1. Introduction
Formic acid (FA) is one of the liquid organic hydrogen carriers (LOHC), which contains 4.4 wt% of hydrogen. This chemical is persistent and thus, can be safely stored, transported, and applied as a source of hydrogen for the needs of hydrogen energetics instead of difficult to handle molecular hydrogen [
1,
2,
3]. Sufficient stability combined with low toxicity and low flammability provides additional advantages for the practical use of FA. It is important that FA can be produced sustainably from biomass [
4,
5] or via reaction of CO
2 with hydrogen, which can be obtained by electrolysis [
6,
7]. Another key point is that FA undergoes facile dehydrogenation over supported noble metal catalysts at mild conditions. In particular, it was reported that the activity of Au/Al
2O
3 catalysts was higher compared to that of Pt/Al
2O
3 catalysts in the gas-phase reaction [
8]. Overall, the catalytic behavior of supported Au in the FA decomposition is influenced by the nature of the support [
9,
10], the gold dispersion [
8,
11], and the use of basic cocatalysts [
12,
13,
14].
In principle, FA can be decomposed through the desired path (1) to afford a mixture of molecular hydrogen and carbon dioxide or alternatively, FA can undergo dehydration to give concurrently highly undesirable carbon monoxide (path 2):
It is noteworthy that despite significant efforts in finding efficient homo- and heterogeneous catalysts to address pathway (1), the achievement of near-theoretical-maximum selectivity of this reaction is still a challenging task. On the other hand, it was demonstrated recently that gold catalysts with the same gold content (~2.5 wt%) and mean Au-particles size (2.4–3.0 nm) supported on MgO, La
2O
3, ZrO
2, CeO
2, and Al
2O
3 have exhibited a clear volcano-type dependence of the FA decomposition along path (1) on the electronegativity of the support’s cation, with the Au/Al
2O
3 on the top [
9]. Moreover, the latter catalyst has demonstrated the best selectivity and practically CO-free hydrogen production. Based on these findings, it was suggested that dehydrogenation of FA should be catalyzed by gold nanoparticles but only in combination with the acid-base activity of the support. It is significant in this context that the acid-base reactivity of metal oxides themselves toward dehydration/dehydrogenation of FA is also generally known [
15,
16,
17]. However, the relative contribution of oxide support as part of the gold catalyst to pathways (1) and (2) remained uncertain.
In order to fill that gap, in this study, we have examined the catalytic behavior of gold catalysts, such as Au/Al2O3, Au/TiO2, and Au/SiO2 in decomposition of FA, and then compared it with the catalytic activity of corresponding supports. It is found that the undesirable pathway (2) results exclusively from the acid-base behavior of supports, regardless of the presence of gold.
2. Materials and Methods
Commercially available Al
2O
3 (A-201 La Roche Industries Inc.), SiO
2 (Merck), and TiO
2 (Aerolyst 7708 Degussa AG) have been used as oxide supports. The deposition of gold on the oxide supports was implemented by a direct ionic exchange method using ammonia according to a procedure elaborated by Ivanova et al. [
18]. An aqueous 5 × 10
−4 M solution of HAuCl
4 (99.9%, ABCR, Darmstadt, Germany) was mixed with a support in a ratio corresponding to the Au concentration of 2 wt%. After stirring at 70 °C for 2 h, a 4 M solution of ammonia was added. The suspension thus obtained was stirred at 70 °C for an additional 1 h, then filtered off and washed with water. The samples were dried overnight at 80 °C and calcined at 300 °C for 4 h.
The specific surface area of the catalysts was determined from N2 adsorption isotherms at 77.3 K on a Quadrosorb evo (Quantachrome Instruments, USA) analyzer after degassing the samples with the use of a FLOVAC Degasser (Quantachrome Instruments, USA) instrument.
