Platinum and Platinum Group Metal-Free Catalysts for Anion Exchange Membrane Fuel Cells

 ,
,  ,
,  , and
, and
Abstract
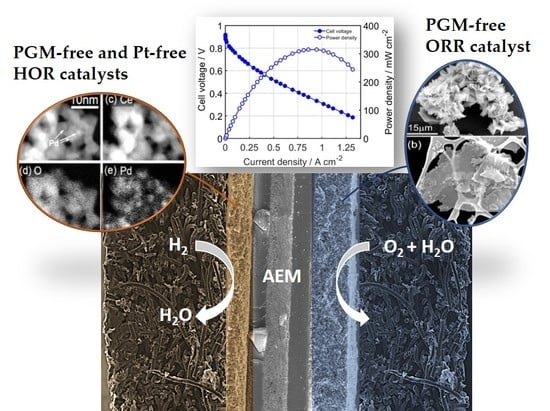
:
1. Introduction
2. Results and Discussion
2.1. Physical Characterization
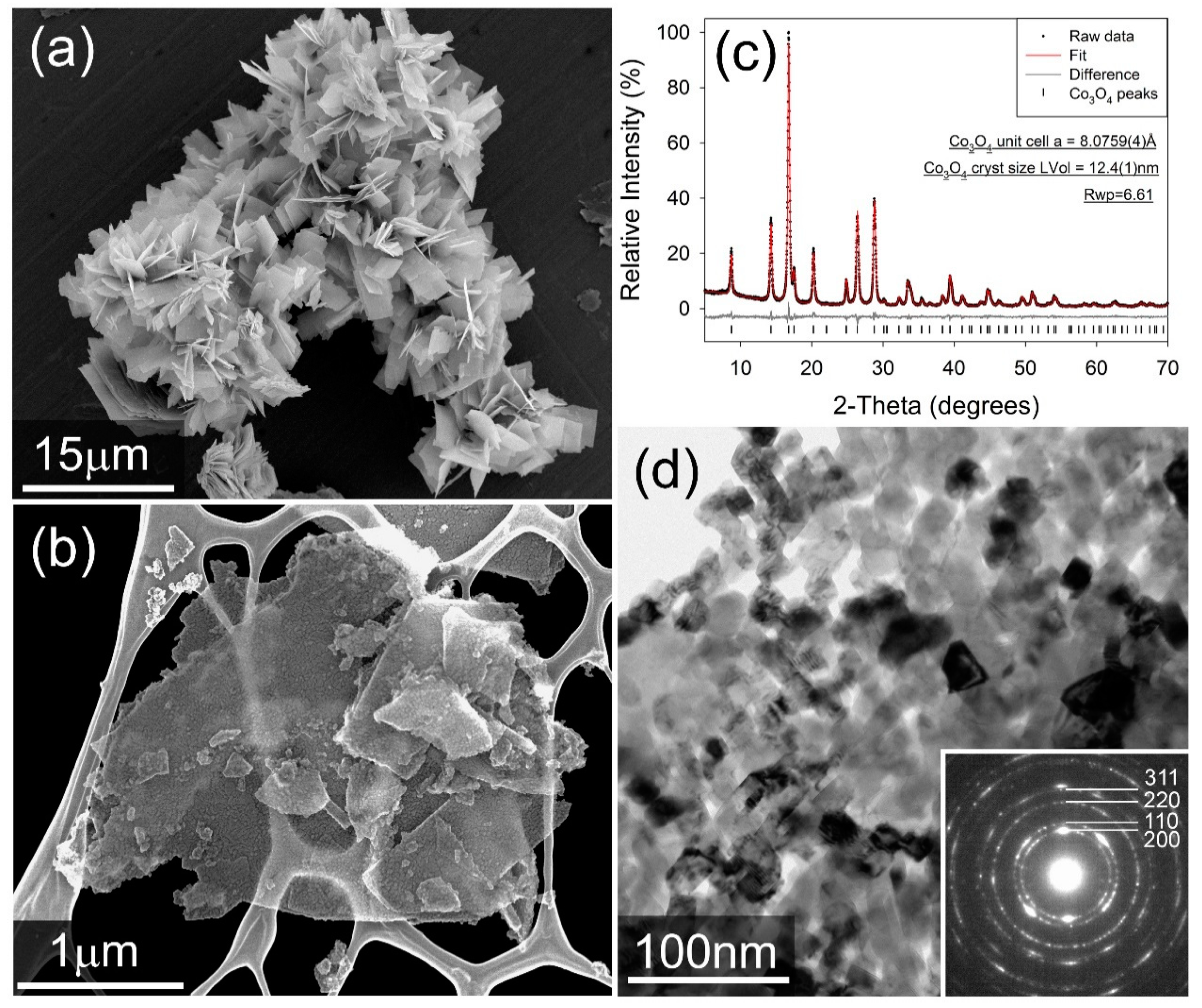
2.1.1. Physical Characterization of the Co3O4 ORR Catalyst
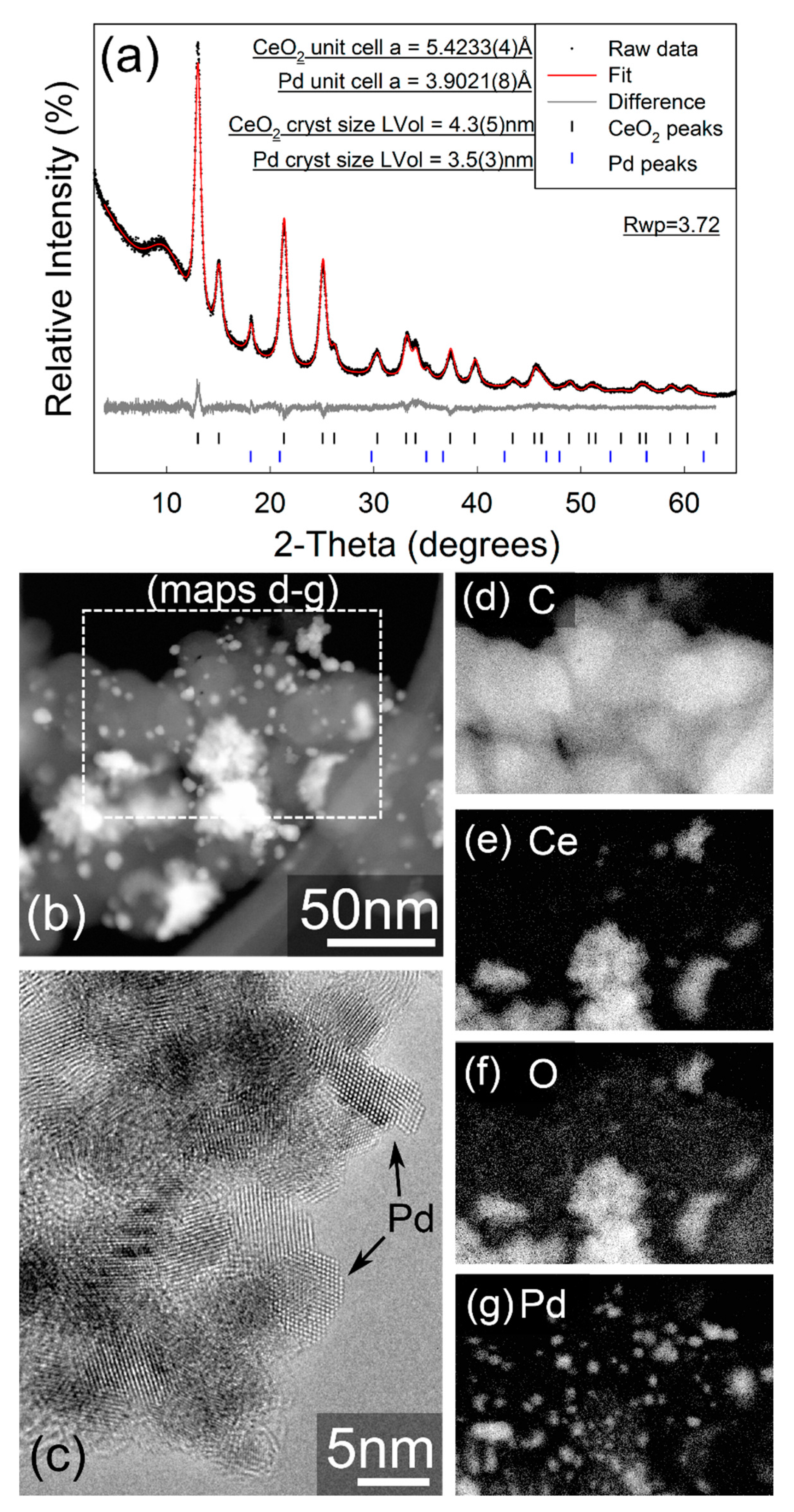
2.1.2. Physical Characterization of the HOR Catalysts
2.2. Electrochemical Characterization
2.2.1. RRDE Characterization of Co3O4
2.2.2. RDE Characterization of HOR Catalysts
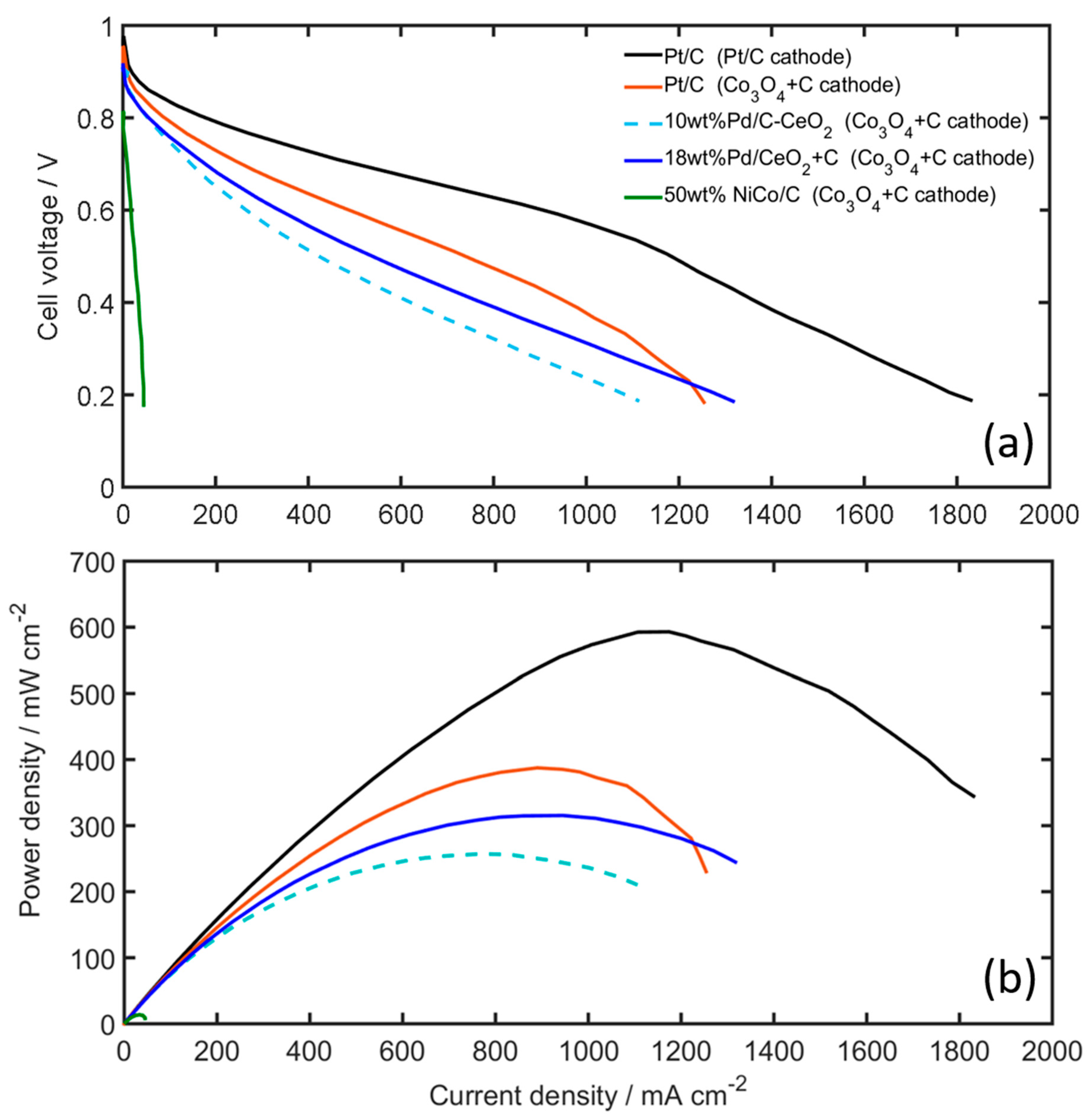
2.3. AEMFC Results
3. Experimental
3.1. Catalyst Synthesis
3.1.1. Synthesis of Co3O4
3.1.2. Synthesis of 10 wt% Pd/C-CeO2 by Wet Chemistry
3.1.3. Synthesis of FSP Pd/CeO2 + C by Flame Spray Pyrolysis (FSP)
3.1.4. Synthesis of NiCo/C
3.2. Physical Characterization
3.2.1. XRD
3.2.2. TEM
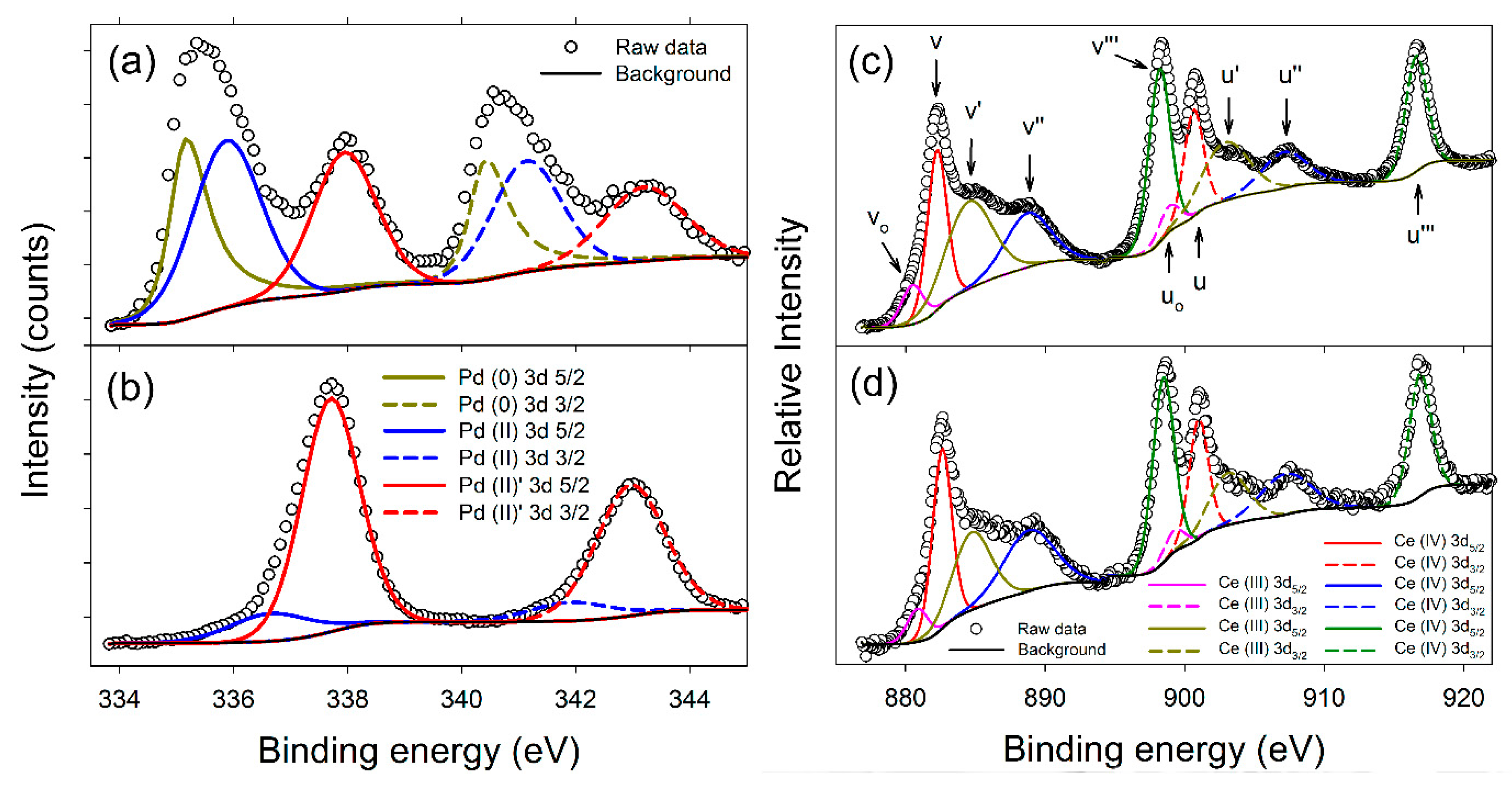
3.2.3. XPS
3.3. Rotating Disk Electrode (RDE) Characterization
3.4. Membrane Electrode Assembly (MEA) Preparation and Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Varcoe, J.R.; Atanassov, P.; Dekel, D.R.; Herring, A.M.; Hickner, M.A.; Kohl, P.A.; Kucernak, A.R.; Mustain, W.E.; Nijmeijer, K.; Scott, K.; et al. Anion-exchange membranes in electrochemical energy systems. Energy Environ. Sci. 2014, 7, 3135–3191. [Google Scholar] [CrossRef] [Green Version]
- Gottesfeld, S.; Dekel, D.R.; Page, M.; Bae, C.; Yan, Y.; Zelenay, P.; Kim, Y.S. Anion exchange membrane fuel cells: Current status and remaining challenges. J. Power Sources 2018, 375, 170–184. [Google Scholar] [CrossRef]
