First-Principles Study of the Structural Stability and Dynamic Properties of Li2MSiO4 (M = Mn, Co, Ni) Polymorphs

Abstract
:1. Introduction
2. Materials and Methods
3. Result and Discussions
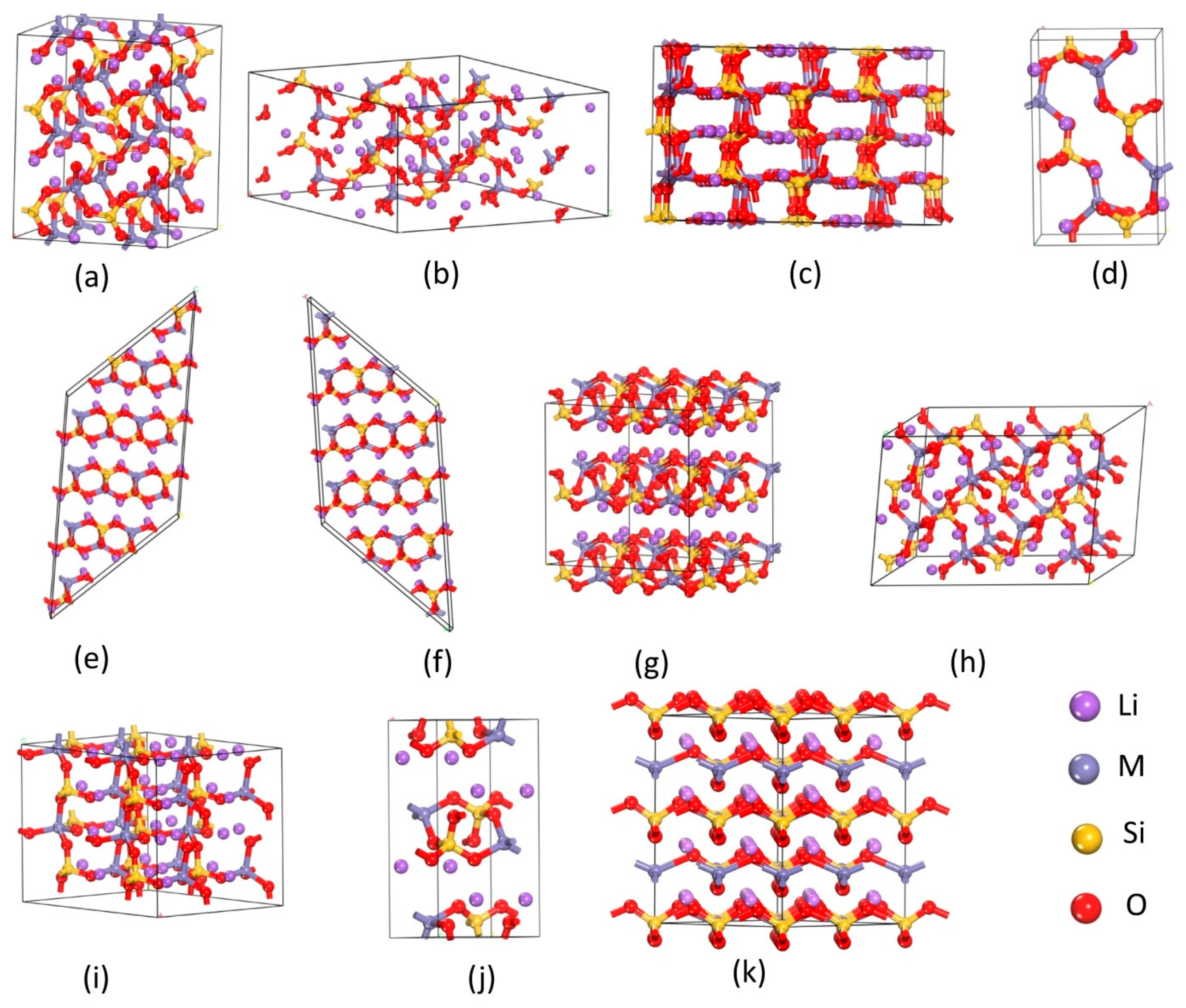
3.1. Structure Models Considered
