1. Introduction
The issue of air pollution caused by the increasing industrialization of the society still remains an unsolved environmental problem. One of the most harmful compounds are nitrogen oxides (NO
x) [
1,
2,
3,
4], mainly due to their strongly climate-changing character that contributes to acid rain and photochemical smog formation or ozone layer depletion [
5,
6,
7,
8,
9]. Over last decades, the public awareness of the environmental subjects greatly increased, resulting in the implementation of political regulations about the emission limits. In order to meet the restrictions imposed by governments, a number of methods of NO
x abatement were developed [
8,
10,
11]. Currently, the common industrial method of nitrogen oxides emission control is selective catalytic reduction with ammonia (NH
3-SCR) [
12,
13,
14]. Among all of the recognized measures, NH
3-SCR is the most effective and reaches even up to 90% of NO
x conversion [
8,
15,
16,
17,
18]. NH
3-SCR assumes the reaction between NO and NH
3 (the reducing agent) that yields molecular nitrogen and water vapor as the desired products [
12]. The presence of the catalyst provides the surface for the reaction to proceed and lowers the activation energy of the process [
19,
20]. The optimum temperature of NH
3-SCR is in the range of 150–450 °C and the majority of the commercial installations utilize vanadium oxide (V
2O
5) supported on titanium oxide (TiO
2) in a form of anatase, wash coated on a honeycomb monolith or deposited on a plate-like structures [
3,
21,
22,
23]. In order to improve the mechanical stability and chemical resistance, the system is usually promoted with tungsten oxide (WO
3) or molybdenum oxide (MoO
3) [
24,
25,
26].
Although the catalyst is highly active, there are some considerable operating problems that limit its efficient application. One of the most important is the narrow temperature window (300–450 °C). Thus, it is required to place the SCR unit in so-called “high-dust” configuration in order to avoid re-heating of the exhausts and in consequence, cut down the costs of DeNO
x installation [
27,
28,
29]. However, despite the high temperature of the flue gas, the position before the desulphurization installation (FGD) and electrostatic precipitator (ESP) can cause severe poisoning by harmful components of the fumes, such as SO
2 and fly ash, especially when the contaminated fuel is burned [
30,
31,
32,
33]. The fly ash of coal-fired power plants contains significant amounts of alkali metals or alkaline earth metal compounds, including K, Na, Mg or Ca [
3,
34,
35,
36]. Another adverse compounds that can be deposited on the catalyst’s surface are non-metallic compounds, such as arsenic or lead [
37,
38,
39,
40]. Additionally, under the influence of elevated temperature and moisture, with the passing of time the material can undergo thermal or hydrothermal aging [
41,
42,
43]. Under typical conditions of NH
3-SCR, the catalyst can also be affected mechanically by sintering, surface masking, fouling or losing of active components and the specific surface area [
44]. All of these aspects can lead to irreversible deterioration of the catalytic performance. Since the lifetime of the commercial catalysts is a crucial issue in the economics of industrial processes, the assurance of the stability and resistance is indispensable. It undoubtedly helps to avoid downtimes and maintain the continuous and efficient work of the purification installation. Additionally, in many cases it is possible to regenerate the poisoned material and restore the activity to some extent. Therefore, the development of the new recovery methods is highly required.
In the following research, we presented the studies on the most adverse compounds that can affect the catalytic performance of the commercial vanadium-titanium-based NH3-SCR system. We analyzed and described the mechanisms proposed in the literature concerning the typical degradations. In order to provide the most reliable description of each mechanism of deactivation, we focused mostly on the findings published in the last 10 years. Our research offers a clear explanation of the interactions of contaminations with the active sites of commercial type of selective catalytic reduction (SCR) catalyst V2O5-TiO2. Additionally, in each paragraph we presented concise and precise description of the origin of each type of poisoning agent in the flue gas.
3. Effect of Water Vapor
The presence of water vapor in the flue gas under industrial conditions of NH
3-SCR reaction is inevitable. Typically, the exhausts contain 10–30 vol.% of H
2O and even if the process takes place in dry conditions, as the product of the reaction, H
2O molecules can cover the active sites of the catalyst. Physically, water can impair the fine structure or lead to the cracking of the catalyst as a result of vaporization and swelling. Moreover, in the presence of alkali metals it can form soluble salts that poison the acid sites of the material [
45]. In general, the chemical deactivation by H
2O can occur according to two routes. The first and reversible interaction of water with the catalyst surface assumes its adsorption on the active sites that can be inhibited after removal of H
2O from the gas stream [
15,
46]. Zhu et al. [
47] analyzed the effect of the presence of 5 vol.% of water in the exhausts on the catalytic performance of 3 wt.% V
2O
5-MoO
3-WO
3-TiO
2. The authors observed that at 200 °C the conversion of NO declined from 83.4% in dry conditions to 63.9% in wet conditions. However, the hampering effect was recovered after switching off the feed of H
2O. The second and irreversible deactivation occurs when the molecules of H
2O undergo chemisorption on the surface and form hydroxyls of very high decomposition temperature [
48]. The major reason of the decreased activity is competitive adsorption of NH
3 or NO and H
2O on the active sites of the catalyst. Additionally the primary studies in that field confirmed that effect is independent of the vanadium loading [
49,
50].
The moisture influences significantly the form of V
2O
5, due to its reconstruction under the in situ conditions of NH
3-SCR [
50]. Jehng et al. [
51] analyzed the impact of moisture on the molecular structure of vanadia using in situ Raman spectroscopy in the temperature range of 120–450 °C. The behavior of the commercial system: 1, 5 or 7 wt.% V
2O
5-TiO
2 was investigated in the presence of oxygen under dry conditions and with 8 vol.% of H
2O in the gas stream. It was observed that in the absence of moisture, V
2O
5 existed in a form of isolated and polymeric species. When water was introduced into the feed above 230 °C, the surface vanadium moieties formed hydrogen bonds with H
2O. The effect of water adsorption at elevated temperature proves that it can interact competitively with NH
3 during NH
3-SCR by the formation of coordinative bonds with surface vanadia species. Additionally, below 230 °C only the monomeric VO
x species were observed to become extensively solvated by the moisture. As a result, the hydrated surface vanadate structures, such as decavanadates were formed. Additionally, the oxygen-18 isotopic labeling experiments confirmed that terminal V=O, bridging V–O–V and V–O–support bonds form the hydrogen bonds with H
2O. Therefore, consumption of active O from the catalyst surface can considerably decrease NO conversion in SCR process.