XPS study was performed with a SPECS Phoibos 150 (Germany) photoelectron spectrometer. The spectra were recorded using an AlKα source with quantum energy of 1486.6 eV. The energy scale was calibrated against the binding energy of gold Au4f7/2 equal to 84.0 eV. A carbon C 1s peak for hydrocarbon impurities at 285.0 eV was used as a reference for the energy scale calibration.
The gold content in the catalysts was determined by inductively coupled plasma optical emission spectrometry (ICP-OES) using an Optima 5300 DV (Perkin-Elmer) instrument. The samples (50 mg) were dissolved in 5 ml of imperial water (2HNO3 and 6HCl). The solutions obtained were diluted with water to 100 ml and then analyzed.
Transmission electron microscopy (TEM) was performed with a Zeiss LEO 912 OMEGA (Germany) microscope at an acceleration voltage of 120 kV. To determine the mean diameter of Au particles, more than 100 particles were chosen.
The gas-phase decomposition of formic acid was performed in a quartz tube flow reactor with an internal diameter of 6 mm. Activity tests were carried out at atmospheric pressure using 20 mg of a catalyst mixed with 0.5 mL of quartz sand (d = 0.25 mm). The overall length of the catalyst layer was ~20 mm. The gas mixture (5 vol% formic acid in He) was fed at a rate of 20 mL min
−1. The experiments were carried out in the temperature-programmed mode. The heating rate was 2 °C min
−1. The reaction temperature was measured with a thermocouple inserted into the catalyst bed. In order to achieve reproducible results, prior to activity measurements, the catalysts were pretreated in the reactor according to a standard procedure: heating at 300 °C in a flow of 5 vol% HCOOH in He for 15 min, which ensured the in situ reduction conditions. Then the catalysts were cooled in the same mixture to the reaction temperature. The progress of the reaction was monitored by gas chromatography, judged by the production of CO and CO
2, as described earlier [
19].
3. Results
3.1. Catalysts Characterization
It is known that the most active gold catalysts contain small gold particles, which are less than 10 nm in diameter, especially on such supports as reducible TiO
2. With this in mind, samples of Au/TiO
2, Au/Al
2O
3, and Au/SiO
2 with a gold content of ~2 wt% and an average gold particle size below 3 nm were prepared. The gold particle size was estimated by TEM. The particle size distribution of Au/TiO
2, Au/Al
2O
3, and Au/SiO
2 samples are shown in
Figure 1. The gold loading in the prepared samples was determined by using ICP-OES. The exact loadings of Au, the surface areas of the catalysts found by BET, and the average sizes of gold particles are stated in
Table 1.
The Au catalysts were also characterized by XPS in order to gain insight into the electronic state of Au. The XPS showed the presence of Au in the metallic state only (Au 4f
7/2~84.0 eV) (
Figure 2), while the nature of the support did not influence notably the electronic state of Au. However, the deconvolution of the XPS spectra revealed the existence of metallic gold with peaks shifted towards lower binding energies. This can be related to divers phenomena—the electron transfer from the support to particles of gold [
20,
21] and/or to a dominant effect of the atoms at low coordinated sites present in small particles, as it was proposed by Radnik et al. [
22,
23].
3.2. Catalytic Activity
The catalytic performance of the oxide supported gold catalysts is shown in
Figure 3. Despite the comparable content of gold (~2 wt%) and mean Au-particles size (~2 nm), the catalytic properties of Au/TiO
2, Au/SiO
2, and Au/Al
2O
3 samples in the FA decomposition are notably different. The relative activity of the catalysts turned out to be as follows: Au/TiO
2 > Au/SiO
2 > Au/Al
2O
3. Complete conversion of FA was achieved at 190 °C for Au/TiO
2, whereas for Au/Al
2O
3 and Au/SiO
2, the FA conversion had attained only 5 and 15% at the same conditions. However, decomposition of FA occurred by different pathways. The most active catalyst, Au/TiO
2, provided mainly undesirable pathway (2) to give CO and H
2O. For this catalyst, the selectivity towards H
2 formation was below 5% at complete conversion of FA. In contrast, decomposition of FA on Au/Al
2O
3 and Au/SiO
2 afforded mostly H
2 and CO
2 with a selectivity of ~80% (at complete conversion of FA).