- Burchardt, T.; Gouérec, P.; Sanchez-Cortezon, E.; Karichev, Z.; Miners, J.H. Alkaline fuel cells: Contemporary advancement and limitations. Fuel 2002, 81, 2151–2155. [Google Scholar] [CrossRef]
- Kostowskyj, M.A.; Gilliam, R.J.; Kirk, D.W.; Thorpe, S.J. Silver nanowire catalysts for alkaline fuel cells. Int. J. Hydrogen Energy 2008, 33, 5773–5778. [Google Scholar] [CrossRef]
- Osgood, H.; Devaguptapu, S.V.; Xu, H.; Cho, J.; Wu, G. Transition metal (Fe, Co, Ni, and Mn) oxides for oxygen reduction and evolution bifunctional catalysts in alkaline media. Nano Today 2016, 11, 601–625. [Google Scholar] [CrossRef]
- Xin, L.; Wang, Z.; Qi, J.; Li, W. Carbon supported Ag nanoparticles as high performance cathode catalyst for H2/O2 anion exchange membrane fuel cell. Front. Chem. 2013, 16, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekel, D.R. Alkaline Membrane Fuel Cell (AMFC) Materials and System Improvement—State-of-the-Art. ECS Trans. 2013, 50, 2051–2052. [Google Scholar] [CrossRef]
- Erikson, H.; Sarapuu, A.; Tammeveski, K. Oxygen Reduction Reaction on Silver Catalysts in Alkaline Media: A Minireview. ChemElectroChem 2019, 6, 73–86. [Google Scholar] [CrossRef]
- Zhao, S.; Yan, L.; Luo, H.; Mustain, W.; Xu, H. Recent progress and perspectives of bifunctional oxygen reduction/evolution catatalyst development for regenerative anion exchange membrane fuel cells. Nano Energy 2018, 47, 172–198. [Google Scholar] [CrossRef]
- Durst, J.; Siebel, A.; Simon, C.; Hasche, F.; Herranz, J.; Gasteiger, H. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 2014, 7, 2255–2260. [Google Scholar] [CrossRef] [Green Version]