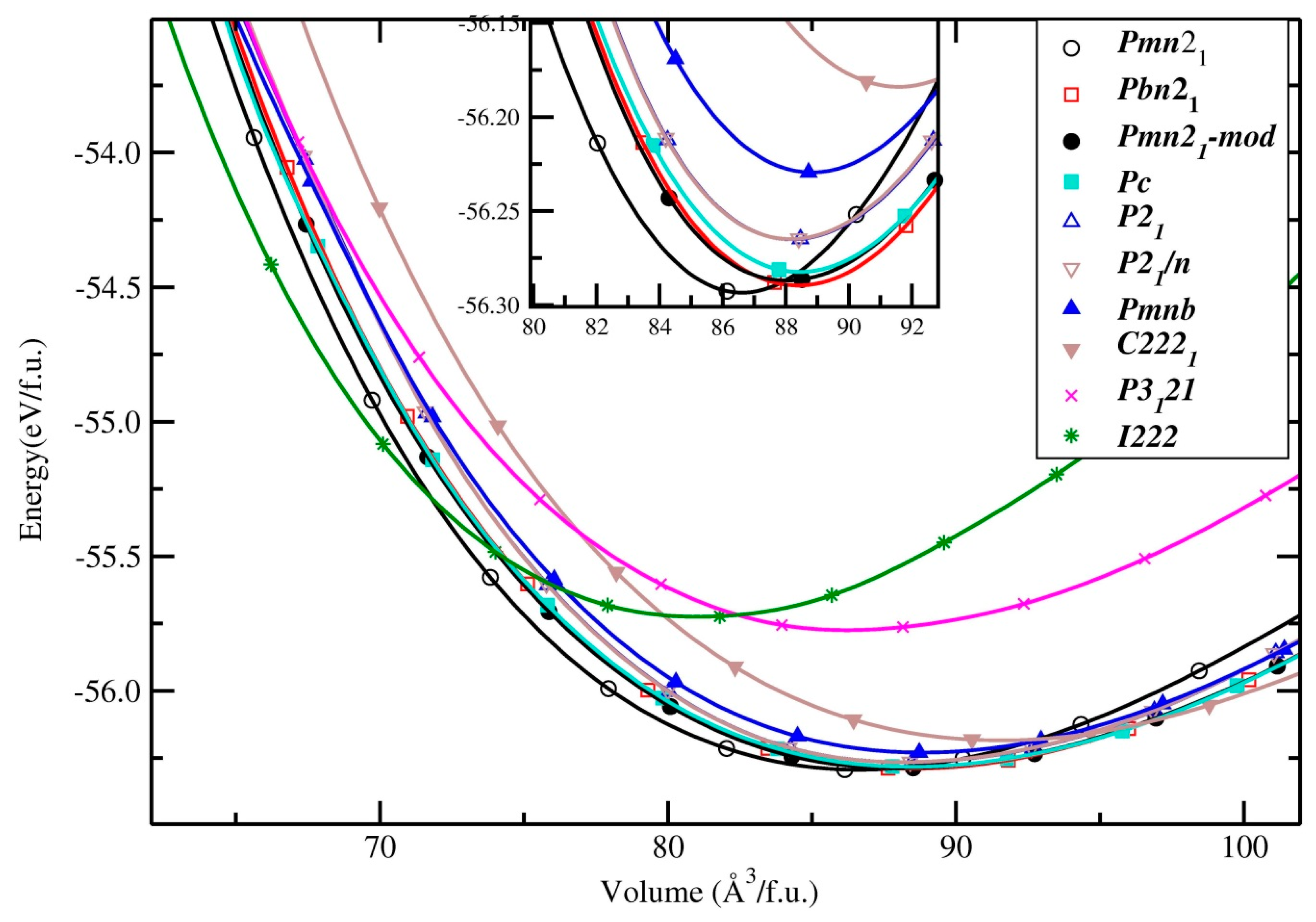
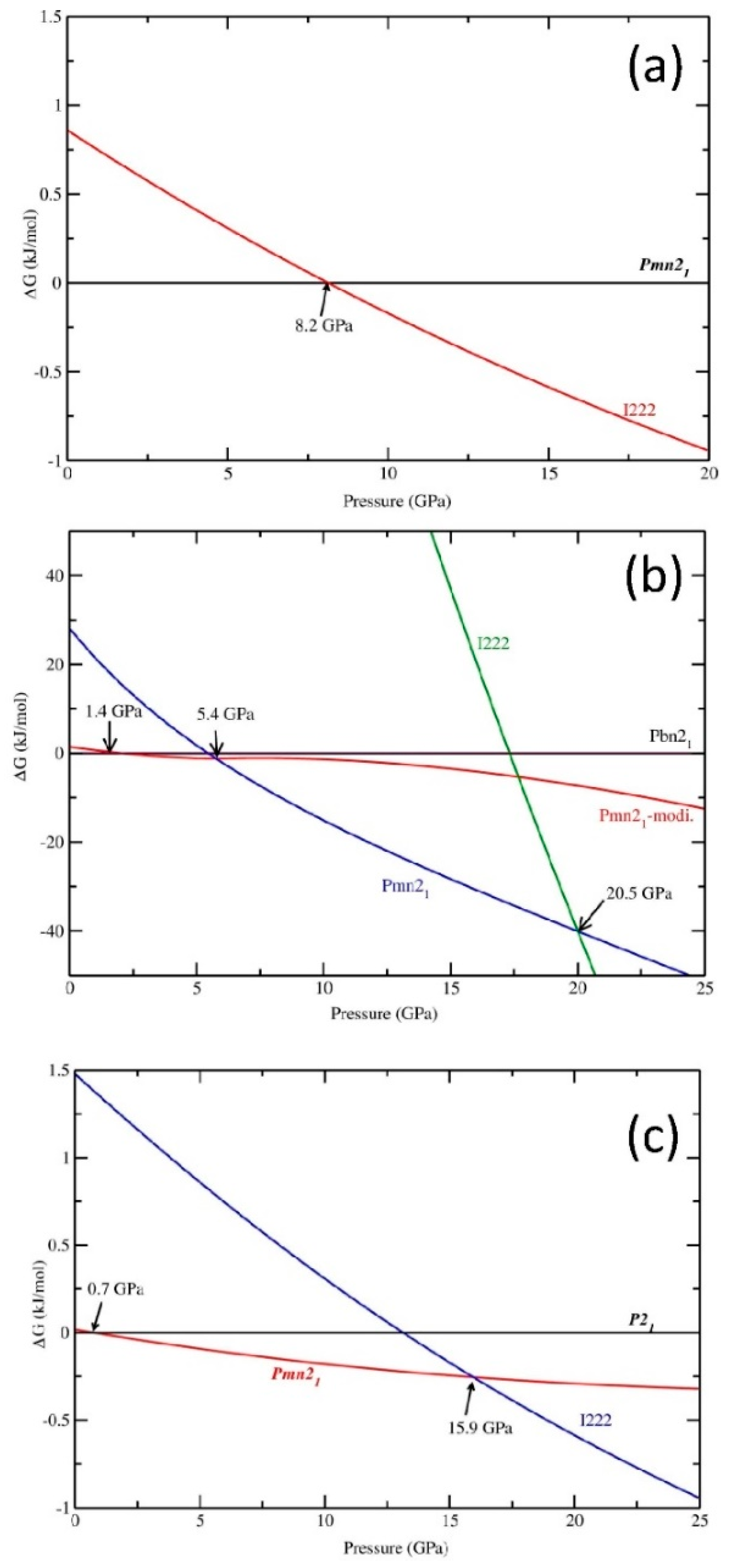
3.1.1. Li2MnSiO4
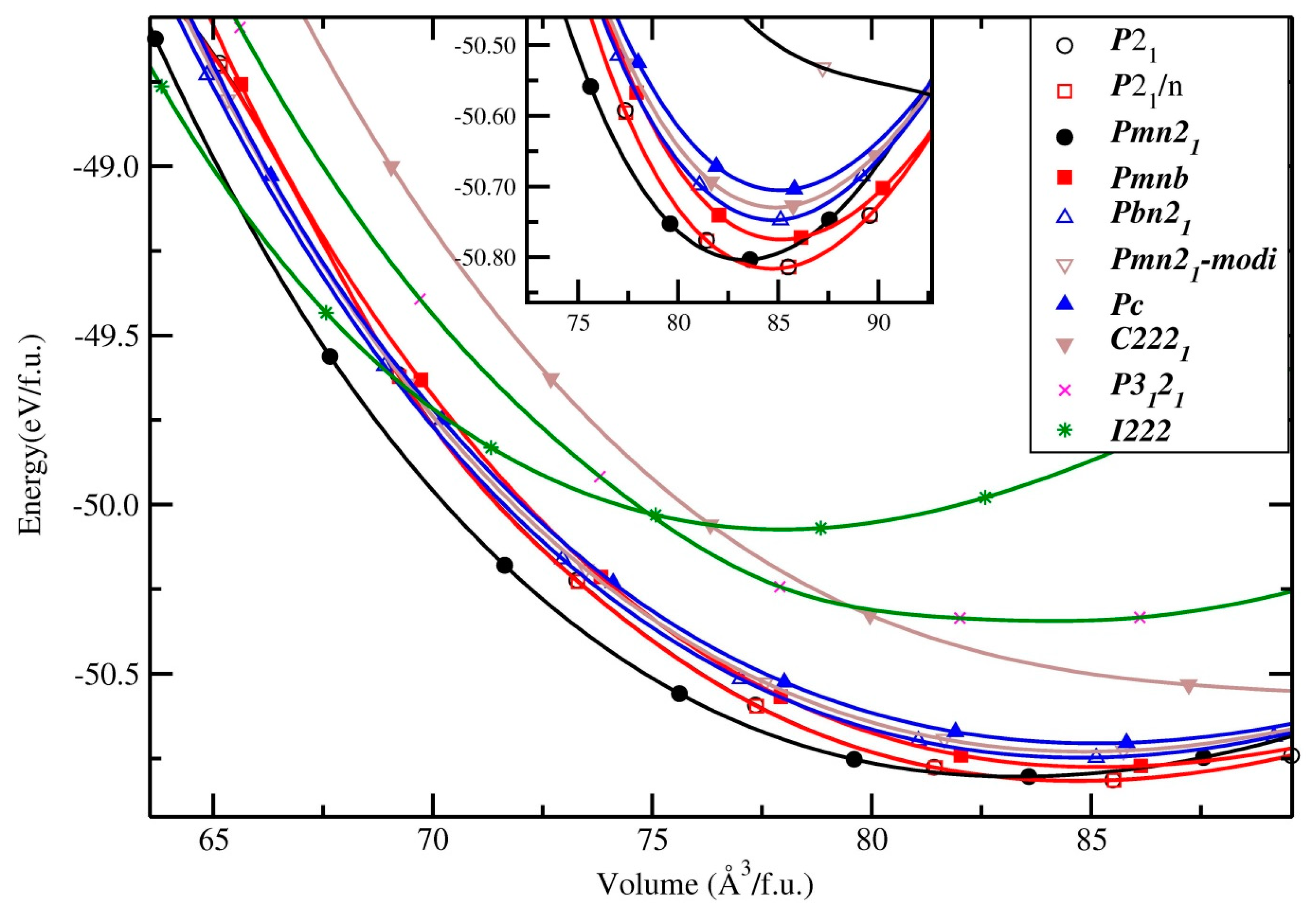
3.1.2. Li2CoSiO4
3.1.3. Li2NiSiO4
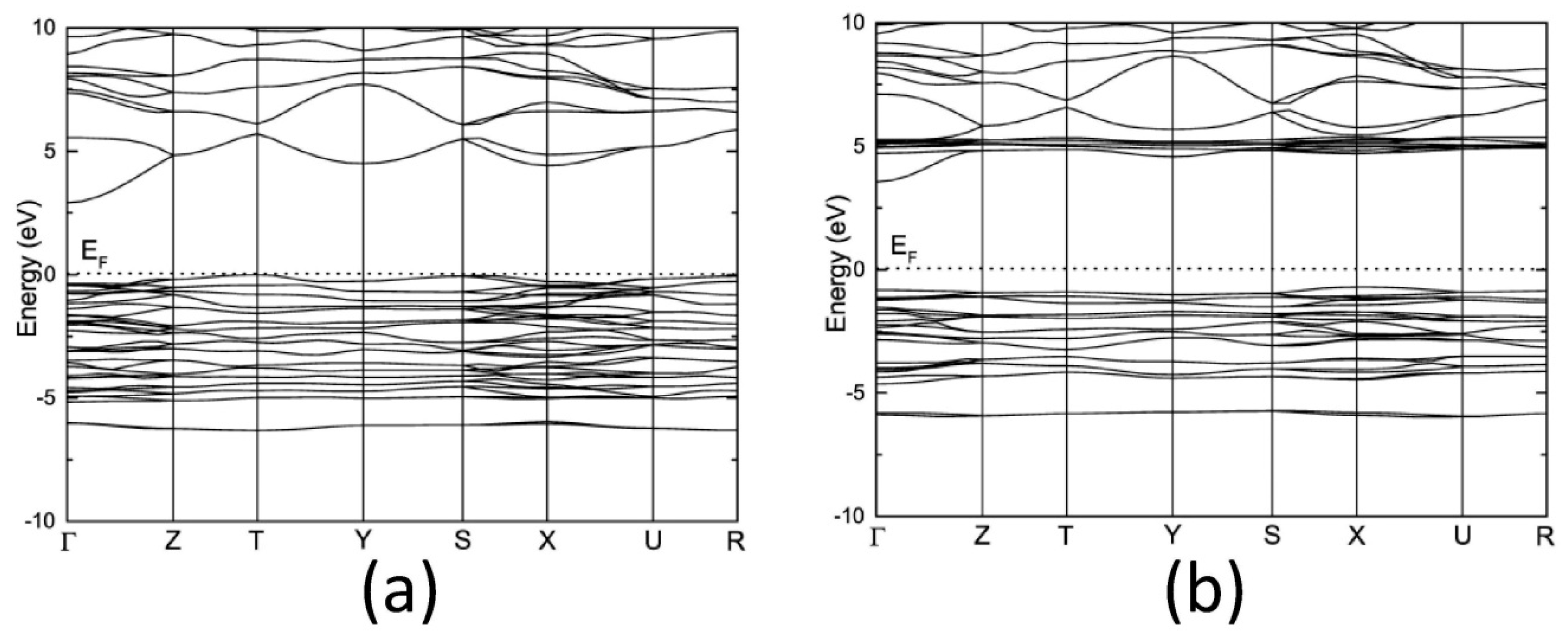
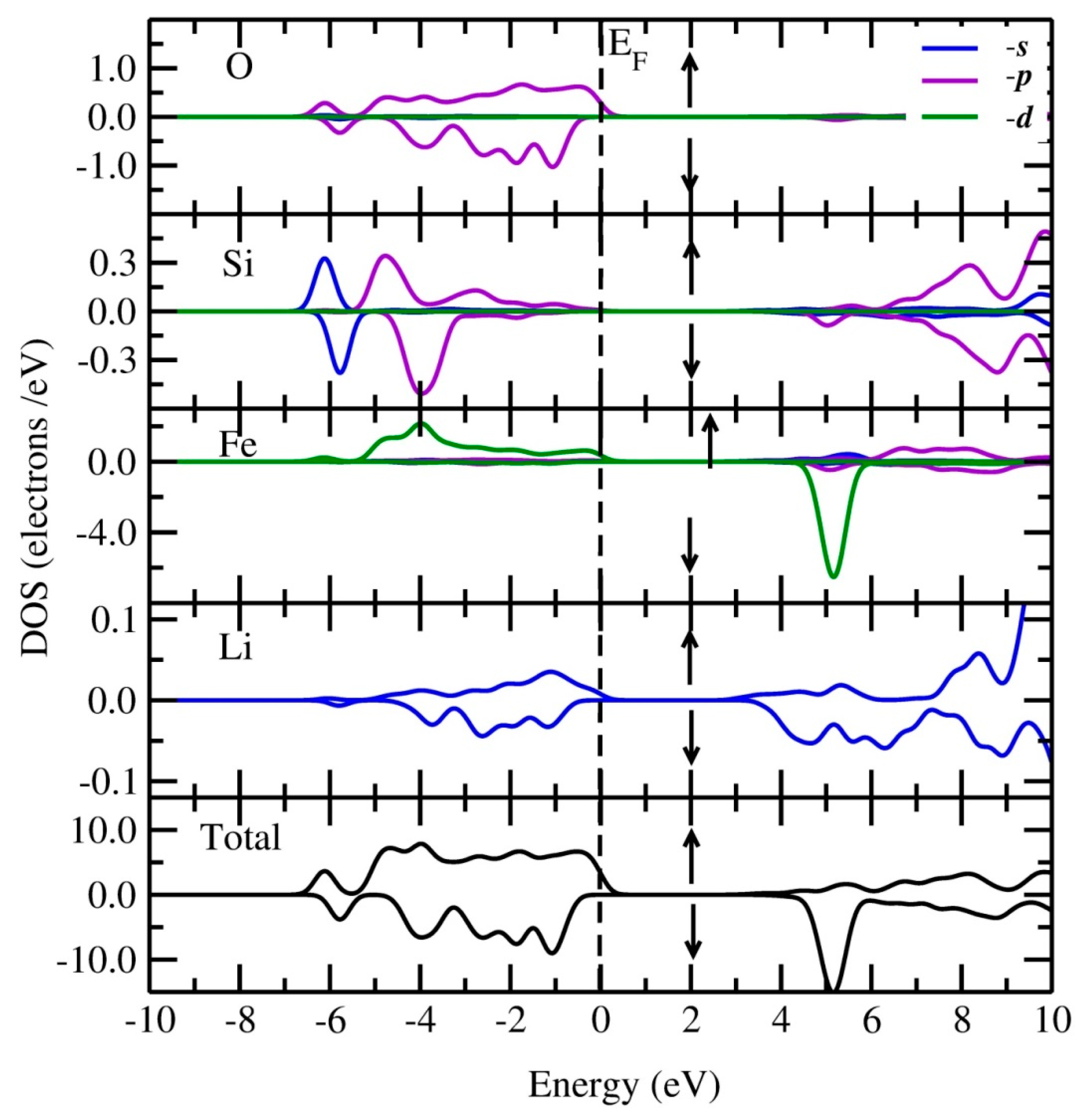