For illustration, the long-term performance of Au/SiO
2 in the FA decomposition is presented in
Figure 4. As can be seen, the catalytic activity and reaction selectivity practically did not change, even after FA dehydrogenation for 5 h at 228 °C.
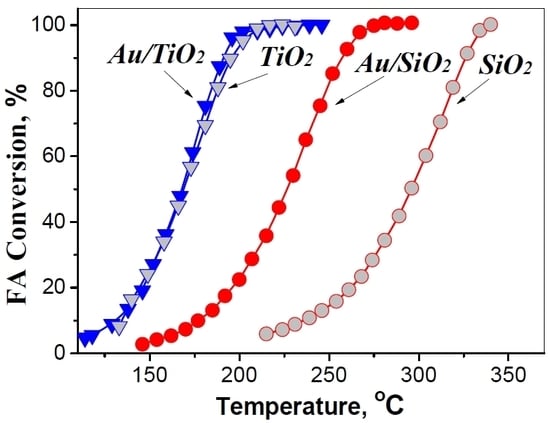
Figure 5 shows the catalytic activity of Au/SiO
2, Au/Al
2O
3, and Au/TiO
2 samples versus that of the corresponding oxide supports. It is definitely seen that the catalytic performance of Au/TiO
2 relies almost entirely on the action of TiO
2 (
Figure 5c). Unfortunately, TiO
2 (and therefore, Au/TiO
2) is more inclined to catalyze dehydration of FA on its acid sites [
17], thus demonstrating comparatively poor catalytic activity towards the desirable path (1).
In contrast, SiO
2 itself has shown much lower catalytic activity compared to Au/SiO
2 (
Figure 5a). The selectivity to FA dehydrogenation on SiO
2 is comparatively low and inversely associated with FA dehydration. Pure Al
2O
3 behaves in a similar way. The selectivity of FA conversion to H
2 and CO
2 is very low (
Figure 5b). Thus, none of the oxide supports has demonstrated significant selectivity for H
2 production. On the contrary, their acid-base properties lead mainly to the undesirable decomposition of FA by pathway (2).
In general, these observations are in accordance with recent findings on the enhanced selectivity to hydrogen formation in FA decomposition over gold and other metals supported on an inert support, such as carbon or carbon doped with nitrogen [
14,
24,
25,
26,
27]. However, these results were considered mainly in terms of the increased catalytic activity of corresponding catalysts toward FA dehydrogenation. In our vision, the oxide supports, such as SiO
2, Al
2O
3, and especially TiO
2, strongly compete with the target reaction, promoting acid-catalyzed dehydration of FA.
4. Conclusions
It is shown that the oxide support (regardless of whether it is TiO2, Al2O3, or SiO2) does not affect the electronic state of the supported nanoparticles of gold, which exist in the metallic state. However, a notable effect of the oxide support on the selectivity of hydrogen production from FA decomposition is found. The target reaction (FA → H2 + CO2) competes strongly with dehydration of FA due to the acid-base properties of the supports in the order given: TiO2 > Al2O3 ~ SiO2. Hence, the results of this study can be applied for the development of supported gold catalysts with the diminished acidity of supports for the selective hydrogen production via FA decomposition.
Author Contributions
Catalytic measurements, data analysis, and text editing, V.S.; data analysis and writing, K.K.; XPS measurements, I.A.