Furthermore, water vapor has a substantial impact on the ratio of Brönsted/Lewis acid sites [
38]. Zhu et al. [
47] examined the distribution and reactivity of ammonia species on the acid sites of V
2O
5-WO
3-TiO
2 in the presence of moisture in the feed gas using time-resolved in situ Fourier transform-infrared (FT-IR). The results of the studies suggested that exposition of the catalyst to the flue gas containing water (8 vol.%) increased the amount of surface NH
4+ species and decreased the density of coordinatively bounded ammonia, especially at 250 °C. It is known that both Brönsted and Lewis acid sites participate in the adsorption of ammonia during NH
3-SCR [
52,
53,
54]. However, according to the turnover frequency (TOF) calculations, the specific activity of surface V
5+ sites of Lewis acidity is higher than that of Brönsted acidity, due to their better thermostability [
55]. Therefore, it was suggested that the domination of less active Brönsted acid sites can be an alternative reason of the diminished catalytic activity of NH
3-SCR in the wet conditions.
In summary, due to the unclear assumptions concerning the mechanism of NH
3-SCR, it is rather difficult to draw unambiguous conclusions about the influence of H
2O in the catalytic performance. On the basis of the suggestions of Topsøe et al. [
50], Brönsted acid sites are the only active centers of the vanadium-based catalyst. The participation of V-OH Brönsted acid sites as the main centers of the process was also confirmed by Janssen et al. [
56]. Therefore, hydration of the active centers by moisture should elevate the catalytic activity. Nevertheless, due to the fact that the studies were performed a few years ago, the outcomes of the analysis may not be fully reliable. According to the more recent postulations of Marberger et al. [
57] and Zhu et al. [
55], Lewis acid sites are the most active and significant in NH
3-SCR. Thus, while Brönsted acid sites are produced in the presence of H
2O, the catalytic activity is significantly diminished. Hence, due to the convoluted mechanism of the process, dependent on many external factors, it is rather complicated to determine the role of water in the catalytic system and the issue definitely deserves further attention.
4. Effect of SOx
In the practical applications of NH
3-SCR, the catalyst is under high risk of being deactivated by sulphur compounds (SO
x). Sulphur appears in the combustion zone due to its presence in fuel and the largest amount of SO
2 is generated in the first stage of incineration. The poisoning effect is observed mainly in the low-temperature range of SCR (below 300 °C). Since vanadium catalysts are commonly used for sulphur dioxide oxidation in the technology of sulphuric acid production, the active phase of commercial NH
3-SCR system is capable to oxidize SO
2 to SO
3 [
46,
58]. The main problem of the exposition of the catalyst to SO
x is the formation of ammonium bisulphates (NH
4HSO
4) and ammonium sulphates ((NH
4)
2SO
4) on its surface [
59]. The extent of deactivation with SO
x is determined by the operating conditions of NH
3-SCR. The prime analysis of the presence of SO
2 in the flue gas was performed by Svachula et al. [
60] and Dunn et al. [
61] who analyzed the influence of O
2, H
2O, NO
x and NH
3 concentration on the oxidation of SO
2 to SO
3 over honeycomb V
2O
5-TiO
2. It was found that the oxidation of SO
2 is almost independent of the partial pressure of O
2 in the flue gas if its concentration is approximately 2% v/v (representative operating conditions of SCR). On the contrary, with the increasing concentration of H
2O or NH
3, the tendency of the catalyst to convert SO
2 was significantly diminished, due to the competitive adsorption of H
2O and SO
2 on the acid sites of the material. Furthermore, the presence of NO
x in the flue gas slightly facilitates the conversion of SO
2. However, it is meaningful only in the low-temperature range of SCR, when the concentration of NO
x is high. The results of more recent studies in the topic of SO
2 presence in the exhausts suggest that SO
2 oxidation depends linearly on the catalyst’s wall thickness and increases with the increasing temperature of the reaction [
30,
62]. The produced SO
3 can react with the steam in the rotary air heater and form corrosive sulphuric acid (H
2SO
4) in the temperature range of 204–426 °C [
63].
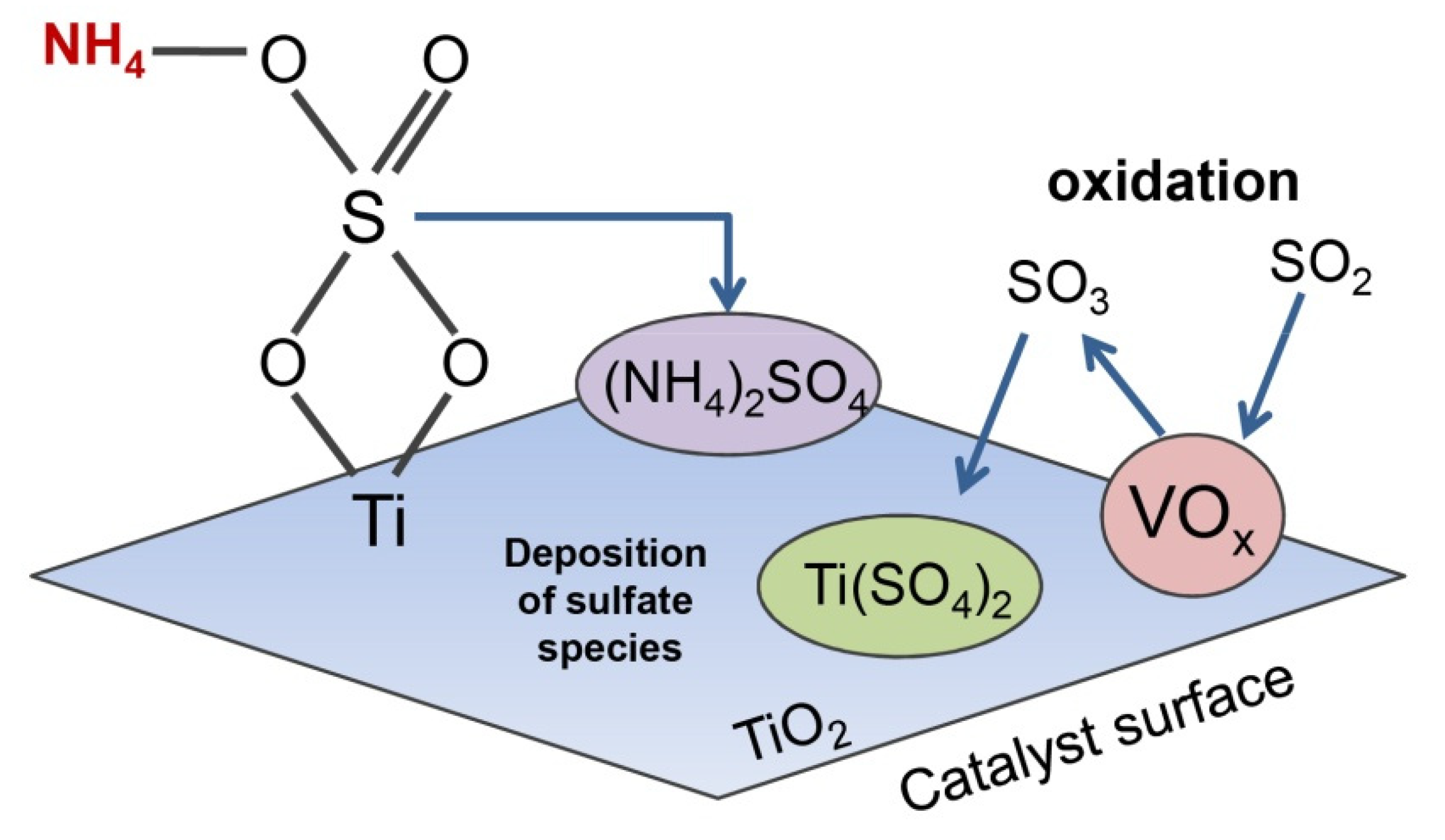