3.2. Electronic Structure
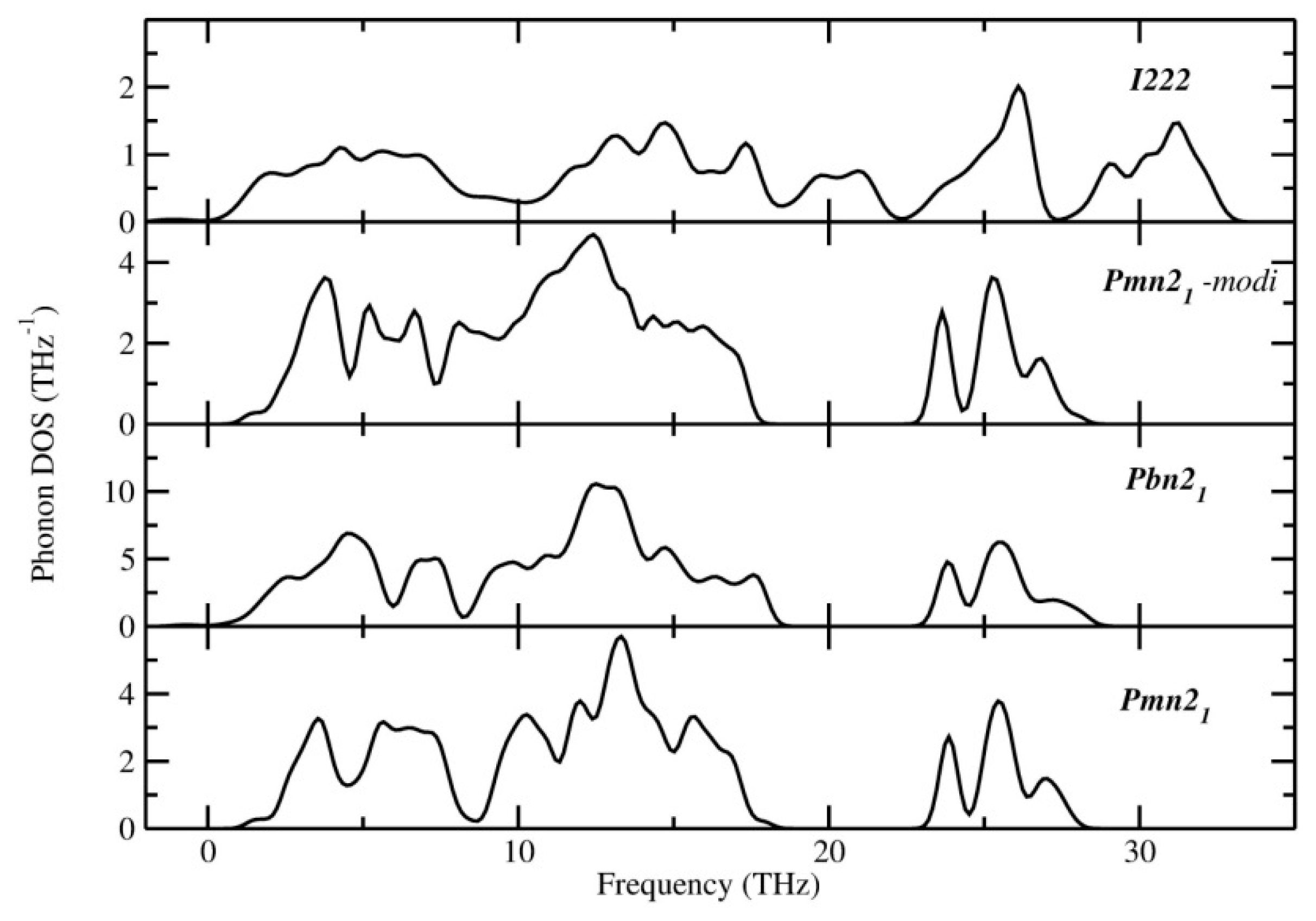
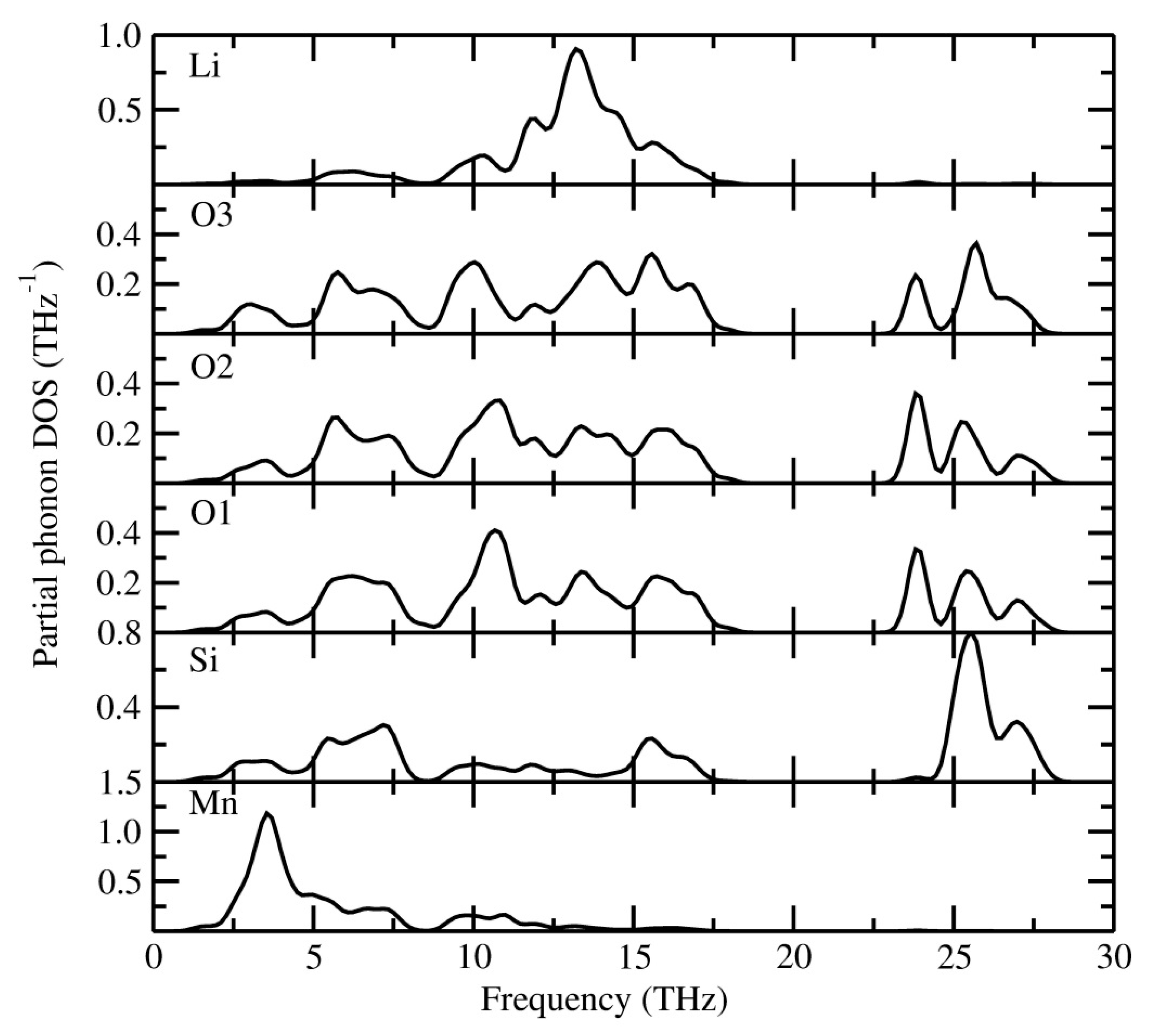
3.3. Dynamical Stability
3.4. Single-Crystal Elastic Constants and Mechanical Stability
- 1)
- The eigenvalues of Cij are positive.
