940–1948. [Google Scholar] [CrossRef]
- Kattula, S.; Byrnes, J.R.; Martin, S.M.; Holle, L.A.; Cooley, B.C.; Flick, M.J.; Wolberg, A.S. Factor XIII in plasma, but not in platelets, mediates red blood cell retention in clots and venous thrombus size in mice. Blood Adv. 2018, 2, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Xie, R.; Zhang, Y.; Liang, H.; Hou, L.; Yu, C.; Zhang, J.; Dong, Z.; Tian, Y.; Bi, Y.; et al. Phosphatidylserine on microparticles and associated cells contributes to the hypercoagulable state in diabetic kidney disease. Nephrol. Dial. Transpl. 2018, 33, 2115–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puddu, P.; Puddu, G.M.; Cravero, E.; Muscari, S.; Muscari, A. The involvement of circulating microparticles in inflammation, coagulation and cardiovascular diseases. Can. J. Cardiol. 2010, 26, 140–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Meijden, P.E.; Van Schilfgaarde, M.; Van Oerle, R.; Renne, T.; ten Cate, H.; Spronk, H.M. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J. Thromb. Haemost. 2012, 10, 1355–1362. [Google Scholar] [CrossRef]
- Whelihan, M.F.; Zachary, V.; Orfeo, T.; Mann, K.G. Prothrombin activation in blood coagulation: The erythrocyte contribution to thrombin generation. Blood 2012, 120, 3837–3845. [Google Scholar] [CrossRef] [Green Version]
- Zwaal, R.F.; Schroit, A.J. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 1997, 89, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, M.; Sirolli, V.; Merciaro, G.; Antidormi, T.; Di Liberato, L.; Brummer, U.; Papponetti, M.; Cappelli, P.; Di Gregorio, P.; Arduini, A. Red blood cells may contribute to hypercoagulability in uraemia via enhanced surface exposure of phosphatidylserine. Nephrol. Dial. Transpl. 2005, 20, 361–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atichartakarn, V.; Angchaisuksiri, P.; Aryurachai, K.; Onpun, S.; Chuncharunee, S.; Thakkinstian, A.; Atamasirikul, K. Relationship between hypercoagulable state and erythrocyte phosphatidylserine exposure in splenectomized haemoglobin E/beta-thalassaemic patients. Br. J. Haematol. 2002, 118, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Lim, K.M.; Noh, J.Y.; Bae, O.N.; Chung, S.M.; Lee, M.Y.; Chung, J.H. Lead-induced procoagulant activation of erythrocytes through phosphatidylserine exposure may lead to thrombotic diseases. Chem. Res. Toxicol. 2007, 20, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.D.; Chauhan, A.K.; Schaeffer, G.V.; Hansen, J.K.; Motto, D.G. Red blood cells mediate the onset of thrombosis in the ferric chloride murine model. Blood 2013, 121, 3733–3741. [Google Scholar] [CrossRef]
- Walker, B.; Towhid, S.T.; Schmid, E.; Hoffmann, S.M.; Abed, M.; Munzer, P.; Vogel, S.; Neis, F.; Brucker, S.; Gawaz, M.; et al. Dynamic adhesion of eryptotic erythrocytes to immobilized platelets via platelet phosphatidylserine receptors. Am. J. Physiol. Cell Physiol. 2014, 306, C291–C297. [Google Scholar] [CrossRef] [PubMed]
- Nergiz-Unal, R.; Rademakers, T.; Cosemans, J.M.; Heemskerk, J.W. CD36 as a multiple-ligand signaling receptor in atherothrombosis. Cardiovasc. Hematol. Agents Med. Chem. 2011, 9, 42–55. [Google Scholar] [CrossRef]
- Klatt, C.; Kruger, I.; Zey, S.; Krott, K.J.; Spelleken, M.; Gowert, N.S.; Oberhuber, A.; Pfaff, L.; Luckstadt, W.; Jurk, K.; et al. Platelet-RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. J. Clin. Investig. 2018, 128, 3906–3925. [Google Scholar] [CrossRef]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Foller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef] [Green Version]