In general, V
2O
5-WO
3-TiO
2 can be deactivated by sulphur compounds according to two routes. The first one, already mentioned, involves the reaction between SO
3 with gaseous NH
3 and H
2O to generate NH
4HSO
4 and (NH
4)
2SO
4. These compounds tend to form deposits in the cold equipment downstream of the SCR reactor and lead to the corrosion of the equipment. Moreover, the accumulation of ammonium sulphates and bisulphates in air-preheater results in the pressure drop and its clogging [
64]. The second route involves the reaction of SO
2 with the active sites of the catalyst and of thermally stable metal sulphites/sulphates that affect redox properties of the material and block the active centers for the adsorption of reactants. The formation of metal sulphites and sulphates can be explained by the difference in the desorption temperature of NH
3 (150–400 °C) and SO
2 (>400 °C). Since the decomposition of (NH
4)
2SO
4 occurs at 150–400 °C, the residual SO
42− species combine easily with the free metal sites left by the desorbed NH
3 and form metal sulphites and sulphates. Due to the fact that the adsorption of SO
2 on TiO
2 is extremely favorable, V
2O
5-TiO
2 can be easily sulphated according to two routes—direct reaction of SO
2 with the anatase surface or its oxidation by VO
x to SO
3 that is subsequently adsorbed on the catalyst’s support [
59,
64]. In summary, the phenomena partly clarify the poisonous influence of SO
2 in the low temperature range of NH
3-SCR [
64]. Furthermore, the inhibited adsorption of NO (according to Langmuir-Hinshelwood mechanism) by the metal sulphites and sulphates causes the suppression of its oxidation to NO
2, lowers NH
3-SCR activity and irreversible deactivation of the catalyst [
32,
65]. The poisoning influence of SO
2 on V
2O
5-TiO
2 is depicted in
Figure 1.
Xu et al. [
62] investigated the effect of
in-situ poisoning with SO
2 and H
2O on V
2O
5-WO
3-TiO
2 by simulating the conditions of flue gas in stationary sources. The reference catalyst reflecting commercial material was prepared by the impregnation method using ammonium vanadate and ammonium tungstate hydrate as the precursors of V and W, respectively. The amounts of the precursors of active phase and the promoter were calculated to obtain 1 wt.% and 5 wt.%, respectively. The catalyst was poisoned for 24 h in a fixed bed quartz reactor, using the inlet gas composed of 500 ppm of NH
3, 500 ppm of SO
2, 5% of H
2O, 5% of O
2 and N
2 as balance. The results of the catalytic tests over the poisoned catalyst indicated that the conversion of NO decreased significantly due to the contact with SO
2, especially below 300 °C. However, the results of low-temperature N
2 sorption measurement indicated only a weak influence of the SO
2 on S
BET of the materials. Therefore, lower catalytic activity of SO
2-poisoned catalysts is not determined by the loss of the specific surface area, which was also confirmed by earlier research in that field [
66]. According to the outcomes of thermogravimetric analysis (TGA), a significant amount of NH
4HSO
4 was formed on the surface of the catalyst, which was suggested to have the major influence on the catalytic performance. Ma et al. [
64] prepared the series of V
2O
5-TiO
2 catalysts with the vanadium content of 1 wt.% and 3 wt.% and doped the materials with W and/or Ce. The authors performed temperature programmed surface reaction (TPSR) and temperature programmed decomposition (TPDC) studies in order to investigate the mechanism of ammonium and metal sulphates formation. On the basis of the obtained results, the highest amount of ammonium sulphates (587.6 μmol·g
cat−1) was generated for the non-promoted V
2O
5-TiO
2, while for the W- and Ce-promoted samples the formation of (NH
4)
2SO
4 was considerably inhibited (to 45.5 μmol·g
cat−1 and 16.7 μmol·g
cat−1, respectively). However, according to the outcomes, the Ce-doped catalyst had high tendency to generate metal sulfates, in contrast to V
2O
5-WO
3-TiO
2. The effect was explained by the high temperature of Ce(SO
3)
2, Ce(S
2O
7)
2, CeOSO
4 and Ce
2(SO
4)
3 decomposition detected by TPDC analysis. In contrast, the addition of WO
x species hindered the formation of Ti(SO
4)
2. The probable reason is the basic nature of ceria and its ability to donate oxygen for SO
2, sulphation of the catalyst’s surface and higher conversion of sulphur dioxide.