- 2)
- Every leading principal (or trailing) minor, including determinants of C is positive.
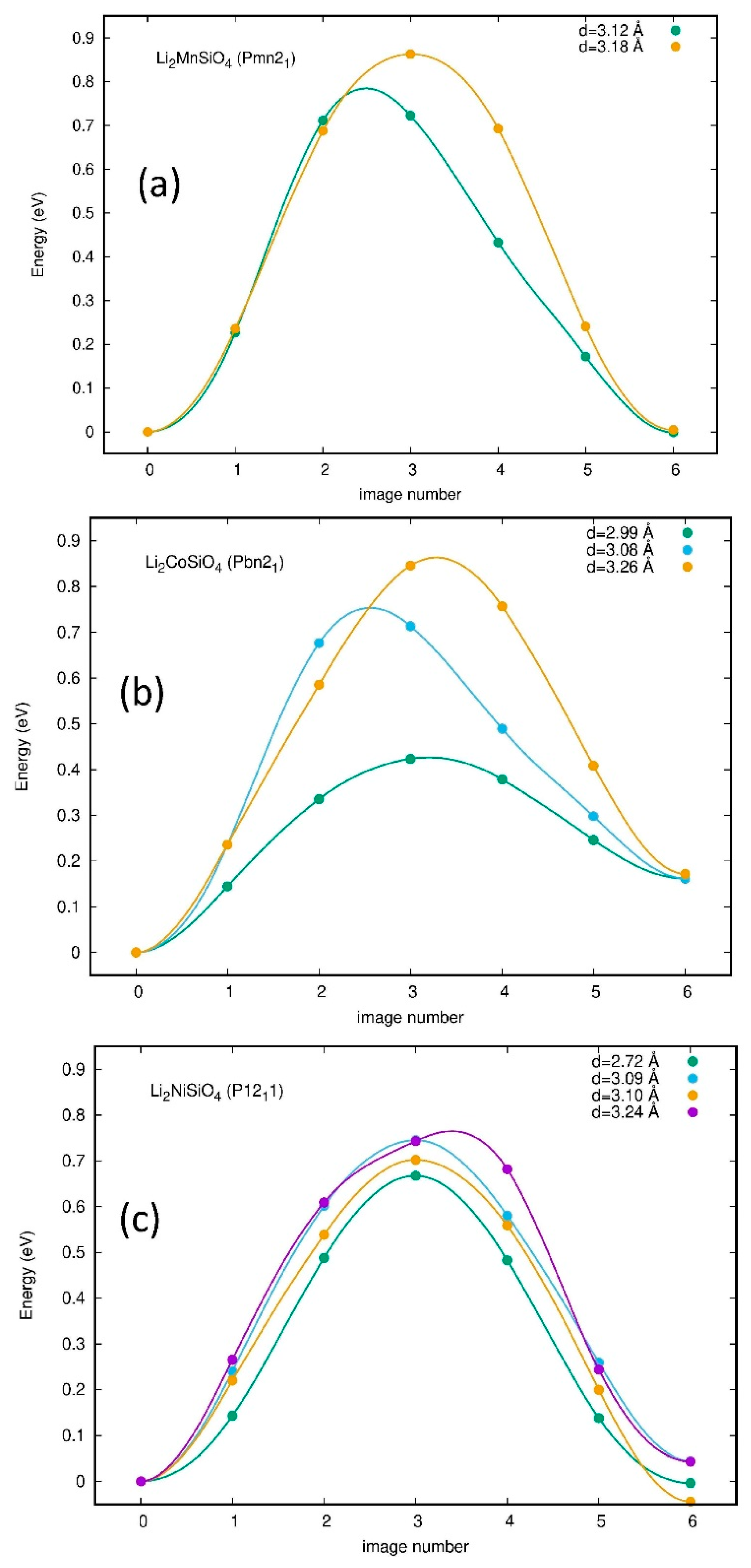
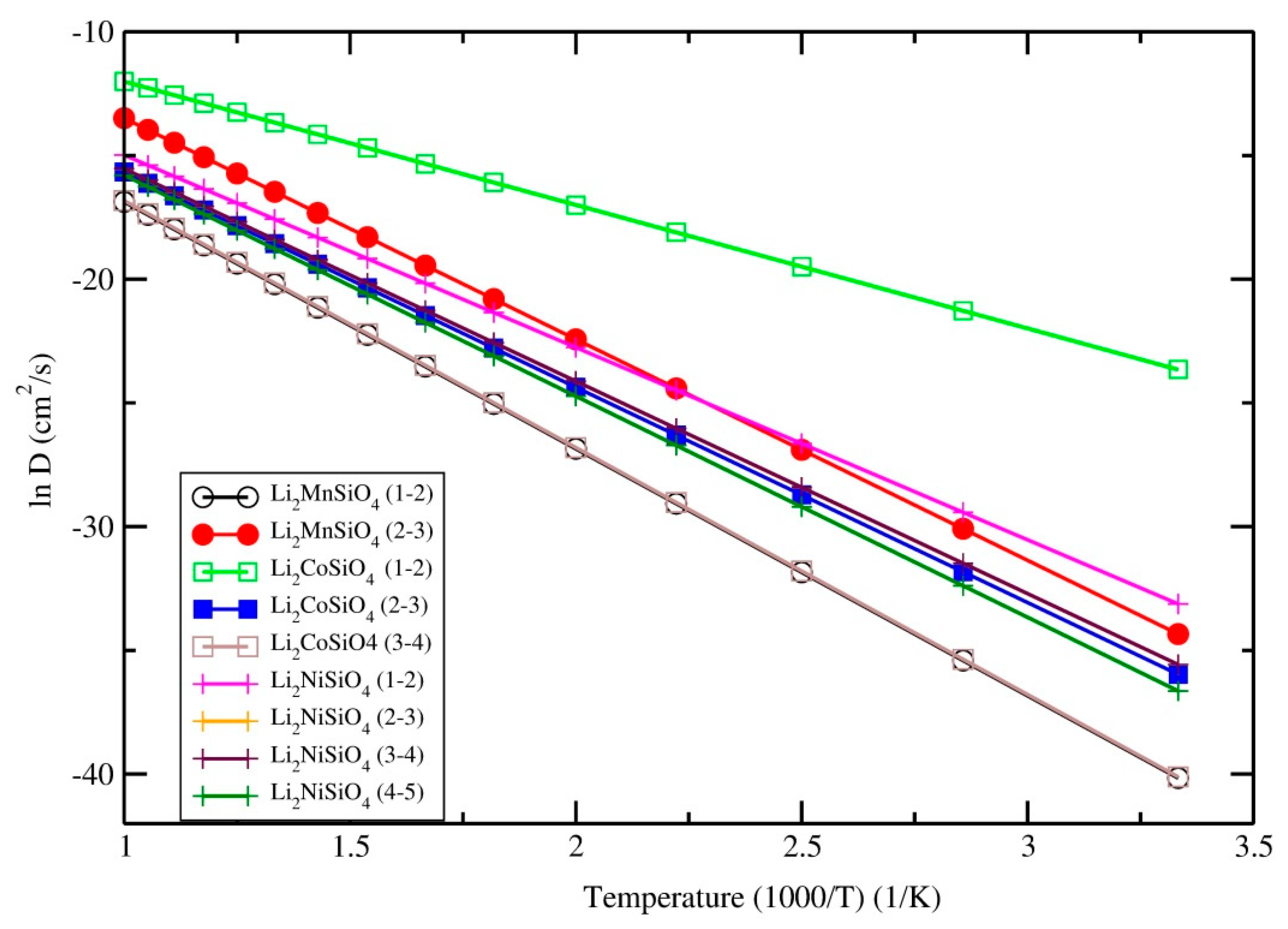
3.5. Diffusion Coefficient and Ionic Conductivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Winter, M.; Brodd, R.J. What Are Batteries, Fuel Cells, and Supercapacitors? Chemical Rev. 2004, 104, 4245–4270. [Google Scholar] [CrossRef] [Green Version]
- Dippel, C.; Krueger, S.; Kloepsch, R.; Niehoff, P.; Hoffmann, B.; Nowak, S.; Passerini, S.; Winter, M.; Li, J. Aging of Li2FeSiO4 cathode material in fluorine containing organic electrolytes for lithium-ion batteries. Electrochim. Acta 2012, 85, 66–71. [Google Scholar] [CrossRef]
- Whittingham, M.S. Lithium batteries and cathode materials. Chem. Rev. 2004, 104, 4271–4301. [Google Scholar] [CrossRef] [PubMed]
- Nytén, A.; Abouimrane, A.; Armand, M.; Gustafsson, T.; Thomas, J.O. Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material. Electrochem. Commun. 2005, 7, 156–160. [Google Scholar] [CrossRef]
- Dominko, R.; Bele, M.; Gaberšček, M.; Meden, A.; Remškar, M.; Jamnik, J. Structure and electrochemical performance of Li2MnSiO4 and Li2FeSiO4 as potential Li-battery cathode materials. Electrochem. Commun. 2006, 8, 217–222. [Google Scholar] [CrossRef]
- Zaghib, K.; Salah, A.; Ravet, N.; Mauger, A.; Gendron, F.; Julien, C. Structural, magnetic and electrochemical properties of lithium iron orthosilicate. J. Power Sources 2006, 160, 1381–1386. [Google Scholar] [CrossRef]
- Quoirin, G.; Taulelle, F.; Dupont, L.; Masquelier, C. Crystal Chemistry and Electrochemical Delithiation of Li2FeSiO4. In Proceedings of the 211th ECS Meeting, Chicago, IL, USA, 6–10 May 2007. [Google Scholar]
- Sirisopanaporn, C.; Boulineau, A.; Hanzel, D.; Dominko, R.; Budic, B.; Armstrong, A.R.; Bruce, P.G.; Masquelier, C. Crystal structure of a new polymorph of Li2FeSiO4. Inorg. Chem. 2010, 49, 7446–7451. [Google Scholar] [CrossRef]
- Arroyo-de Dompablo, M.E.; Armand, M.; Tarascon, J.M.; Amador, U. On-demand design of polyoxianionic cathode materials based on electronegativity correlations: An exploration of the Li2MSiO4 system (M = Fe, Mn, Co, Ni). Electrochem. Commun. 2006, 8, 1292–1298. [Google Scholar] [CrossRef]
- Islam, M.S.; Dominko, R.; Masquelier, C.; Sirisopanaporn, C.; Armstrong, A.R.; Bruce, P.G. Silicate cathodes for lithium batteries: Alternatives to phosphates? J. Mater. Chem. 2011, 21, 9811–9818. [Google Scholar] [CrossRef]
- Longo, R.C.; Xiong, K.; Cho, K. First Principles Study of Multicomponent Silicate Materials for Rechargeable Li-Ion Batteries. ECS Trans. 2012, 41, 75–85. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, S.; Fu, B.L.; Yang, S.Y.; Ma, L. Characterization of Li2MnSiO4 and Li2FeSiO4 cathode materials synthesized via a citric acid assisted sol–gel method. Mater. Chem. Phys. 2010, 120, 14–17. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, N.; Lian, F.; Song, Y.; Li, Y. First principle calculation of lithiation/delithiation voltage in Li-ion battery materials. Chin. Sci. Bull. 2011, 56, 3229. [Google Scholar] [CrossRef]
- Tsubakiyama, K.; Kudo, Y.; Yamashita, K. First-Principles Study on Li2MSiO4 (M = Mn, Fe, Co, Ni) as Cathode Materials of Lithium Ion Battery. ECS Trans. 2013, 53, 25–34. [Google Scholar] [CrossRef]
- Larsson, P.; Ahuja, R.; Nytén, A.; Thomas, J.O. An ab initio study of the Li-ion battery cathode material Li2FeSiO4. Electrochem. Commun. 2006, 8, 797–800. [Google Scholar] [CrossRef]
- Nyten, A.; Kamali, S.; Haggstrom, L.; Gustafsson, T.; Thomas, J.O. The lithium extraction/insertion mechanism in Li2FeSiO4. J. Mater. Chem. 2006, 16, 2266–2272. [Google Scholar] [CrossRef]
- Romain, G.; Pluton, P.; François-Xavier, C. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar]
- Vajeeston, P.; Fjellvag, H. First-principles study of structural stability, dynamical and mechanical properties of Li2FeSiO4 polymorphs. RSC Adv. 2017, 7, 16843–16853. [Google Scholar] [CrossRef]
- Longo, R.C.; Xiong, K.; Wang, W.; Cho, K. Influence of the exchange-correlation potential on the electrochemical properties of multicomponent silicate cathode materials. Electrochim. Acta 2012, 80, 84–89. [Google Scholar] [CrossRef]