- Dekel, D.R. Unraveling mysteries of hydrogen electrooxidation in anion exchange membrane fuel cells. Curr. Opin. Electrochem. 2018, 12, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Gao, P.; Zhao, T. Non-precious Co3O4 nano-rod electrocatalyst for oxygen reduction reaction in anion-exchange membrane fuel cells. Energy Environ. Sci. 2012, 5, 5333–5339. [Google Scholar] [CrossRef]
- Wang, D.-C.; Huang, N.-B.; Sun, Y.; Zhan, S.; Zhang, J.-J. GO clad Co3O4 (Co3O4@GO) as ORR catalyst of anion exchange membrane fuel cell. Int. J. Hydrogen Energy 2017, 42, 20216–20223. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.; Wang, H.; Zhou, J.; Wang, J.; Regier, T.; Dai, H. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 2011, 10, 780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamlouk, M.; Kumar, S.S.; Gouerec, P.; Scott, K. Electrochemical and fuel cell evaluation of Co based catalyst for oxygen reduction in anion exchange polymer membrane fuel cells. J. Power Sources 2011, 196, 7594–7600. [Google Scholar] [CrossRef]
- Truong, V.M.; Wang, C.-L.; Yang, M.; Yang, H. Effect of tunable hydrophobic level in the gas diffusion substrate and microporous layer on anion exchange membrane fuel cells. J. Power Sources 2018, 402, 301–310. [Google Scholar] [CrossRef]
- Davydova, E.S.; Mukerjee, S.; Jaouen, F.; Dekel, D.R. Electrocatalysts for hydrogen oxidation reaction in alkaline electrolytes. ACS Catal. 2018, 8, 6665–6690. [Google Scholar] [CrossRef]