Undoubtedly, the formation of ammonium and metal sulphates and sulphites severely affects the catalytic activity of V
2O
5- TiO
2. The early studies on the interaction between SO
x and the catalyst’s surface were carried out by Orsenigo et al. [
67]. The researchers suggested that the sulphation occurs firstly on vanadia sites and later on tungsten and titania sites. On the contrary, Amiridis et al. [
49] and Choo et al. [
68] found that TiO
2 is sulphated first. Nevertheless, the studies were not confirmed by the full surface analysis [
67] or the sulphate species were introduced artificially by impregnation [
49]. Guo et al. [
69] performed the in situ experimental investigation of the interaction between SO
2 and vanadia-titania catalyst and monitored the reaction by
operando FT-IR spectroscopy. The results of the research evidenced that the surface sulphate species were formed rather upon the interaction with titania than with vanadia. Interestingly, the results of NH
3-SCR catalytic tests indicated that the sulphated 1 wt.% V
2O
5-TiO
2 exhibited 200% higher intrinsic rate than non-sulphated sample. It was concluded that the formation of S–OH groups attached to the support introduced new Brönsted acid sites which accelerated NO conversion.
There is a general agreement that the oxidation of SO
2 to SO
3 over V
2O
5-WO
3-TiO
2 is promoted by the increasing loading of V
2O
5 and thus higher aggregation degree of vanadium on the catalyst surface [
24]. When vanadium content on the catalyst is high, the predominant species are polymeric vanadyls (–V(=O)–O–O–V(=O)–) that tend to form aggregates on the catalyst surface. Kamata et al. [
66] investigated the relationship between the amount and structure of vanadium oxide and the catalytic activity in SO
2 oxidation. The outcomes of the studies indicated that the oxidation rate increased from 0.002 μmol·m
−1·s
−1 to 0.008 μmol·m
−1·s
−1 while the loading of V
2O
5 was increased from 1.5 wt.% to 5 wt.%, respectively. The infrared analysis (DRIFT) carried out over the catalysts suggested that both V=O and V–O–V species are involved in the adsorption of SO
2 and desorption of SO
3. On the other hand, on the basis of the reaction turnover frequency (TOF) measurement, Dunn et al. [
61] reported that both the bridging V–O–V and terminal V=O do not play a crucial role in the oxidation of SO
2. The authors assumed that only vanadium species attached to the support (V–O–Ti) are active towards SO
2 oxidation. It is in agreement with the conclusion that sulphur species have significantly higher affinity to the species containing TiO
2. A few years later, the availability of more advanced techniques opened up new possibilities to analyze the mechanism of the SO
2 oxidation over V
2O
5-TiO
2. Du et al. [
59] confirmed that polymeric vanadate species very active in SO
2 oxidation and for that reason, the commercial SCR catalyst should contain small amount of V
2O
5 (below 2.5%). According to the density functional theory (DFT) calculations performed by the authors, the energy barrier of SO
2 adsorption and oxidation to SO
3 is almost equal for both vanadium monomers and dimers. Three possible routes of SO
2 adsorption and oxidation on the SCR catalyst were considered. The first one involves the adsorption on TiO
2 uncovered by the active phase. The results of the calculations based on the projected model catalyst indicated that sulphur dioxide can interact with Ti–O–Ti sites due to the escape of bridge oxygen from the structure and its strong bonding with sulphur atom to form Ti(SO
3)Ti– configurations. However, the high energy barrier of SO
3 desorption needs to be overcome to break the structure of the complex (~100 kcal mol
−1). Thus, the formation of harmful SO
3 and subsequent deactivation can hardly happen due to the low reducibility of Ti
4+ species. The study confirmed the earlier assumptions of Dunn et al. [
61] that the coexistence of Ti–OH and vanadia monomer species facilitate capturing of SO
2. Nonetheless, DFT calculations indicated that in this case, the most favorable is the formation of stable Ti–OSOOH intermediates and the exchange of Ti–OH Brönsted acidic sites with S–OH sites. The second path that was appraised, involved the interaction of SO
2 with a vanadia monomer. In this case, sulphur dioxide can be oxidized by bridge oxygen of V–O–Ti or terminal oxygen of V=O. According to the authors, the direct release of SO
3 from this site is hampered by the high energy needed for desorption and Ti(SO
4)
4 deposits that are formed. In contrast, it was also found that for the terminal oxygen of V=O the oxidation process passes via sulphation of the vanadia site and not by direct oxidation. The phenomenon was explained by the reduction of energy barrier, while SO
2 reacts with active sites of the catalyst surface first. This results in the close interaction of SO
2 with the catalyst-detached oxygen. Herein, tetrahedral –V(SO
4)– species are formed and SO
3 can be simply released. Additionally, the analysis revealed that for the vanadia dimer, the energy barrier for SO
2 oxidation is slightly higher (about 4–5 kcal mol
−1) than for vanadia monomer. The assumptions presented by Du et al. [
59] on the influence of vanadium content on the activity in SO
2 oxidation were in agreement with those postulated by Ma et al. [
64]. The latter authors found that the formation of polymeric vanadium species resulted in higher reducibility of the catalyst and facilitated activity towards SO
2 oxidation. However, the increased loading with vanadium inhibited the formation of (NH
4)
2SO
4 deposits, probably due to the higher catalytic activity and increased consumption of NH
3 provided by the abundance of polyvanadates. Thus, the main role in the mechanism of SO
2 oxidation is played not only by the loading of vanadium on the catalyst surface but also by the nature of oxygen in the vanadium species. Additionally, due to the acidic character of V
2O
5, the SO
2 adsorption capacity is poor and vanadia sites oxidize SO
2 to SO
3 by the sulphation of the catalyst’s surface.