- Wu, S.Q.; Zhu, Z.Z.; Yang, Y.; Hou, Z.F. Structural stabilities, electronic structures and lithium deintercalation in LixMSiO4 (M = Mn, Fe, Co, Ni): A GGA and GGA + U study. Comput. Mater. Sci. 2009, 44, 1243–1251. [Google Scholar] [CrossRef]
- Liivat, A.; Thomas, J.O. Li-ion migration in Li2FeSiO4-related cathode materials: A DFT study. Solid State Ion. 2011, 192, 58–64. [Google Scholar] [CrossRef]
- Zhong, G.; Li, Y.; Yan, P.; Liu, Z.; Xie, M.; Lin, H. Structural, Electronic, and Electrochemical Properties of Cathode Materials Li2MSiO4 (M = Mn, Fe, and Co): Density Functional Calculations. J. Phys. Chem. C 2010, 114, 3693–3700. [Google Scholar] [CrossRef]
- Li, L.; Zhu, L.; Xu, L.-H.; Cheng, T.-M.; Wang, W.; Li, X.; Sui, Q.-T. Site-exchange of Li and M ions in silicate cathode materials Li2MSiO4 (M = Mn, Fe, Co and Ni): DFT calculations. J. Mater. Chem. A 2014, 2, 4251–4255. [Google Scholar] [CrossRef]
- Kalantarian, M.M.; Asgari, S.; Capsoni, D.; Mustarelli, P. An ab initio investigation of Li2M0.5N0.5SiO4 (M, N = Mn, Fe, Co Ni) as Li-ion battery cathode materials. Phys. Chem. Chem. Phys. 2013, 15, 8035–8041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, X.D.; Yu, S.; Wu, S.Q.; Zhu, Z.Z.; Yang, Y. Effects of Na Substitution on Li Ion Migration in Li2CoSiO4 Cathode Material. J. Electrochem. Soc. 2013, 160, A658–A661. [Google Scholar] [CrossRef]
- Bianchini, F.; Fjellvåg, H.; Vajeeston, P. First-principles study of the structural stability and electrochemical properties of Na2MSiO4 (M = Mn, Fe, Co and Ni) polymorphs. Phys. Chem. Chem. Phys. 2017, 19, 14462–14470. [Google Scholar] [CrossRef]
- Santamaría-Pérez, D.; Amador, U.; Tortajada, J.; Dominko, R.; Arroyo-de Dompablo, M.E. High-Pressure Investigation of Li2MnSiO4 and Li2CoSiO4 Electrode Materials for Lithium-Ion Batteries. Inorg. Chem. 2012, 51, 5779–5786. [Google Scholar] [CrossRef]
- Armand, M. Method for Synthesis of Carbon-Coated Redox Materials with Controlled Size. World Patent WO02/27823, 4 April 2002. [Google Scholar]
- Feng, Y.; Ji, R.; Ding, Z.; Zhang, D.; Liang, C.; Chen, L.; Ivey, D.G.; Wei, W. Understanding the Improved Kinetics and Cyclability of a Li2MnSiO4 Cathode with Calcium Substitution. Inorg. Chem. 2018, 57, 3223–3231. [Google Scholar] [CrossRef]
- Wang, C.; Xu, Y.; Sun, X.; Zhang, B.; Chen, Y.; He, S. Enhanced electrochemical properties of F-doped Li2MnSiO4/C for lithium ion batteries. J. Power Sources 2018, 378, 345–352. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Z.; Zhang, X.; Wu, D.; Li, J. P-doping Li2CoSiO4/C cathode material: A joint experimental and theoretical study. Electrochim. Acta 2018, 264, 166–172. [Google Scholar] [CrossRef]
- Du, H.; Zhang, X.; Chen, Z.; Wu, D.; Zhang, Z.; Li, J. Carbon coating and Al-doping to improve the electrochemistry of Li2CoSiO4 polymorphs as cathode materials for lithium-ion batteries. RSC Adv. 2018, 8, 22813–22822. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Liechtenstein, A.I.; Anisimov, V.I.; Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467–R5470. [Google Scholar] [CrossRef] [Green Version]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Szotek, Z.; Temmerman, W.M.; Sutton, A.P. Electronic Structure and Elastic Properties of Strongly Correlated Metal Oxides from First Principles: LSDA + U, SIC-LSDA and EELS Study of UO2 and NiO. Phys. Status Solidi A 1998, 166, 429–443. [Google Scholar] [CrossRef]
- Vinet, P.; Rose, J.H.; Ferrante, J.; Smith, J.R. Temperature effects on the universal equation of state of solids. J. Phys. Condens. Matter 1989, 1, 1941–1963. [Google Scholar] [CrossRef]
- Vajeeston, P.; Ravindran, P.; Fjellvåg, H. Novel High Pressure Phases of β-AlH3: A Density-Functional Study. Chem. Mater. 2008, 20, 5997–6002. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Jonsson, H.; Mills, G.; Jacobsen, K.W. Nudged elastic band method for finding minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 2011; pp. 385–404. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Inorganic Crystal Structure Database (ICSD); Version 2011/2; Fachinformationszentrum Karlsruhe: Karlsruhe, Germany, 2011.