- Miller, H.A.; Lavacchi, A.; Vizza, F.; Marelli, M.; Di Benedetto, F.; D’Acapito, F.; Paska, Y.; Page, M.; Dekel, D.R. A Pd/C-CeO2 Anode Catalyst for High-Performance Platinum-Free Anion Exchange Membrane Fuel Cells. Angew. Chem. Int. Ed. 2016, 55, 6004–6007. [Google Scholar] [CrossRef]
- Alesker, M.; Page, M.; Shviro, M.; Paska, Y.; Gershinsky, G.; Dekel, D.R.; Zitoun, D. Palladium/nickel bifunctional electrocatalyst for hydrogen oxidation reaction in alkaline membrane fuel cell. J. Power Sources 2016, 304, 332–339. [Google Scholar] [CrossRef]
- Dekel, D.R. Review of cell performance in anion exchange membrane fuel cells. J. Power Sources 2018, 375, 158–169. [Google Scholar] [CrossRef]
- Omasta, T.J.; Peng, X.; Miller, H.A.; Vizza, F.; Wang, L.; Varcoe, J.R.; Dekel, D.R.; Mustain, W.E. Beyond 1.0 W cm−2 performance without platinum: The beginning of a new era in anion exchange membrane fuel cells. J. Electrochem. Soc. 2018, 165, J3039–J3044. [Google Scholar] [CrossRef]
- Davydova, E.; Zaffran, J.; Dhaka, K.; Toroker, M.; Dekel, D. Hydrogen Oxidation on Ni-Based Electrocatalysts: The Effect of Metal Doping. Catalysts 2018, 8, 454. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Pan, J.; Huang, A.; Zhuang, L.; Lu, J. Alkaline polymer electrolyte fuel cells completely free from noble metal catalysts. Proc. Natl. Acad. Sci. USA 2008, 105, 20611–20614. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Li, G.; Pan, J.; Tan, L.; Lu, J.; Zhuang, L. Alkaline polymer electrolyte fuel cell with Ni-based anode and Co-based cathode. Int. J. Hydrogen Energy 2013, 38, 16264–16268. [Google Scholar] [CrossRef]