In summary, both previous and more recent studies on the presence of SO
2 in the flue gas and its influence on the catalytic performance of SCR reaction confirm that the oxidation to SO
3 and poisoning by the sulfate and sulfite compounds is influenced by the composition of the flue gas, geometry of the catalyst and temperature of NO reduction process. There is a general agreement that on two routes of deactivation of the catalyst by the sulfur compounds confirmed by primary and most recent studies on that topic. However, the explanation of the mechanism of poisoning evaluated significantly among the last few years. Most of the original studies carried out in 90s of XX century and at the beginning of XXI century confirm that the main role in the sulphation of the catalyst is played by TiO
2. Indeed, more recent studies postulate that the stable Ti-OSOOH intermediates are formed with the participation of Ti-OH Brönsted sites. Nonetheless, in general the energy barrier of SO
3 desorption from this configuration is too high to overcome and instead the presence of both mono- and polyvanadate species is the main reason for SO
2 oxidation and formation of (NH
4)
2SO
4 and NH
4HSO
4 by the reaction with NH
3, which leads to the formation of deposits. The summary of the most important findings about the deactivation of V
2O
5-TiO
2 with sulphur compounds discussed in the section is presented in
Table 1.
Interestingly, according to a number of studies, the exposition of the catalyst to SO
2 results in the formation of additional acid sites provided by the generation of SO
42−. Therefore, the commercial SCR catalyst contains about 0.5–1.0 wt.% of sulphur, mainly in the form of surface sulphate, in order to promote adsorption of NH
3 and NO reduction [
70]. The role of sulphate groups in the catalytic activity in NH
3-SCR was widely discussed by the scientists in recent times [
70,
71,
72,
73]. According to some studies, surface sulphate groups can act as the reservoir for the adsorbed NH
3 [
70]. Nevertheless, the issue of the beneficial effect of sulphation of V
2O
5-TiO
2 is still unclear and remains under intensive investigation.
5. Effect of Alkali Metals
Alkali metals and alkaline-earth metal oxides are one of the strongest poisons of NH
3-SCR catalyst. The large amount of alkaline metals in the fly ash of coal-fired power plants results in their deposition on the catalyst surface, especially while it is placed in the “high dust” configuration. Additionally, the strict legislations regarding air pollution control popularized the renewable energy sources, such as biomass [
74,
75]. In fact, the utilization of biomass as an energy source can reduce the combustion of fossil fuel but biomass contains a large amount of alkali metal compounds and the fly ash produced during its combustion can severely contaminate DeNO
x catalyst [
76,
77].
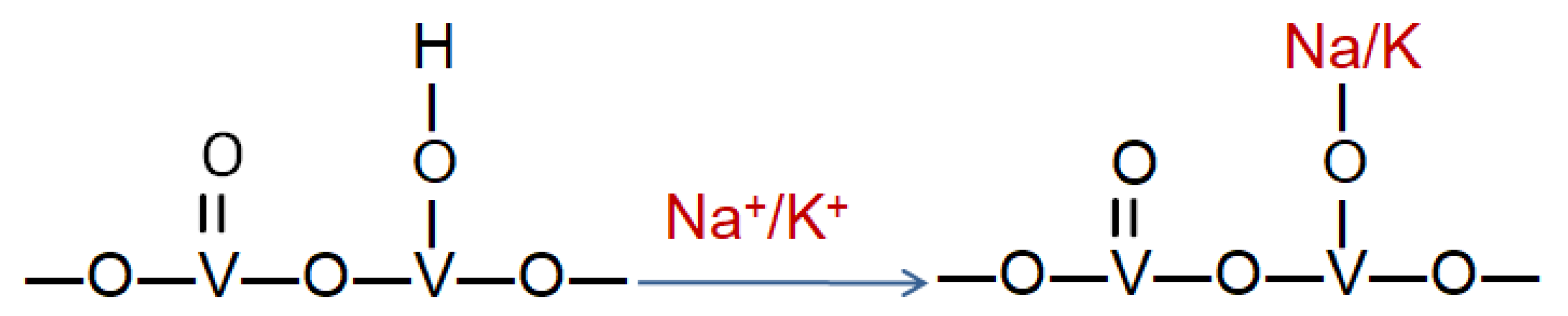
The main reason of the strongly poisoning impact of these compounds on the catalyst is their basic character. Therefore, when adsorbed on the acidic sites of the active phase, they reduce NH
3 adsorption capacity and decrease the catalytic activity. Most of the studies performed so far assumed that the poisoning by the elements of basic character is caused by the formation of alkali—vanadium compounds (such as NaVO
3, KVO
3, RbVO
3) upon acid-base reactions that change the properties of the catalyst’s surface. These formations tend to block the pores of the catalyst and adsorb as deposits causing strong deactivation of the active phase [
31,
78]. The schematic representation of the chemical poisoning of V
2O
5-TiO
2 by alkali metals is presented in
Figure 2.
Evaporation of the alkali metal compounds during combustion and further condensation when the temperature decreases results in the formation of submicron solid particles that are hard to remove from the exhausts [
75]. Most of the studies concluded that the alkalis of IA group (Na and K) are stronger poisons than those belonging to IIA group (Ca and Mg) [
72].
The deactivation of V
2O
5-TiO
2 by alkali metal compounds was extensively investigated both on a pilot-scale and in lab experiments at the beginning of XXI century [
75,
79,
80,
81,
82]. The primary study in that field was carried out in 1990 by Chen et al. [
83]. It was suggested that the strength of the poison follows the order of basicity—Cs
2O > Rb
2O > K
2O > Na
2O > Li
2O. The authors also analyzed the influence of atomic ratios of the alkali metal-vanadium species on the poisoning degree and it was found that one atom of Cs deactivates ca. 14 atoms of V. Furthermore, the poisoning effect of CaO was found to be considerably weaker in comparison to the alkali metal oxides of IA group, which is consistent with the scale of basicity of the metal oxides. The poisoning effect of alkali metals and their compounds on the SCR catalyst was extensively studied in further times. Zheng et al. [
75] suggested that both chemical and physical deactivation of the catalyst is caused by the interaction of alkali metals with active sites but the former is more severe and more difficult to reverse. Moradi et al. [
82] analyzed the behavior of the vanadium catalyst contaminated with various alkali metal-aerosol particles. It was observed that the deactivating effect was accelerated when the temperature of the process was elevated. Generally, according to most of the studies, the poisoning by alkali metals is caused by their interaction with the active phase-V
2O
5 via blocking the Brönsted active sites (V–OH). Besides, the latest investigations confirmed that the decreased catalytic activity may be correlated with the lowered reducibility of vanadium and tungsten species under the influence of alkaline compounds [
84]. Chang et al. [
34] analyzed the influence of different alkali metal cations (Na
+, K
+ and Ca
2+) in the form of bromides on the deactivation of a commercial SCR catalyst. In comparison to the fresh material, the samples treated with alkali metals exhibited lower NO conversion above 350 °C and slightly diminished selectivity to N
2 in the temperature range of 150–450 °C. The most noticeable decrease in catalytic activity (24% of NO conversion at 450 °C) was observed for the material poisoned with potassium. Moreover, the shift of NH
3 desorption temperature to lower value for all of the considered materials indicated that the strength of acidic sites was affected by alkali metals. CO
2-TPD analysis confirmed the formation of new basic sites, especially after addition of potassium. Doping with alkali metals had only negligible effect on the specific surface area. Therefore, it can be concluded that the poisoning effect is correlated only with the changes in the chemical properties of the catalysts.
