Density Functional Theory-Based Molecular Modeling: Verification of Decisive Roles of Van der Waals Aggregation of Triiodide Ions for Effective Electron Transfer in Wet-Type N3-Dye-Sensitized Solar Cells



Abstract
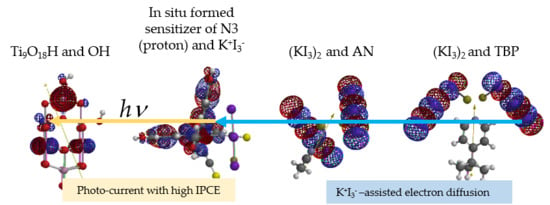
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. DFT-Based Electrostatic Potential Map (EPM) and Van der Waals Aggregation
2.2. Verification of KI3 Formation by Van der Waals Aggregation of I2 and KI in Electrolytes
2.3. Van der Waals Aggregation of Potassium Triiodide (KI3) and 4-Tertbyutylpyridine (TB) Effect on DSSC Electrolytes
2.4. Electrostatic Potential Map (ESPM) of TiO2 Anatase Nanoparticle and Conductivity
2.5. Electrostatic Potential Map (ESPM) of Sensitizing N3 Dyes and Perspective of Van der Waals Aggregation
2.6. Verification of Van der Waals Aggregation of N3 Dye with Anatase TiO2 Model
2.7. Van der Waals Aggregation of N3 Dye with Potassium Triiodide
3. Discussion
4. Conclusions
5. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| vdW | van der Waals |
| MM | Molecular modelling |
| LUMO | Lowest-unoccupied molecular orbital |
| ESPM | Electrostatic potential map |
| MMFF | Merck molecular force field |
| DFT | Density functional theory |
| HOMO | Highest-occupied molecular orbital |
| DSSC | Dye-sensitized solar cell |
| NP | Nanoparticle |
References
- Yanagida, S.; Yanagisawa, S.; Segawa, H. Molecular Orbital-Based Verification of Conductivity of Tetramethylammonium Pentaiodide and Pentaiodide-Based Electrolytes in Dye-Sensitized Solar Cells. J. Electrochem. Soc. 2015, 162, E263–E270. [Google Scholar] [CrossRef]
- Yanagida, S.; Yanagisawa, S.; Segawa, H. Computational Verification of So-Called Perovskite Solar Cells as PbI64−-Aligned Solar Cells. J. Electrochem. Soc. 2017, 164, E3598–E3605. [Google Scholar] [CrossRef] [Green Version]
- Yanagida, S.; Yanagisawa, S.; Yanagida, M.; Segawa, H. Validity of density-functional-theory-based molecular modeling for UV/visible spectroscopy and rationale of panchromatic PbI6 4−(MeNH3+) 4-structured molecular solar cells. Jpn. J. Appl. Phys. 2018, 57, 121602. [Google Scholar] [CrossRef]
- Yanagida, S.; Osabe, K.; Nagai, T.; Murakami, N. Quantum chemistry molecular modeling for longevity: Importance of antioxidative effects in mitochondria as battery of cells. Integr. Mol. Med. 2019, 6, 1–6. [Google Scholar] [CrossRef]
- Yanagida, S.; Kaname, A.; Murakami, N. Quantum chemistry-based verification of antioxidative action of iodide in mitochondria. Integr. Mol. Med. 2019, 6, 1–6. [Google Scholar] [CrossRef]
- Yanagida, S.; Yanagisawa, S.; Murakami, N. Quantum chemistry molecular modelling for mitochondria targeted chemotherapy: Verification of oxidative stress on mitochondria and anticancer medicines. Integr. Mol. Med. 2020, 7, 1–7. [Google Scholar] [CrossRef]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Jiang, J.-K.; Yanagida, S. Optimization of Redox Mediators and Electrolysis. In Dye-sensitized Solar Cells; EPFL Press (Distributed by CRC Press): Lausanne, Switzerland, 2010; Chapter 4; pp. 117–144. [Google Scholar]
- Xia, J.; Yanagida, S. Strategy to improve the performance of dye-sensitized solar cells: Interface engineering principle. Sol. Energy 2011, 85, 3143–3159. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, X.; Zhang, K.; Chen, H.; Yanagida, M.; Han, L. Effects of 4-tert-butylpyridine on the quasi-Fermi levels of TiO2 films in the presence of different cations in dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2011, 13, 19310–19313. [Google Scholar] [CrossRef] [PubMed]
- Boschloo, G.; Haggman, L.; Hagfeldt, A. Quantification of the effect of 4-tert-butylpyridine addition to I-/I3-redox electrolytes in dye-sensitized nanostructured TiO2 solar cells. J. Phys. Chem. B 2006, 110, 13144–13150. [Google Scholar] [CrossRef]
- Zhang, S.; Yanagida, M.; Yang, X.; Han, L. Effect of 4-tert-butylpyridine on the quasi-Fermi level of dye-sensitized TiO2 films. Appl. Phys. Express 2011, 4, 042301. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, S.; Sodeyama, K.; Yang, X.; Chen, H.; Yanagida, M.; Tateyama, Y.; Han, L. A new factor affecting the performance of dye-sensitized solar cells in the presence of 4-tert-butylpyridine. Appl. Phys. Express 2012, 5, 042303. [Google Scholar] [CrossRef]