- Kabir, S.; Lemire, K.; Artyushkova, K.; Roy, A.; Odgaard, M.; Schlueter, D.; Oshchepkov, A.; Bonnefont, A.; Savinova, E.; Sabarirajan, D.C. Platinum group metal-free NiMo hydrogen oxidation catalysts: High performance and durability in alkaline exchange membrane fuel cells. J. Mater. Chem. A 2017, 5, 24433–24443. [Google Scholar] [CrossRef]
- Roy, A.; Talarposhti, M.R.; Normile, S.J.; Zenyuk, I.V.; De Andrade, V.; Artyushkova, K.; Serov, A.; Atanassov, P. Nickel–copper supported on a carbon black hydrogen oxidation catalyst integrated into an anion-exchange membrane fuel cell. Sustain. Energy Fuels 2018, 2, 2268–2275. [Google Scholar] [CrossRef]
- Knop, O.; Reid, K.; Sutarno; Nakagawa, Y. Chalkogenides of the transition elements. VI. X-Ray, neutron, and magnetic investigation of the spinels Co3O4, NiCo2O4, Co3S4, and NiCo2S4. Can. J. Chem. 1968, 46, 3463–3476. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, V.K.; Shcherbakov, A.B.; Usatenko, A. Structure-sensitive properties and biomedical applications of nanodispersed cerium dioxide. Russ. Chem. Rev. 2009, 78, 855. [Google Scholar] [CrossRef]
- Paparazzo, E. On the curve-fitting of XPS Ce(3d) spectra of cerium oxides. Mater. Res. Bull. 2011, 46, 323–326. [Google Scholar] [CrossRef]
- Burroughs, P.; Hamnett, A.; Orchard, A.F.; Thornton, G. Satellite structure in the X-ray photoelectron spectra of some binary and mixed oxides of lanthanum and cerium. J. Chem. Soc. Dalton Trans. 1976, 17, 1686–1698. [Google Scholar] [CrossRef]