- Vajeeston, P.; Ravindran, P.; Kjekshus, A.; Fjellvåg, H. Crystal structure of KAlH4 from first principle calculations. J. Alloy. Compd. 2004, 363, L8–L12. [Google Scholar] [CrossRef]
- Vajeeston, P.; Ravindran, P.; Hauback, B.C.; Fjellvag, H. Prediction of crystal structure, lattice dynamical, and mechanical properties of CaB2H2. Int. J. Hydrogen Energy 2011, 36, 10149–10158. [Google Scholar] [CrossRef]
- Vajeeston, P.; Ravindran, P.; Kjekshus, A.; Fjellvåg, H. First-principles investigations of the MMgH3 (M = Li, Na, K, Rb, Cs) series. J. Alloy. Compd. 2008, 450, 327–337. [Google Scholar] [CrossRef]
- Sirisopanaporn, C.; Masquelier, C.; Bruce, P.G.; Armstrong, A.R.; Dominko, R. Dependence of Li2FeSiO4 electrochemistry on structure. J. Am. Chem. Soc. 2011, 133, 1263–1265. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Yamakawa, Y.; Yoshii, K.; Komaba, S. Hydrothermal Synthesis and Characterization of Li2FeSiO4 as Positive Electrode Materials for Li-Ion Batteries. Electrochemistry 2010, 78, 363–366. [Google Scholar] [CrossRef]
- Huang, X.; Li, X.; Wang, H.; Pan, Z.; Qu, M.; Yu, Z. Synthesis and electrochemical performance of Li2FeSiO4/carbon/carbon nano-tubes for lithium ion battery. Electrochim. Acta 2010, 55, 7362–7366. [Google Scholar] [CrossRef]
- Qu, L.; Fang, S.H.; Yang, L.; Hirano, S. Synthesis and characterization of high capacity Li2MnSiO4/C cathode material for lithium-ion battery. J. Power Sources 2014, 252, 169–175. [Google Scholar] [CrossRef]
- Shao, B.; Taniguchi, I. Synthesis of Li2MnSiO4/C nanocomposites for lithium battery cathode employing sucrose as carbon source. Electrochim. Acta 2014, 128, 156–162. [Google Scholar] [CrossRef]
- Choi, J.-J.; Kim, S.; Im, W.-B.; Chang, W.; Cho, B.-W.; Kim, J.H.; Choi, H.-L.; Chung, K.Y. Synthesis of nanostructured Li2MnSiO4/C using a microwave assisted sol–gel process with water as a base solvent. J. Electroceramics 2013, 31, 176–180. [Google Scholar] [CrossRef]
- Ghosh, P.; Mahanty, S.; Basu, R.N. Improved Electrochemical Performance of Li2MnSiO4/C Composite Synthesized by Combustion Technique. J. Electrochem. Soc. 2009, 156, A677–A681. [Google Scholar] [CrossRef]
- Politaev, V.V.; Petrenko, A.A.; Nalbandyan, V.B.; Medvedev, B.S.; Shvetsova, E.S. Crystal structure, phase relations and electrochemical properties of monoclinic Li2MnSiO4. J. Solid State Chem. 2007, 180, 1045–1050. [Google Scholar] [CrossRef]
- Duncan, H.; Kondamreddy, A.; Mercier, P.H.J.; Le Page, Y.; Abu-Lebdeh, Y.; Couillard, M.; Whitfield, P.S.; Davidson, I.J. Novel Pn Polymorph for Li2MnSiO4 and Its Electrochemical Activity As a Cathode Material in Li-Ion Batteries. Chem. Mater. 2011, 23, 5446–5456. [Google Scholar] [CrossRef]
- Zhang, S.; Deng, C.; Fu, B.L.; Yang, S.Y.; Ma, L. Doping effects of magnesium on the electrochemical performance of Li2FeSiO4 for lithium ion batteries. J. Electroanal. Chem. 2010, 644, 150–154. [Google Scholar] [CrossRef]
- Arroyo-de Dompablo, M.; Dominko, R.; Gallardo-Amores, J.; Dupont, L.; Mali, G.; Ehrenberg, H.; Jamnik, J.; Moran, E. On the Energetic Stability and Electrochemistry of Li2MnSiO4 Polymorphs. Chem. Mater. 2008, 20, 5574–5584. [Google Scholar] [CrossRef]
- Eames, C.; Armstrong, A.R.; Bruce, P.G.; Islam, M.S. Insights into Changes in Voltage and Structure of Li2FeSiO4 Polymorphs for Lithium-Ion Batteries. Chem. Mater. 2012, 24, 2155–2161. [Google Scholar] [CrossRef]
- Saracibar, A.; Van der Ven, A.; Arroyo-de Dompablo, M.E. Crystal Structure, Energetics, And Electrochemistry of Li2FeSiO4 Polymorphs from First Principles Calculations. Chem. Mater. 2012, 24, 495–503. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Akatsuka, K.; Setoguchi, M.; Takaki, Y. Structure of cobalt dilithium silicate βII-Li2CoSiO4. Acta Crystallogr. Sect. B 1979, 35, 2680–2682. [Google Scholar] [CrossRef]
- Arroyo y de Dompablo, M.E.; Amador, U.; Gallardo-Amores, J.M.; Morán, E.; Ehrenberg, H.; Dupont, L.; Dominko, R. Polymorphs of Li3PO4 and Li2MSiO4 (M = Mn, Co): The role of pressure. J. Power Sources 2009, 189, 638–642. [Google Scholar] [CrossRef]
- West, A.R.; Glasser, F.P. Preparation and crystal chemistry of some tetrahedral Li3PO4-type compounds. J. Solid State Chem. 1972, 4, 20–28. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Akatsuka, K.; Setoguchi, M. Structure of Dilithium Zinc Silicate GammaII-Li2ZnSiO4. Acta Crystallogr. Sect. B 1979, 35, 2678–2680. [Google Scholar] [CrossRef]
- Lyness, C.; Delobel, B.; Armstrong, A.R.; Bruce, P.G. The lithium intercalation compound Li2CoSiO4 and its behaviour as a positive electrode for lithium batteries. Chem. Commun. 2007, 4890–4892. [Google Scholar] [CrossRef]
- Keffer, C.; Mighell, A.; Mauer, F.; Swanson, H.; Block, S. Crystal Structure of Twinned Low-Temperature Lithium Phosphate. Inorg. Chem. 1967, 6, 119–125. [Google Scholar] [CrossRef]
- Kieu My, B.; Van An, D.; Takahisa, O. Hybrid functional study on diffusion of silicate cathode material Li2NiSiO4. J. Phys. Conf. Ser. 2013, 454, 012061. [Google Scholar]
- Ashcroft, N.W.; Mermin, N.D. Solid State Physics; Holt, Rinehart and Winston: New York, NY, USA, 1976. [Google Scholar]
- Nye, J.F. Physical Properties of Crystals; Oxford University Press: Oxford, UK, 1985. [Google Scholar]
- Watt, J.P. Hashin-Shtrikman bounds on the effective elastic moduli of polycrystals with orthorhombic symmetry. J. Appl. Phys. 1980, 51, 1520. [Google Scholar] [CrossRef]
- Haines, J.; Leger, J.; Bocquillon, G. Synthesis and Design of Superhard Materials. Annu. Rev. Mater. Res. 2001, 31, 1–23. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Pugh, S.F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Deyirmenjian, V.B.; Heine, V.; Payne, M.C.; Milman, V.; Lynden-Bell, R.M.; Finnis, M.W. Ab Initio Atomistic Simulation of the Strength of Defective Aluminum and Tests of Empirical Force Models. Phys. Rev. B 1995, 52, 15191–15207. [Google Scholar] [CrossRef]