- Shimaoka, T.; Nakayama, T.; Fukumoto, N.; Kume, N.; Takahashi, S.; Yamaguchi, J.; Minami, M.; Hayashida, K.; Kita, T.; Ohsumi, J.; et al. Cell surface-anchored SR-PSOX/CXC chemokine ligand 16 mediates firm adhesion of CXC chemokine receptor 6-expressing cells. J. Leukoc. Biol. 2004, 75, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Yipp, B.G.; Anand, S.; Schollaardt, T.; Patel, K.D.; Looareesuwan, S.; Ho, M. Synergism of multiple adhesion molecules in mediating cytoadherence of Plasmodium falciparum-infected erythrocytes to microvascular endothelial cells under flow. Blood 2000, 96, 2292–2298. [Google Scholar] [CrossRef]
- Wandersee, N.J.; Olson, S.C.; Holzhauer, S.L.; Hoffmann, R.G.; Barker, J.E.; Hillery, C.A. Increased erythrocyte adhesion in mice and humans with hereditary spherocytosis and hereditary elliptocytosis. Blood 2004, 103, 710–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wautier, J.L.; Wautier, M.P. Cellular and Molecular Aspects of Blood Cell-Endothelium Interactions in Vascular Disorders. Int. J. Mol. Sci. 2020, 21, 5315. [Google Scholar] [CrossRef] [PubMed]
- Grossin, N.; Wautier, M.P.; Wautier, J.L. Red blood cell adhesion in diabetes mellitus is mediated by advanced glycation end product receptor and is modulated by nitric oxide. Biorheology 2009, 46, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; Heron, E.; Picot, J.; Colin, Y.; Hermine, O.; Wautier, J.L. Red blood cell phosphatidylserine exposure is responsible for increased erythrocyte adhesion to endothelium in central retinal vein occlusion. J. Thromb. Haemost. 2011, 9, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, J.P.; Thorn, V.; Daniel, C.; Amann, K.; Siraskar, B.; Lang, F.; Hillgruber, C.; Goerge, T.; Hoffmann, S.; Gorzelanny, C.; et al. Cellular stress induces erythrocyte assembly on intravascular von Willebrand factor strings and promotes microangiopathy. Sci. Rep. 2018, 8, 10945. [Google Scholar] [CrossRef] [Green Version]
- Catan, A.; Turpin, C.; Diotel, N.; Patche, J.; Guerin-Dubourg, A.; Debussche, X.; Bourdon, E.; Ah-You, N.; Le Moullec, N.; Besnard, M.; et al. Aging and glycation promote erythrocyte phagocytosis by human endothelial cells: Potential impact in atherothrombosis under diabetic conditions. Atherosclerosis 2019, 291, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Collado, A.; Sun, C.; Tratsiakovich, Y.; Mahdi, A.; Winter, H.; Chernogubova, E.; Seime, T.; Narayanan, S.; Jiao, T.; et al. Downregulation of Erythrocyte miR-210 Induces Endothelial Dysfunction in Type 2 Diabetes. Diabetes 2022, 71, 285–297. [Google Scholar] [CrossRef]
- Mahdi, A.; Tengbom, J.; Alvarsson, M.; Wernly, B.; Zhou, Z.; Pernow, J. Red Blood Cell Peroxynitrite Causes Endothelial Dysfunction in Type 2 Diabetes Mellitus via Arginase. Cells 2020, 9, 1712. [Google Scholar] [CrossRef]
- Zhou, Z.; Mahdi, A.; Tratsiakovich, Y.; Zahoran, S.; Kovamees, O.; Nordin, F.; Uribe Gonzalez, A.E.; Alvarsson, M.; Ostenson, C.G.; Andersson, D.C.; et al. Erythrocytes From Patients with Type 2 Diabetes Induce Endothelial Dysfunction Via Arginase I. J. Am. Coll. Cardiol. 2018, 72, 769–780. [Google Scholar] [CrossRef]
- Herance, J.R.; Ciudin, A.; Lamas-Domingo, R.; Aparicio-Gomez, C.; Hernandez, C.; Simo, R.; Palomino-Schatzlein, M. The Footprint of Type 1 Diabetes on Red Blood Cells: A Metabolomic and Lipidomic Study. J. Clin. Med. 2023, 12, 556. [Google Scholar] [CrossRef] [PubMed]
- Palomino-Schatzlein, M.; Lamas-Domingo, R.; Ciudin, A.; Gutierrez-Carcedo, P.; Mares, R.; Aparicio-Gomez, C.; Hernandez, C.; Simo, R.; Herance, J.R. A Translational In Vivo and In Vitro Metabolomic Study Reveals Altered Metabolic Pathways in Red Blood Cells of Type 2 Diabetes. J. Clin. Med. 2020, 9, 1619. [Google Scholar] [CrossRef]
- Whillier, S.; Raftos, J.E.; Kuchel, P.W. Glutathione synthesis by red blood cells in type 2 diabetes mellitus. Redox Rep. 2008, 13, 277–282. [Google Scholar] [CrossRef]