- Sharma, S.; Mukri, B.D.; Hegde, M. Direct evidence of redox interaction between metal ion and support oxide in Ce0.98Pd0.02O2−δ by a combined electrochemical and XPS study. Dalton Trans. 2011, 40, 11480–11489. [Google Scholar] [CrossRef] [PubMed]
- Brun, M.; Berthet, A.; Bertolini, J. XPS, AES and Auger parameter of Pd and PdO. J. Electron Spectrosc. Relat. Phenom. 1999, 104, 55–60. [Google Scholar] [CrossRef]
- Shafeev, G.; Themlin, J.M.; Bellard, L.; Marine, W.; Cros, A. Enhanced adherence of area-selective electroless metal plating on insulators. J. Vac. Sci. Technol. A Vac. Surf. Film. 1996, 14, 319–326. [Google Scholar] [CrossRef]
- Kim, K.S.; Gossmann, A.; Winograd, N. X-ray photoelectron spectroscopic studies of palladium oxides and the palladium-oxygen electrode. Anal. Chem. 1974, 46, 197–200. [Google Scholar] [CrossRef]
- Li, L.; Zhang, N.; He, H.; Zhang, G.; Song, L.; Qiu, W. Shape-controlled synthesis of Pd nanocrystals with exposed {110} facets and their catalytic applications. Catal. Today 2019, 327, 28–36. [Google Scholar] [CrossRef]
- Priolkar, K.; Bera, P.; Sarode, P.; Hegde, M.; Emura, S.; Kumashiro, R.; Lalla, N. Formation of Ce1−xPdxO2−δ Solid Solution in Combustion-Synthesized Pd/CeO2 Catalyst: XRD, XPS, and EXAFS Investigation. Chem. Mater. 2002, 14, 2120–2128. [Google Scholar] [CrossRef]
- Mason, M.; Gerenser, L.; Lee, S.-T. Electronic structure of catalytic metal clusters studied by X-ray photoemission spectroscopy. Phys. Rev. Lett. 1977, 39, 288. [Google Scholar] [CrossRef]
- Zhou, W.P.; Lewera, A.; Larsen, R.; Masel, R.I.; Bagus, P.S.; Wieckowski, A. Size effects in electronic and catalytic properties of unsupported palladium nanoparticles in electrooxidation of formic acid. J. Phys. Chem. B 2006, 110, 13393–13398. [Google Scholar] [CrossRef]