- Marlo, M.; Milman, V. Density-functional study of bulk and surface properties of titanium nitride using different exchange-correlation functionals. Phys. Rev. B 2000, 62, 2899–2907. [Google Scholar] [CrossRef]
- Milman, V.; Warren, M.C. Elastic properties of TiB2 and MgB2. J. Phys. Condens. Matter 2001, 13, 5585–5595. [Google Scholar] [CrossRef]
- Ravindran, P.; Vajeeston, P.; Vidya, R.; Kjekshus, A.; Fjellvåg, H. Detailed electronic structure studies on superconducting MgB2 and related compounds. Phys. Rev. B 2001, 64, 224509. [Google Scholar] [CrossRef]
- de, A.; Manassidis, I.; Lin, J.S.; Gillan, M.J. The energetics of Frenkel defects in Li2O from 1st principles. Europhys. Lett. 1992, 19, 605–610. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Karki, B.B.; Clark, S.J.; Warren, M.C.; Hsueh, H.C.; Ackland, G.J.; Crain, J. Ab initio elasticity and lattice dynamics of AgGaSe2. J. Phys. Condens. Matter 1997, 9, 375–380. [Google Scholar] [CrossRef]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. ZAMM 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. A 1952, 65, 350. [Google Scholar] [CrossRef]
- Voigt, W. Lehrbuch der Kristallphysik; B.G. Teubner: Leipzig, Germany, 1928. [Google Scholar]
- Horn, R.A.; Johnson, C.R. Matrix Analysis; Cambridge University Press: Cambridge, UK, 2012; p. 664. [Google Scholar]
- Luo, S.H.; Wang, M.; Sun, W.N. Fabricated and improved electrochemical properties of Li2MnSiO4 cathodes by hydrothermal reaction for Li-ion batteries. Ceram. Int. 2012, 38, 4325–4329. [Google Scholar] [CrossRef]
- Vajeeston, Ionic conductivity enhancement by particle size reduction in Li2FeSiO4. Mater. Lett. 2018, 218, 313–316. [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Lattice Parameter | Eg (eV) | Bo | Bo’ | |||
|---|---|---|---|---|---|---|---|
| a | b | c | β(deg) | ||||
| Li2MnSiO4-P21/n | 6.4102 (6.3360) a | 10.9932 (10.9146) a | 5.1034 (5.0730) a | 91.1 (90.99) a | 2.94 | 47 | 3.8 |
| Li2MnSiO4-Pbn21 | 6.3382 | 10.9504 | 5.0512 | 90 | 2.90 | 46 | 3.8 |
| Li2MnSiO4-Pmn21 | 6.3250 (6.3109) b | 5.3842 (5.3454) b | 5.0031 (4.9624) b | 90 | 2.97 | 47 | 4.0 |
| Li2MnSiO4-I222 | 4.4033 | 4.4033 | 6.4288 | 90 | 1.56 | 58 | 4.1 |
| Li2CoSiO4-Pbn21 | 6.2782 (6.253) c | 10.084 (10.685) c | 5.282 (4.929) c | 90 | 2.39 | 44 | 3.9 |
| Li2CoSiO4-Pn | 8.2320 (8.231) b | 5.0168 (5.022) b | 8.2348 (8.232) b | 99.18 (99.27) | 3.15 | 42 | 4.0 |
| Li2CoSiO4-I222 | 4.2823 | 4.2823 | 6.6211 | 90 | 2.2 | 58 | 3.7 |
| Li2NiSiO4-P21 | 8.4536 | 5.0858 | 8.4682 | 100.25 | 2.35 | 43 | 4.0 |
| Li2NiSiO4-Pmn21 | 6.3210 (6.294) d | 5.4755 (5.3702) d | 5.1002 (4.9137) d | 90 | 1.3 | 42 | 4 |
| Li2NiSiO4-I222 | 4.4203 | 4.4203 | 6.9204 | 90 | 1.72 | 56 | 3.6 |
| Li2NiSiO4-P21 | 8.4536 | 5.0858 | 8.4682 | 100.25 | 2.35 | 43 | 4.0 |
| Compound | Space Group | Positional Parameters |
|---|---|---|
| Li2MnSiO4 | Pmn21 | Li (4b): 0.7490, 0.3281, 0.9420; Mn (2a): ½, 0.175, 0.4384; Si (2a): 0, 0.1766, 0.4502; O1 (4b): 0.71224, 0.6813, 0.8404; O2 (2a): ½, 0.1175, 0.8496; O3 (2a): ½, 0.8205, 0.2820 |
| Pbn21 | Li (4a): −0.1607, 0.0034, 0.2580; Li (4a): −0.4233, 0.7444, 0.2582; Mn (4a): −0.1660, 0.4965, 0.2589; Si (4a): −0.4141, 0.2524, 0.2640; O1 (4a): −0.4122, 0.2544, 0.5911; O2 (4a): −0.5566, 0.2520, 0.1557; O2 (4a): −0.3312, 0.0360, 0.1641; O2 (4a): −0.3445, 0.4668, 0.1528 | |
| I222 | Li (4d): −1/2, 0, ¼; Mn (2b): −1/2, ½, 0; Si (2a): 0, 0, 0; O (8i): −0.2132, 0.2133, 0.1288 | |
| Li2CoSiO4 | Pbn21 | Li (4a): −0.1596, −0.0012, 0.2548; Li (4a): −0.4255, 0.750, 0.2541; Co (4a): −0.1697, 0.4864, 0.2545; Si (4a): −0.4129, 0.2418, 0.2646; O1 (4a): −0.4090, 0.2357, 0.5960; O2 (4a): −0.5547, 0.2418, 0.1641; O2 (4a): −0.3392, 0.0319, 0.1503; O2 (4a): −0.3473, 0.64625, 0.1641 |
| Pn | Li (2a): −0.5, 0.1602, −0.9993; Li (2a): 0.2469, 0.3262, −0.4990; Ni (2a): -0.2437, 0.3332, −0.4951 Si (2a): 0.9877, 0.1662, −0.0099; O1 (2a): −0.3438, 1719, −0.3454; O3 (2a): 0.0879, 0.1233, −0.4081; O3 (2a): −0.1232, 0.6891, −0.4081; O4 (2a): −0.6863, 0.3080, −0.8978 | |
| I222 | Li (4d): 0, ½,¼; Co (2b): 0, 0, ½; Si (2a): 0, 0, 0; O1(8i): 0.2784, 0.2784, 0.6304 | |
| Li2NiSiO4 | P21 | Li (2a): 0.6593, 0.9166, 0.6623; Li (2a): 0.1598, 0.8134, 0.1621; Li (2a): 0.5818, 0.4112, 0.0884; Li (2a): 0.0815, 0.383, 0.5883; Ni (2a): 0.2879, 0.9167, 0.5358; Ni (2a): 0.7886, 0.8136, 0.0363; Si (2a): 0.0444, 0.8106, 0.7953; Si (2a): 0.5449, 0.9200, 0.2955; O1 (2a): 0.8656, 0.9064, 0.8318; O2 (2a): 0.3659, 0.8241, 0.3316; O3 (2a): 0.4258, 0.3121, 0.8823; O4 (2a): 0.9263, 0.4183, 0.3824; O5 (2a): 0.6875, 0.8084, 0.4409; O6 (2a): 0.1873, 0.9218, 0.9408; O7 (2a): 0.9514, 0.9829, 0.2049; O8 (2a): 0.4505, 0.7475, 0.7047 |
| Pmn21 | Li (4b): 0.751, 0.3256, 0.9385; Ni (2a): ½, 0.1818, 0.4359; Si (2a): 0, 0.16204, 0.4497; O1 (4b): 0.7119, 0.6922, 0. 8502; O2 (2a): ½, 0.1275, 0.8372; O3 (2a): ½, 0.8439, 0.2851 | |
| I222 | Li (4d): 0, ½, ¼; Co (2b): 0, 0, ½; Si (2a): 0, 0, 0; O1 (8i): 0.2709, 0.2709, 0.6270 |
| Properties | Phase | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Li2MnSiO4 | Li2CoSiO4 | Li2NiSiO4 | |||||||
| Pmn21 | Pbn21 | I222 | Pbn21 | Pmn21-mod | I222 | P21 | Pmn21 | I222 | |
| Cij | C11 = 192 | C11 = 124 | C11 = 312 | C11 = 119 | C11 = 96 | C11 = 336 | C11 = 135 | C11 = 203 | C11 = 310 |
| C12 = 66 | C12 = 65 | C12 = 165 | C12 = 50 | C12 = 36 | C12 = 230 | C12 = 38 | C12 = 47 | C12 = 214 | |
| C13 = 70 | C13 =55 | C13 = 141 | C13 = 40 | C13 = 33 | C13 = 199 | C13 = 64 | C13 = 50 | C13 = 122 | |
| C22 = 128 | C22 = 157 | C22 = 320 | C22 = 141 | C15 = −16 | C22 = 339 | C15 = 0 | C22 = 119 | C22 = 378 | |
| C23 = 42 | C23 = 60 | C23 = 183 | C23 = 38 | C22 = 123 | C23 = 249 | C22 = 112 | C23 = 36 | C23 = 185 | |
| C33 = 150 | C33 = 55 | C33 = 270 | C33 = 131 | C23 = 29 | C33 = 301 | C23 = 43 | C33 = 132 | C33 = 229 | |
| C44 = 47 | C44 = 37 | C44 = 34 | C44 = 47 | C25 = 0 | C44 = 9 | C25= 0 | C44 = 47 | C44 = 44 | |
| C55 = 37 | C55 = 37 | C55 = 59 | C55 = 37 | C33 = 115 | C55 = 38 | C33 = 157 | C55 = 39 | C55 = 52 | |