Most of the studies focused on the influence of alkaline metals on the catalytic performance of V
2O
5-TiO
2 in NH
3-SCR concentrated on the surface acidity of the active material. However, the key step of the reaction is the oxidative dehydrogenation of ammonia (following Eley-Rideal mechanism of SCR) by vanadia species, which was suggested in the most original studies of the mechanism of SCR reaction with ammonia over vanadium-based catalyst [
85,
86] and confirmed by the updated research [
87,
88]. The phenomenon is strongly correlated with the reducibility of the active phase on the anatase support, which can be affected by alkali metals. Tang et al. [
89] prepared 3.87 wt.% V
2O
5-TiO
2 using impregnation method and poisoned the catalyst with Na
+ and Ca
2+ cations. The results of H
2-TPR experiments carried out over the poisoned materials indicated that the presence of sodium or calcium cations shifts the reduction temperature peak from 535 °C to about 560 °C, especially when alkali metal/vanadium ratio is higher than 0.05. In UV-vis DR spectra it was observed that the deposition of sodium caused the decrease in the position of absorption band from 518 nm to 515 and 507 nm, suggesting that Na
+ lowers the polymerization degree of vanadia species which results in lower catalytic activity in NH
3-SCR. On the contrary, no significant changes were observed in the spectra obtained for Ca
2+-poisoned samples, regardless its content. The results of the catalytic tests confirmed that Na
+ species exhibit significantly stronger poisoning effect in comparison to Ca
2+.
Thus the poisoning effect of alkali metals is diversified and depends on many number of factors. Nevertheless, both primary and recent studies over the deactivation by alkali metal-containing deposits are in agreement and confirm that the poisoning influence is strictly correlated with the consumption of acid sites and inhibition of the adsorption of NH3. Nevertheless, it is worth to emphasize that among K, Na and Ca, every particular compound undergoes various interactions with the catalyst surface. Hence, in the next subchapters special attention is paid to the influence of specific alkali metal on the catalytic performance of promoted or non-promoted V2O5-TiO2. In order to present various points of view and evolution of the studies and the understanding of the interactions, chronological review over the poisoning with alkali metals was presented.
5.1. The Effect of Potassium
Potassium, belonging to the IA group, was confirmed to react actively with the Brönsted acid sites of the catalyst and thus inhibit the adsorption of ammonia during NH
3-SCR. The element occurs in the oxide form (K
2O) or inorganic salts (KCl or K
2SO
4), mainly in the exhaust gas produced by the combustion of biomass [
90] and, according to the studies carried out by Zheng, Jensen and Johnsson in 2004, the average amount of potassium in straw oscillates between 0.2 to 1.9 wt.% [
74]. The authors also found that in the presence of potassium, the Brönsted center is affected by K
+ and the amount of adsorbed NH
3 decreases with the increasing content of alkali metal. Moreover, the authors suggested that raising the operation temperature cannot inhibit deactivating effect of potassium. Thus, the most probable consequence of deactivation with potassium is the interruption in the SCR mechanism involving Brönsted acid sites.
Kong et al. [
76] suggested that the vanadium content can play a key role in the level of deactivation by potassium, which is in disagreement with the conclusions drawn on the basis of earlier studies [
74]. The former authors investigated KNO
3-poisoned V
2O
5-WO
3-TiO
2 with various loadings of vanadium and potassium (1, 3, 5 wt.% and 0.8, 0.45 and 2.4 wt.%, respectively). NH
3-SCR catalytic tests over the poisoned samples showed that the material containing 3 wt.% of V
2O
5 exhibited the highest activity and resistance to K-poisoning. When the vanadium loading was increased to 5 wt.%, significant deactivation of the catalyst was observed, especially above 450 °C. The effect was explained by the combined oxidation of NH
3 at elevated temperature and adsorption of K
+ on V–OH polymeric active sites generated due to high content of vanadium. The mechanism of deactivation with potassium and the influence of vanadium content was explained basing on three factors—(1) decreased amount and strength of the acid sites (2) lower reducibility of vanadium species as a result of KVO
3 formation and (3) intensified formation of polymeric forms of V–OH sites with the increasing vanadium content and competitive adsorption of K
+ and NH
3 on the Brönsted centers. On the basis of the obtained results, it can be assumed that an appropriate content of vanadium can reduce harmful influence of potassium on the active sites and thus, result in maintaining, to some extent, satisfactory catalytic activity.