- Kostara, C.E.; Tsiafoulis, C.G.; Bairaktari, E.T.; Tsimihodimos, V. Altered RBC membrane lipidome: A possible etiopathogenic link for the microvascular impairment in Type 2 diabetes. J. Diabetes Complicat. 2021, 35, 107998. [Google Scholar] [CrossRef]
- Bukowiecka-Matusiak, M.; Burzynska-Pedziwiatr, I.; Sansone, A.; Malachowska, B.; Zurawska-Klis, M.; Ferreri, C.; Chatgilialoglu, C.; Ochedalski, T.; Cypryk, K.; Wozniak, L.A. Lipid profile changes in erythrocyte membranes of women with diagnosed GDM. PLoS ONE 2018, 13, e0203799. [Google Scholar] [CrossRef]
- Cho, Y.I.; Mooney, M.P.; Cho, D.J. Hemorheological disorders in diabetes mellitus. J. Diabetes Sci. Technol. 2008, 2, 1130–1138. [Google Scholar] [CrossRef] [Green Version]
- Le Devehat, C.; Vimeux, M.; Khodabandehlou, T. Blood rheology in patients with diabetes mellitus. Clin. Hemorheol. Microcirc. 2004, 30, 297–300. [Google Scholar] [PubMed]
- Mantskava, M.; Momtselidze, N.; Pargalava, N.; McHedlishvili, G. Hemorheological disorders in patients with type 1 or 2 diabetes mellitus and foot gangrene. Clin. Hemorheol. Microcirc. 2006, 35, 307–310. [Google Scholar]
- Soma, P.; Pretorius, E. Interplay between ultrastructural findings and atherothrombotic complications in type 2 diabetes mellitus. Cardiovasc. Diabetol. 2015, 14, 96. [Google Scholar] [CrossRef] [Green Version]
- Keymel, S.; Heiss, C.; Kleinbongard, P.; Kelm, M.; Lauer, T. Impaired red blood cell deformability in patients with coronary artery disease and diabetes mellitus. Horm. Metab. Res. 2011, 43, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Antonova, N.; Velcheva, I.; Paskova, V. Hemorheological and microvascular disturbances in patients with type 2 diabetes mellitus. Clin. Hemorheol. Microcirc. 2022, 81, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Tzounakas, V.L.; Anastasiadi, A.T.; Lekka, M.E.; Papageorgiou, E.G.; Stamoulis, K.; Papassideri, I.S.; Kriebardis, A.G.; Antonelou, M.H. Deciphering the Relationship Between Free and Vesicular Hemoglobin in Stored Red Blood Cell Units. Front. Physiol. 2022, 13, 840995. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.R.; Mohandas, N.; Shohet, S.B. Osmotic gradient ektacytometry: Comprehensive characterization of red cell volume and surface maintenance. Blood 1983, 61, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Le Devehat, C.; Khodabandehlou, T.; Vimeux, M. Relationship between hemorheological and microcirculatory abnormalities in diabetes mellitus. Diabete Metab. 1994, 20, 401–404. [Google Scholar] [PubMed]
- Maulucci, G.; Cordelli, E.; Rizzi, A.; De Leva, F.; Papi, M.; Ciasca, G.; Samengo, D.; Pani, G.; Pitocco, D.; Soda, P.; et al. Phase separation of the plasma membrane in human red blood cells as a potential tool for diagnosis and progression monitoring of type 1 diabetes mellitus. PLoS ONE 2017, 12, e0184109. [Google Scholar] [CrossRef] [Green Version]
- Cahn, A.; Livshits, L.; Srulevich, A.; Raz, I.; Yedgar, S.; Barshtein, G. Diabetic foot disease is associated with reduced erythrocyte deformability. Int. Wound J. 2016, 13, 500–504. [Google Scholar] [CrossRef]
- Schwartz, R.S.; Madsen, J.W.; Rybicki, A.C.; Nagel, R.L. Oxidation of spectrin and deformability defects in diabetic erythrocytes. Diabetes 1991, 40, 701–708. [Google Scholar] [CrossRef]