| C66 = 49 | C66 = 43 | C66 = 33 | C66 = 44 | C35 = 0 | C66 = 13 | C35 = 0 | C66 = 45 | C66 = 40 | |
| C44 = 417 | C44 = 46 | ||||||||
| C46 = 0 | C46 = 0 | ||||||||
| C55 = 34 | C55 = 42 | ||||||||
| C66 = 44 | C66 = 54 | ||||||||
| BV | 92 | 89 | 208 | 72 | 59 | 77 | 80 | ||
| BR | 86 | 88 | 181 | 72 | 59 | 71 | 75 | ||
| BVRH | 89 | 88 | 195 | 72 | 59 | 74 | 77 | ||
| GV | 46 | 40 | 53 | 43 | 40 | 46 | 48 | ||
| GR | 44 | 40 | 589 | 43 | 39 | 44 | 46 | ||
| GVRH | 45.5 | 40 | 321 | 43 | 39 | 45 | 47 | ||
| E | 117 | 104 | 621 | 107 | 96 | 112 | 117 | ||
| Compressibility | 0.01 | 0.01 | 0.005 | 0.01 | 0.01 | 0.01 | 0.01 | ||
| Ductility | 0.5 | 0.5 | 1.64 | 0.6 | 0.7 | 0.6 | 0.609 | ||
| Lamè constant | 59 | 61 | −19 | 43 | 33 | 44 | 46 | ||
| σ | 0.28 | 0.30 | −0.03 | 0.25 | 0.23 | 0.25 | 0.247 | ||
| L | 6973 | 6846 | 13709 | 6332 | 5889 | 6452 | 6527 | ||
| T | 3844 | 3643 | 9841 | 3651 | 3499 | 3732 | 3784 | ||
| 4284 | 4070 | 10646 | 4053 | 3874 | 4142 | 4199 | |||
| ΘD | 1573 | 1485 | 4008 | 1498 | 1430 | 1532 | 1564 | ||
| Phase | Leading Minors (GPan) | Trailing Minors (GPan) | Eigenvalues (GPa) |
|---|---|---|---|
| Li2MnSiO4-Pmn21 | 1.92 × 102 2.02 × 104 | 2.09 × 1011 1.47 × 109 | 2.83 × 102 8.64 × 101 |
| 2.45 × 106 1.15 × 108 | 1.28 × 107 8.51 × 104 | 1.00 × 102 4.72 × 101 | |
| 4.29 × 109 2.09 × 1011 | 1.81 × 103 4.86 × 101 | 3.72 × 101 4.86 × 101 | |
| Li2MnSiO4-Pmn21-mod | 1.19 × 102 1.51 × 104 | 1.01 × 1011 1.06 × 109 | 2.34 × 102 7.92 × 101 |
| 1.76 × 106 6.53 × 107 | 7.98 × 106 5.76 × 104 | 9.54 × 101 4.24 × 101 | |
| 2.38 × 109 1.01 × 1011 | 1.55 × 103 4.26 × 101 | 3.53 × 101 3.83 × 101 | |
| Li2MnSiO4-Pbn21 | 1.24 × 102 1.52 × 104 | 1.09 × 1011 1.20 × 109 | 2.67 × 102 7.24 × 101 |
| 1.88 × 106 6.85 × 107 | 9.05 × 106 5.81 × 104 | 9.70 × 101 3.65 × 101 | |
| 2.56 × 109 1.09 × 1011 | 1.59 × 103 4.25 × 101 | 3.74 × 101 4.25 × 101 | |
| Li2MnSiO4-I222 | 3.12 × 102 7.24 × 104 | 1.39 × 1010 2.09 × 108 | 6.37 × 102 1.64 × 102 |
| 1.13 × 107 1.09 × 108 | 2.42 × 106 2.93 × 104 | 1.35 × 102 8.07 × 101 | |
| 6.12 × 109 1.39 × 1010 | 8.74 × 102 3.31 × 101 | 1.24 9.87 | |
| Li2CoSiO4-Pmn21 | 1.83 × 102 1.94 × 104 | 1.85 × 1011 1.23 × 109 | 2.46 × 102 1.02 × 102 |
| 2.20 × 106 1.04 × 108 | 1.10 × 107 8.39 × 104 | 8.73 × 101 4.75 × 101 | |
| 4.30 × 109 1.85 × 1011 | 1.77 × 103 4.29 × 101 | 4.12 × 101 4.29 × 101 | |
| Li2CoSiO4-Pmn21-modi | 9.64 × 101 1.05 × 104 | 6.57 × 1010 8.20 × 108 | 1.78 × 102 4.33 × 101 |
| 1.07 × 106 4.45 × 107 | 7.11 × 106 6.19 × 104 | 6.74 × 101 8.94 × 101 | |
| 1.51 × 109 6.57 × 1010 | 1.48 × 103 4.37 × 101 | 3.38 × 101 4.19 × 101 | |
| Li2CoSiO4-Pbn21 | 1.19 × 102 1.44 × 104 | 1.24 × 1011 1.29 × 109 | 2.16 × 102 7.74 × 101 |
| 1.64 × 106 7.66 × 107 | 9.90 × 106 7.53 × 104 | 9.81 × 101 4.66 × 101 | |
| 2.84 × 109 1.24 × 1011 | 1.62 × 103 4.36 × 101 | 3.71 × 101 4.36 × 101 | |
| Li2CoSiO4-I222 | 2.02 × 102 3.72 × 104 | 2.66 × 1011 2.49 × 109 | 3.09 × 102 1.42 × 102 |
| 7.88 × 105 4.72 × 107 | 2.19 × 107 3.38 × 105 | 1.80 × 101 6.00 × 101 | |
| 3.55 × 109 2.66 × 1011 | 5.64 × 103 7.51 × 101 | 7.51 × 101 7.51 × 101 | |
| Li2NiSiO4-P21 | 1.35 × 102 1.37 × 104 | 1.59 × 1011 1.56 × 109 | 2.41 × 102 4.87 × 101 |
| 1.66 × 106 7.54 × 107 | 1.55 × 107 1.03 × 105 | 8.11 × 101 8.79 × 101 | |
| 3.15 × 109 1.59 × 1011 | 2.28 × 103 5.43 × 101 | 4.75 × 101 4.01 × 101 | |
| Li2NiSiO4-P21/n | 1.37 × 102 1.34 × 104 | 1.48 × 1011 1.39 × 109 | 2.29 × 102 4.77 × 101 |
| 1.53 × 106 6.69 × 107 | 1.43 × 107 1.05 × 105 | 8.13 × 101 8.57 × 101 | |
| 2.97 × 109 1.48 × 1011 | 2.41 × 103 5.40 × 101 | 4.74 × 101 4.11 × 101 | |
| Li2NiSiO4-Pmn21 | 2.03 × 102 2.20 × 104 | 2.12 × 1011 1.21 × 109 | 2.54 × 102 1.12 × 102 |
| 2.53 × 106 1.19 × 108 | 1.11 × 107 8.39 × 104 | 8.91 × 101 4.72 × 101 | |
| 4.67 × 109 2.12 × 1011 | 1.78 × 103 4.54 × 101 | 3.92 × 101 4.54 × 101 | |
| Li2NiSiO4-I222 | 2.01 × 102 2.66 × 104 | 7.86 × 1010 1.36 × 109 | 2.89 × 102 1.07 × 102 |
| 2.43 × 105 1.80 × 107 | 1.58 × 107 3.24 × 105 | 7.83 7.40 × 101 | |
| 1.17 × 109 7.86 × 1010 | 4.38 × 103 6.74 × 101 | 6.49 × 101 6.74 × 101 |
| Phase | Barrier Height | Li-Li Distance | Diffusion Coefficient at 300 K |
|---|---|---|---|
| Li2MnSiO4-Pmn21 | 0.78 | 3.12 | 7.6 × 10−16 |
| 0.86 | 3.18 | 3.6 × 10−18 | |
| Li2MnSiO4-Pbn21 | 0.68 | 3.03 | 3.4 × 10−15 |
| 0.70 | 3.09 | 1.6 × 10−15 | |
| 0.73 | 3.12 | 5.3 × 10−16 | |
| Li2MnSiO4-I222 | 1.2 | 3.12 | 6.7 × 10−24 |
| 1.23 | 3.20 | 2.2 × 10−24 | |
| Li2CoSiO4-Pbn21 | 0.43 | 2.99 | 5.3 × 10−10 |
| 0.75 | 3.08 | 2.4 × 10−15 | |
| 0.86 | 3.26 | 3.8 × 10−17 | |
| Li2CoSiO4-Pmn21-mod | 0.74 | 3.24 | 3.9 × 10−16 |
| 0.79 | 3.33 | 5.9 × 10−17 | |
| Li2CoSiO4-I222 | 1.1 | 3.03 | 3.0 × 10−22 |
| 1.31 | 3.31 | 1.08 × 10−25 | |
| Li2NiSiO4-P21 | 0.67 | 2.72 | 4.1 × 10−15 |
| 0.74 | 3.09 | 3.5 × 10−16 | |
| 0.74 | 3.10 | 3.5 × 10−16 | |
| 0.77 | 3.24 | 1.2 × 10−16 | |
| Li2NiSiO4-Pmn21 | 0.85 | 3.12 | 5.1 × 10−18 |
| 0.89 | 3.27 | 1.2 × 10−18 | |
| Li2NiSiO4-I222 | 1.07 | 3.12 | 1.0 × 10−21 |
| 1.12 | 3.27 | 1.6 × a10−22 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vajeeston, P.; Bianchini, F.; Fjellvåg, H. First-Principles Study of the Structural Stability and Dynamic Properties of Li2MSiO4 (M = Mn, Co, Ni) Polymorphs. Energies 2019, 12, 224. https://doi.org/10.3390/en12020224
Vajeeston P, Bianchini F, Fjellvåg H. First-Principles Study of the Structural Stability and Dynamic Properties of Li2MSiO4 (M = Mn, Co, Ni) Polymorphs. Energies. 2019; 12(2):224. https://doi.org/10.3390/en12020224
Chicago/Turabian StyleVajeeston, Ponniah, Federico Bianchini, and Helmer Fjellvåg. 2019. "First-Principles Study of the Structural Stability and Dynamic Properties of Li2MSiO4 (M = Mn, Co, Ni) Polymorphs" Energies 12, no. 2: 224. https://doi.org/10.3390/en12020224
APA StyleVajeeston, P., Bianchini, F., & Fjellvåg, H. (2019). First-Principles Study of the Structural Stability and Dynamic Properties of Li2MSiO4 (M = Mn, Co, Ni) Polymorphs. Energies, 12(2), 224. https://doi.org/10.3390/en12020224