Funding
This work was conducted within the framework of the budget project for the Boreskov Institute of Catalysis (No. AAAA-A17-117041710083-5).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gianotti, E.; Taillades-Jacquin, M.; Roziere, J.; Jones, D.J. High-Purity Hydrogen Generation Via Dehydrogenation of Organic Carriers: A Review on the Catalytic Process. ACS Catal. 2018, 8, 4660–4680. [Google Scholar] [CrossRef]
- Niermann, M.; Beckendorff, A.; Kaltschmitt, M.; Bonhoff, K. Liquid Organic Hydrogen Carrier (LOHC)—Assessment Based on Chemical and Economic Properties. Int. J. Hydrogen Energy 2019, 44, 6631–6654. [Google Scholar] [CrossRef]
- Zhong, H.; Iguchi, M.; Chatterjee, M.; Himeda, Y.; Xu, Q.; Kawanami, H. Formic Acid-Based Liquid Organic Hydrogen Carrier System with Heterogeneous Catalysts. Adv. Sustain. Syst. 2018, 2, 1700161. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R.H. Towards Sustainable Production of Formic Acid. ChemSusChem 2018, 11, 821–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preuster, P.; Albert, J. Biogenic Formic Acid as a Green Hydrogen Carrier. Energy Technol. 2018, 6, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Bulushev, D.A.; Ross, J.R.H. Heterogeneous Catalysts for Hydrogenation of CO2 and Bicarbonates to Formic Acid and Formates. Catal. Rev. 2018, 60, 566–593. [Google Scholar] [CrossRef]
- Wang, W.H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef]
- Ojeda, M.; Iglesia, E. Formic Acid Dehydrogenation on Au-Based Catalysts at near-Ambient Temperatures. Angew. Chem. Int. Ed. 2009, 48, 4800–4803. [Google Scholar] [CrossRef]
- Zacharska, M.; Chuvilin, A.L.; Kriventsov, V.V.; Beloshapkin, S.; Estrada, M.; Simakov, A.; Bulushev, D.A. Support Effect for Nanosized Au Catalysts in Hydrogen Production from Formic Acid Decomposition. Catal. Sci. Technol. 2016, 6, 6853–6860. [Google Scholar] [CrossRef]
- Gazsi, A.; Bansagi, T.; Solymosi, F. Decomposition and Reforming of Formic Acid on Supported Au Catalysts: Production of CO-Free H2. J. Phys. Chem. C 2011, 115, 15459–15466. [Google Scholar] [CrossRef]
- Singh, S.; Li, S.; Carrasquillo-Flores, R.; Alba-Rubio, A.C.; Dumesic, J.A.; Mavrikakis, M. Formic Acid Decomposition on Au Catalysts: DFT, Microkinetic Modeling, and Reaction Kinetics Experiments. AIChE J. 2014, 60, 1303–1319. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Zacharska, M.; Guo, Y.; Beloshapkin, S.; Simakov, A. CO-Free Hydrogen Production from Decomposition of Formic Acid over Au/Al2O3 Catalysts Doped with Potassium Ions. Catal. Commun. 2017, 92, 86–89. [Google Scholar] [CrossRef]
- Jia, L.; Bulushev, D.A.; Beloshapkin, S.; Ross, J.R.H. Hydrogen Production from Formic Acid Vapour over a Pd/C Catalyst Promoted by Potassium Salts: Evidence for Participation of Buffer-Like Solution in the Pores of the Catalyst. Appl. Catal. B Environ. 2014, 160, 35–43. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Sobolev, V.I.; Pirutko, L.V.; Starostina, A.V.; Asanov, I.P.; Modin, E.; Chuvilin, A.L.; Gupta, N.; Okotrub, A.V.; Bulusheva, L.G. Hydrogen Production from Formic Acid over Au Catalysts Supported on Carbon: Comparison with Au Catalysts Supported on SiO2 and Al2O3. Catalysts 2019, 9, 376. [Google Scholar] [CrossRef]