It is known that potassium can be released as a gas phase, aerosols or in the form of condensed compounds [
75]. Additionally, the influence of K was found to depend on the quantity of the poison and its precursor, as well as on the introduction pathway. Due to that, Lei et al. [
84] compared the deactivating effect of KCl introduced onto V
2O
5-TiO
2 by vapor deposition, solid diffusion and wet impregnation, in order to reflect the three major routes of deactivation by potassium in the industrial conditions. The results of the inductively coupled plasma analysis (ICP) over the poisoned samples showed that vapor deposition resulted in the lowest concentration of potassium on the catalysts’ surface, while comparable contents were obtained for the samples treated by solid diffusion and wet impregnation. NH
3-SCR catalytic tests showed that the deactivation followed the order—wet impregnation < solid diffusion ≤ vapor deposition. Basing on the outcomes of the X-ray photoelectron spectroscopy analysis (XPS), the reason for the highest deactivation after poisoning of the catalyst by vapor deposition was concluded to be the formation of eutectic V
2O
5-K
2S
2O
7 that significantly decreased the specific surface area of the catalyst. Additionally, H
2-TPR experiment showed that the temperature of V
5+ reduction was shifted to the higher values for the impregnated materials. The effect was explained by the deeper penetration of the catalyst’s channels with KCl and stronger interaction with vanadium species. For all of the analyzed materials vanadium was present in a form of V
5+, V
4+ and V
3+ species. Thus, all of the procedures of K
+ deposition negatively influenced the redox properties of the catalysts and interrupted the catalytic cycle of SCR. Despite the fact that the K-diffused samples adsorbed more NH
3 than the impregnated one, it exhibited lower catalytic activity. Thus, it was concluded that not NH
3 adsorption capacity but rather the interaction of potassium with vanadium species is the main factor in terms of NO conversion.
The formation of V
2O
5-K
2S
2O
7 eutectic as the major reason of deactivation of the catalyst by potassium was confirmed also by Li et al. [
91]. The authors poisoned V
2O
5-TiO
2 with KCl by impregnation and obtained 0.02, 0.1, 0.3 molar ratios of K/V. In order to reflect the real conditions of NH
3-SCR, the catalytic tests were carried out in the presence of SO
2 with a long running time of 140 h. It was observed that the precursor of potassium determined the level of chemical deactivation. The results of XPS analysis indicated the formation of V
2O
5-K
2S
2O
7 eutectic at K/V ratio of 0.1 and 0.02 and K
2SO
4 for K/V ratio of 0.3. NH
3-TPD and NH
3-TPO experiments confirmed that the presence of V
2O
5-K
2S
2O
7 results in lower catalytic activity due to the decreased Brönsted acidity and oxidation ability.
Kong et al. [
92] analyzed the effect of different potassium species on the deactivation of V
2O
5-WO
3-TiO
2. In order to elucidate the influence of different precursors on the catalytic behavior, a fresh catalyst containing 1 wt.% or 5 wt.% of V
2O
5 was poisoned with the solutions of K
2SO
4, KCl and KNO
3 (as K
2O precursor) by wet impregnation procedure. The results of the studies indicated that the deactivation rate is determined strongly by the precursor of potassium—the introduction of SO
42− anions was beneficial for the adsorption of NH
3 and behaved as a weak Brönsted acid site. In contrast, despite acidic character of Cl
− it was recognized as inactive in NO conversion. Additionally, when the catalyst was poisoned with KCl the vanadium species reached the highest temperature of reduction and the lowest activity in NH
3-SCR. Deposition of K
2O resulted in the substitution of hydrogen from V–OH species for K
+ and blocking the Brönsted active sites.
The most important assumptions regarding the deactivating effect of potassium on the catalytic properties of V
2O
5-TiO
2 in NH
3-SCR are presented in
Table 2.
5.2. The Effect of Sodium
According to most studies, Na is placed in the second position in terms of harmful influence on the catalyst between potassium and calcium [
34,
93]. In coal, sodium occurs in the highest amounts in a form of sodium oxide (Na
2O), sodium hydroxide (NaOH), sodium chloride (NaCl) and sodium sulfate (Na
2SO
4) [
93] and exhibits a tendency to adsorb competitively with NH
3 on the acid centers of the catalyst. Moreover, it influences the reducibility of the vanadium species and hinders surface dehydrogenation of ammonia which is a key step of NH
3-SCR [
89,
94].
Du et al. [
93] investigated the influence of sodium on V
2O
5-WO
3-TiO
2 by its impregnation with the solutions of NaCl, NaOH and Na
2SO
4. It was found that NaOH is the most severe agent, since less than 15% of NO conversion was obtained for sodium hydroxide-poisoned catalyst in the whole temperature range. It was assigned to the high alkalinity of the poison that removed the majority of acidic sites of the catalyst. On the other hand, NaCl caused negligible deactivation, while the catalytic performance of Na
2SO
4-doped material exhibited the highest catalytic activity. Therefore, not only the alkali metal cation but also the coexistent anion determines the level of the catalyst’s deactivation.
Hu et al. [
94] investigated the resistance of V
2O
5-WO
3-TiO
2 to poisoning with Na deposited as NaCl and Na
2O. It was found that the level of deactivation depended on the alkali metal loading. When the ratio of Na/V was below 1, the conversion of NO decreased only slightly, while for Na/V above 1 it was significantly lowered. Additionally, the poisoning effect of NaCl was smaller than Na
2O in the temperature range of 200–500 °C. The main reason was assigned to the formation of strongly basic NaOH on the catalyst’s surface in the presence of water of the flue gas. Additionally, despite adverse influence of Cl
− on the vanadium catalyst, its coexistence with Na
+ can neutralize the basic character of sodium cations. As a consequence, the total amount of acidic sites detected for NaCl-doped samples was higher than that for Na
2O-poisoned ones. The authors suggested two main reasons for the deactivation with sodium. Firstly, in the presence of sodium, the O
α/(O
α + O
β) ratio (where O
α—surface chemisorbed oxygen; O
β— lattice oxygen) significantly decreased, inhibiting the effective oxidation of ammonia in the NH
3-SCR cycle. Secondly, sodium tends to lower the stability and the amount of acidic sites, especially Brönsted centers. It was proposed that the addition of ceria can hinder the negative effect of sodium on V
2O
5-WO
3-TiO
2, due to its capacity to store and release oxygen and form of new Brönsted acid sites. Similar experiments concerning poisoning of the vanadium catalyst with Na
2O were performed by Gao et al. [
45]. According to the authors, sodium changes the environment of vanadium species and blocks V–OH acid sites by the formation of V–ONa deposits. Additionally, the results of XPS measurement of the amount of surface active oxygen species were in agreement with that carried out by Hu et al. [
94]. Interestingly, in comparison to the K
2O-doped sample, the one with Na
2O exhibited significantly worse catalytic performance, which contradicted the generally established regularity of alkali metal poisoning impact [
34,
95]. The summary of the most important assumptions about deactivation of V
2O
5-TiO
2 with sodium are presented in
Table 3.