- Bizjak, D.A.; Brinkmann, C.; Bloch, W.; Grau, M. Increase in Red Blood Cell-Nitric Oxide Synthase Dependent Nitric Oxide Production during Red Blood Cell Aging in Health and Disease: A Study on Age Dependent Changes of Rheologic and Enzymatic Properties in Red Blood Cells. PLoS ONE 2015, 10, e0125206. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, C.; Bizjak, D.A.; Bischof, S.; Latsch, J.; Brixius, K.; Bloch, W.; Grau, M. Endurance training alters enzymatic and rheological properties of red blood cells (RBC) in type 2 diabetic men during in vivo RBC aging. Clin. Hemorheol. Microcirc. 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Bareford, D.; Jennings, P.E.; Stone, P.C.; Baar, S.; Barnett, A.H.; Stuart, J. Effects of hyperglycaemia and sorbitol accumulation on erythrocyte deformability in diabetes mellitus. J. Clin. Pathol. 1986, 39, 722–727. [Google Scholar] [CrossRef]
- Robey, C.; Dasmahapatra, A.; Cohen, M.P.; Suarez, S. Sorbinil partially prevents decreased erythrocyte deformability in experimental diabetes mellitus. Diabetes 1987, 36, 1010–1013. [Google Scholar] [CrossRef]
- Huang, Q.; Liu, Q.; Ouyang, D. Sorbinil, an Aldose Reductase Inhibitor, in Fighting Against Diabetic Complications. Med. Chem. 2019, 15, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Le Jeune, S.; Sadoudi, S.; Charue, D.; Abid, S.; Guigner, J.M.; Helley, D.; Bihan, H.; Baudry, C.; Lelong, H.; Mirault, T.; et al. Low grade intravascular hemolysis associates with peripheral nerve injury in type 2 diabetes. PLoS ONE 2022, 17, e0275337. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Inada, S.; Ijima, H.; Jinnouchi, H.; Ono, Y.; Iwasaka, T.; Okumiya, T. Shortened mean erythrocyte age in female patients with type 2 diabetes mellitus. J. Clin. Lab. Anal. 2019, 33, e22681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloutier, G.; Zimmer, A.; Yu, F.T.; Chiasson, J.L. Increased shear rate resistance and fastest kinetics of erythrocyte aggregation in diabetes measured with ultrasound. Diabetes Care 2008, 31, 1400–1402. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, L.; Li, Y. Enhanced RBC Aggregation in Type 2 Diabetes Patients. J. Clin. Lab. Anal. 2015, 29, 387–389. [Google Scholar] [CrossRef]
- Deng, Y.; Papageorgiou, D.P.; Li, X.; Perakakis, N.; Mantzoros, C.S.; Dao, M.; Karniadakis, G.E. Quantifying Fibrinogen-Dependent Aggregation of Red Blood Cells in Type 2 Diabetes Mellitus. Biophys. J. 2020, 119, 900–912. [Google Scholar] [CrossRef]
- Chung, S.M.; Oh, J.H.; Moon, J.S.; Kim, Y.K.; Yoon, J.S.; Won, K.C.; Lee, H.W. Author Correction: Critical Shear Stress is Associated with Diabetic Kidney Disease in Patients with Type 2 Diabetes. Sci. Rep. 2018, 8, 6995. [Google Scholar] [CrossRef]
- Lee, S.; Lee, M.Y.; Nam, J.S.; Kang, S.; Park, J.S.; Shin, S.; Ahn, C.W.; Kim, K.R. Hemorheological Approach for Early Detection of Chronic Kidney Disease and Diabetic Nephropathy in Type 2 Diabetes. Diabetes Technol. Ther. 2015, 17, 808–815. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, A.; Bissinger, R.; Shamaa, H.; Patel, S.; Bourne, L.; Artunc, F.; Qadri, S.M. Pathophysiology of Red Blood Cell Dysfunction in Diabetes and Its Complications. Pathophysiology 2023, 30, 327-345. https://doi.org/10.3390/pathophysiology30030026
Williams A, Bissinger R, Shamaa H, Patel S, Bourne L, Artunc F, Qadri SM. Pathophysiology of Red Blood Cell Dysfunction in Diabetes and Its Complications. Pathophysiology. 2023; 30(3):327-345. https://doi.org/10.3390/pathophysiology30030026
Chicago/Turabian StyleWilliams, Alyssa, Rosi Bissinger, Hala Shamaa, Shivani Patel, Lavern Bourne, Ferruh Artunc, and Syed M. Qadri. 2023. "Pathophysiology of Red Blood Cell Dysfunction in Diabetes and Its Complications" Pathophysiology 30, no. 3: 327-345. https://doi.org/10.3390/pathophysiology30030026
APA StyleWilliams, A., Bissinger, R., Shamaa, H., Patel, S., Bourne, L., Artunc, F., & Qadri, S. M. (2023). Pathophysiology of Red Blood Cell Dysfunction in Diabetes and Its Complications. Pathophysiology, 30(3), 327-345. https://doi.org/10.3390/pathophysiology30030026