- Trillo, J.M.; Munera, G.; Criado, J.M. Catalytic decomposition of formic acid on metal oxides. Catal. Rev. 1972, 7, 51–86. [Google Scholar] [CrossRef]
- Patermarakis, G. The parallel dehydrative and dehydrogenative catalytic action of γ-Al2O3 pure and doped by MgO. Appl. Catal. A 2003, 252, 231–241. [Google Scholar] [CrossRef]
- Sobolev, V.I.; Koltunov, K.Y. Oxidative and non-oxidative degradation of C1–C3 carboxylic acids over V2O5/TiO2 and MoVTeNb oxides: A comparative study. Appl. Catal. A Gen. 2013, 466, 45–50. [Google Scholar] [CrossRef]
- Ivanova, S.; Pitchon, V.; Zimmermann, Y.; Petit, C. Preparation of Alumina Supported Gold Catalysts: Influence of Washing Procedures, Mechanism of Particles Size Growth. Appl. Catal. A Gen. 2006, 298, 57–64. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Chuvilin, A.L.; Sobolev, V.I.; Stolyarova, S.G.; Shubin, Y.V.; Asanov, I.P.; Ishchenko, A.V.; Magnani, G.; Ricco, M.; Okotrub, A.V.; et al. Copper on Carbon Materials: Stabilization by Nitrogen Doping. J. Mater. Chem. A 2017, 5, 10574–10583. [Google Scholar] [CrossRef]
- Claus, P.; Bruckner, A.; Mohr, C.; Hofmeister, H. Supported Gold Nanoparticles from Quantum Dot to Mesoscopic Size Scale: Effect of Electronic and Structural Properties on Catalytic Hydrogenation of Conjugated Functional Groups. J. Am. Chem. Soc. 2000, 122, 11430–11439. [Google Scholar] [CrossRef]
- Sanchez, A.; Abbet, S.; Heiz, U.; Schneider, W.-D.; Hakkinen, H.; Barnett, R.N.; Landman, U. When Gold Is Not Noble: Nanoscale Gold Catalysts. J. Phys. Chem. A 1999, 103, 9573–9578. [Google Scholar] [CrossRef]
- Radnik, J.; Mohr, C.; Claus, P. On the Origin of Binding Energy Shifts of Core Levels of Supported Gold Nanoparticles and Dependence of Pretreatment and Material Synthesis. Phys. Chem. Chem. Phys. 2003, 5, 172–177. [Google Scholar] [CrossRef]
- Pohl, M.-M.; Radnik, J.; Schneider, M.; Bentrup, U.; Linke, D.; Bruckner, A.; Ferguson, E. Bimetallic PdAu–KOac/SiO2 catalysts for vinyl acetate monomer (VAM) synthesis: Insights into deactivation under industrial conditions. J. Catal. 2009, 262, 314–323. [Google Scholar] [CrossRef]
- Tang, C.; Surkus, A.-E.; Chen, F.; Pohl, M.-M.; Agostini, G.; Schneider, M.; Junge, H.; Beller, M. A Stable Nanocobalt Catalyst with Highly Dispersed CoNx Active Sites for the Selective Dehydrogenation of Formic Acid. Angew. Chem. Int. Ed. 2017, 56, 16616–16620. [Google Scholar] [CrossRef]
- Sanchez, F.; Alotaibi, M.H.; Motta, D.; Chan-Thaw, C.E.; Rakotomahevitra, A.; Tabanelli, T.; Roldan, A.; Hammond, C.; He, Q.; Davies, T.; et al. production from formic acid decomposition in the liquid phase using Pd nanoparticles supported on CNFs with different surface properties. Sustain. Energy Fuels 2018, 2, 2705–2716. [Google Scholar] [CrossRef]
- Sanchez, F.; Motta, D.; Roldan, A.; Hammond, C.; Villa, A.; Dimitratos, N. Hydrogen Generation from Additive-Free Formic Acid Decomposition Under Mild Conditions by Pd/C: Experimental and DFT Studies. Top. Catal. 2018, 61, 254–266. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, F.; Motta, D.; Bocelli, L.; Albonetti, S.; Roldan, A.; Hammond, C.; Villa, A.; Dimitratos, N. Investigation of the Catalytic Performance of Pd/CNFs for Hydrogen Evolution from Additive-Free Formic Acid Decomposition. C J. Carbon Res. 2018, 4, 26. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}