- Jono, R.; Fujisawa, J.; Segawa, H.; Yamashita, K. Theoretical study of the surface complex between TiO2 and TCNQ showing interfacial charge-transfer transitions. J. Phys. Chem. Lett. 2011, 2, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, S.; Yanagisawa, S.; Yamashita, K.; Jono, R.; Segawa, H. Theoretical Verification of Photoelectrochemical Water Oxidation Using Nanocrystalline TiO2 Electrodes. Molecules 2015, 20, 9732–9744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yum, J.-H.; Nazzeeruddin, M.K. Molecular Engineering of Sensitizers for Conversion of Solar Energy into Electricity. In Dye-sensitized Solar Cells; EPFL Press (Distributed by CRC Press): Lausanne, Switzerland, 2010; Chapter 3; pp. 84–116. [Google Scholar]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Persson, P.; Lundqvist, M.J. Calculated structural and electronic interactions of the ruthenium dye N3 with a titanium dioxide nanocrystal. J. Phys. Chem. B 2005, 109, 11918–11924. [Google Scholar] [CrossRef] [PubMed]
- Labat, F.; Ciofini, I.; Hratchian, H.P.; Frisch, M.J.; Raghavachari, K.; Adamo, C. Insights into working principles of ruthenium polypyridyl dye-sensitized solar cells from first principles modeling. J. Phys. Chem. C 2011, 115, 4297–4306. [Google Scholar] [CrossRef]
- Angelis, F.D.; Fantacci, S. Theoretical and Model. System Calculation. In Dye-Sensitized Solar Cells; EPFL Press (Distributed by CRC Press): Lausanne, Switzerland, 2010; Chapter 13; pp. 555–591. [Google Scholar]
- Küpper, F.C.; Feiters, M.C.; Olofsson, B.; Kaiho, T.; Yanagida, S.; Zimmermann, M.B.; Carpenter, L.J.; Luther, G.W., III; Lu, Z.; Jonsson, M.; et al. Commemorating Two Centuries of Iodine Research: An Interdisciplinary Overview of Current Research. Angew. Chem. Int. Ed. 2011, 50, 11598–11620. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.K.; Kar, S.; Leszczynski, J. Insight into the optoelectronic properties of designed solar cells efficient tetrahydroquinoline dye-sensitizers on TiO2 (101) surface: First principles approach. Sci. Rep. 2018, 8, 10997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zych, D.; Slodek, A. Sensitizers for DSSC containing triazole motif with acceptor/donor substituents—Correlation between theoretical and experimental data in prediction of consistent photophysical parameters. J. Mol. Struct. 2020, 1207, 127771. [Google Scholar] [CrossRef]
- Halgren, A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490. [Google Scholar] [CrossRef]
- Burke, K.; Werschnik, J.; Gross, E.K.U. Time-dependent density functional theory: Past, present, and future. J. Chem. Phys. 2005, 123, 062206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreuw, A.; Head-Gordon, M. Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. Rev. 2005, 105, 4009–4037. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]














© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanagisawa, S.; Yanagida, S. Density Functional Theory-Based Molecular Modeling: Verification of Decisive Roles of Van der Waals Aggregation of Triiodide Ions for Effective Electron Transfer in Wet-Type N3-Dye-Sensitized Solar Cells. Energies 2020, 13, 3027. https://doi.org/10.3390/en13113027
Yanagisawa S, Yanagida S. Density Functional Theory-Based Molecular Modeling: Verification of Decisive Roles of Van der Waals Aggregation of Triiodide Ions for Effective Electron Transfer in Wet-Type N3-Dye-Sensitized Solar Cells. Energies. 2020; 13(11):3027. https://doi.org/10.3390/en13113027
Chicago/Turabian StyleYanagisawa, Susumu, and Shozo Yanagida. 2020. "Density Functional Theory-Based Molecular Modeling: Verification of Decisive Roles of Van der Waals Aggregation of Triiodide Ions for Effective Electron Transfer in Wet-Type N3-Dye-Sensitized Solar Cells" Energies 13, no. 11: 3027. https://doi.org/10.3390/en13113027
APA StyleYanagisawa, S., & Yanagida, S. (2020). Density Functional Theory-Based Molecular Modeling: Verification of Decisive Roles of Van der Waals Aggregation of Triiodide Ions for Effective Electron Transfer in Wet-Type N3-Dye-Sensitized Solar Cells. Energies, 13(11), 3027. https://doi.org/10.3390/en13113027