5.3. The Effect of Calcium
Calcium is one of the alkali metals commonly present in the low-rank fuels, such as lignite or subbituminous coals used for the generation of electricity in power plants [
96,
97]. Some studies on the impact of alkali metals on the catalytic performance of V
2O
5-TiO
2 in NH
3-SCR proved that the poisoning effect of calcium is much lower than that of potassium or sodium [
62]. The primary studies carried out in 1994 on the influence of calcium oxide on the efficiency of the work of commercial SCR catalyst confirmed that CaO narrows the operating temperature window of V
2O
5-TiO
2 and inhibits the effective conversion of nitrogen oxides [
98]. Additionally, the coexistence of Ca and other compounds present in flue gases, such as CO
2, H
2O or SO
2 results in the formation of CaO, CaSO
4 or CaCO
3 that are hard to remove and tend to accumulate on the catalyst’s surface. A few years later, Benson et al. [
99] suggested that the main reason of the deactivation of the catalyst with calcium is the blocking of pores of the catalyst and hindering of the diffusion of NO and NH
3 to the active sites. A number of the most recent studies in that field have confirmed that ammonia can be adsorbed on the surface of CaO and dissociate to the–NH
2 intermediates that react with surface oxygen and produce secondary NO [
96].
Li et al. [
97] investigated the deactivating effect of Ca on the commercial vanadium-based catalyst. The honeycomb V
2O
5-WO
3-TiO
2 was shredded and poisoned with calcium by ultrasonic-assisted equivalent-volume impregnation with Ca(NO
3)
2 to obtain the 10 wt.% of calcium loading. According to the results of NH
3-SCR catalytic tests, the activity of the poisoned material decreased to less than 50% in the whole temperature range. Despite the fact that SEM and EDX analysis confirmed the presence of Ca-containing sediments on the catalyst’s surface, the lowered catalytic activity was not attributed to the structural or textural changes that occurred. NH
3-TPD experiments demonstrated that the major reason of deactivation was the interaction of CaO with weak and strong acid sites and competitive adsorption of calcium oxide and ammonia. Additionally, the lack of the V=O bond on the FT-IR spectrum of the poisoned sample suggested that the presence of Ca caused transformation of these groups into V–OH species and increase of Brönsted active sites. Hence, considering the mechanism of NH
3-SCR, the presence of calcium can cause disruptions in both acid-basic and redox reactions involved in the catalytic cycle of NH
3-SCR [
57,
90,
100].
For the application of DeNO
x installations on an industrial scale, the influence of calcium-containing compounds, such as CaO, CaSO
4 and CaCO
3 must be taken into account, especially in coal-fired power plants that emit large amounts of SO
2 and CO
2. Li et al. [
96] deactivated the V
2O
5-WO
3-TiO
2 with 2 wt.% of calcium oxide, calcium carbonate and calcium sulfate. The results of NH
3-SCR catalytic tests showed that CaCO
3 had the most severe influence on the activity in NO conversion and the declined formation of N
2O. The effect was probably caused by its agglomeration and plugging of the catalyst’s pores and channels. On the other hand, the poisoning effect of CaSO
4 on the catalytic performance was minor, which was explained by the formation of additional Brönsted acid sites in the catalyst’s surface by SO
42−. The outcomes of the structural analysis suggested that for all of the materials the specific surface area decreased after doping with Ca-containing compounds. Moreover, according to XPS and X-ray diffraction (XRD) results, the surface tungsten species of the catalyst react with calcium and form CaWO
4 that leads to poorer dispersion of the promoter and diminishes the activity of the catalyst. Apart from the interaction with the active species of the catalyst, the studies on the surface acidity indicated that the strength and amount of acid sites were the determining factors in the declined catalytic activity. Brönsted as well as Lewis, acid sites were significantly influenced by CaO and CaCO
3. According to in situ DRIFTS experiments, for CaO- and CaCO
3-doped samples only the remaining Lewis acid sites exhibited activity in the adsorption of NH
3, while for CaSO
4-doped sample both the coordinated and protonated ammonia took part in the NH
3-SCR cycle.
The formation of CaWO
4 and bulk tungsten species was acknowledged to be one of the main reasons of V
2O
5-WO
3-TiO
2 deactivation with Ca. Li et al. [
101] poisoned V
2O
5-WO
3-TiO
2 with Ca(OH)
2 in order to obtain 4 wt.% of CaO and obtained the maximum conversion of NO below 25% at 450 °C. XRD and Raman spectroscopy analysis of the poisoned material showed that a significant amount of CaWO
4 and aggregated CaO species were formed on the catalyst’s surface. On the basis of H
2-TPR studies, it was concluded that these deposits were the main reason of the increased temperature of reduction of V
5+ to V
4+ and W
6+ to W
4+. Due to that, the completion of catalytic cycle of SCR was suppressed. Additionally, it was suggested that the addition of CaO leads to the irreversible changes in the interaction between vanadium and tungsten and in the ratio of W=O/V=O. As the latter one is crucial for the effective adsorption and activation of NH
3 in the initial step of NH
3-SCR, the changes lead to disruption of the catalytic cycle.
More detailed understanding of the deactivating effect of calcium-containing deposits on V
2O
5-TiO
2 can be provided by the analysis of the interaction between CaO with ammonia and nitrogen oxide. As it was already emphasized, one of the key steps of NH
3-SCR cycle is the abstraction of hydrogen from NH
4+ ions or coordinated NH
3 molecules attached to the acidic sites, so called “activation of ammonia.” Yang et al. [
102] found that calcium oxide activates ammonia to the–NH surface species, while calcium sulfate promotes the formation of–NH
2 form. Additionally, the presence of SO
42− was confirmed to increase the amount of surface chemisorbed oxygen, resulting in the formation of NO and N
2O due to the oxidation of ammonia. Correlating the findings with the mechanism of NH
3-SCR, it can be assumed that even though SO
42− supplies the catalyst with the additional Brönsted sites, its presence can lead to undesired reactions, formation of side-products and consumption of the reducing agent for NO abatement. The essential findings on the interaction of calcium with the surface of V
2O
5-TiO
2 during NH
3-SCR reaction are summarized in
Table 4.









{kind=link}
{kind=link}
{kind=link}
{kind=link}