- Nishizawa, T.; Ishida, K. The Co−Ni (Cobalt-Nickel) system. Bull. Alloy Phase Diagr. 1983, 4, 390–395. [Google Scholar] [CrossRef]
- Houska, C.R.; Averbach, B.L.; Cohen, M. The cobalt transformation. Acta Metall. 1960, 8, 81–87. [Google Scholar] [CrossRef]
- Kitakami, O.; Sato, H.; Shimada, Y.; Sato, F.; Tanaka, M. Size Effect on the Crystal Phase of Cobalt Fine. Phys. Rev. B 1997, 56, 13849. [Google Scholar] [CrossRef]
- Moreau, L.M.; Ha, D.-H.; Bealing, C.R.; Zhang, H.; Hennig, R.G.; Robinson, R.D. Unintended phosphorus doping of nickel nanoparticles during synthesis with TOP: A discovery through structural analysis. Nano Lett. 2012, 12, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Grosvenor, A.P.; Biesinger, M.C.; Smart, R.S.C.; McIntyre, N.S. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 2006, 600, 1771–1779. [Google Scholar] [CrossRef]
- Roberts, M.W.; Smart, R.S.C. The defect structure of nickel oxide surfaces as revealed by photoelectron spectroscopy. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1984, 80, 2957–2968. [Google Scholar] [CrossRef]
- Carley, A.; Jackson, S.; O’shea, J.; Roberts, M. The formation and characterisation of Ni3+—an X-ray photoelectron spectroscopic investigation of potassium-doped Ni (110)–O. Surf. Sci. 1999, 440, L868–L874. [Google Scholar] [CrossRef]
- Li-Shing, H.; Williams, R.S. Electronic-structure study of the NiGa and the NiIn intermetallic compounds using X-ray photoemission spectroscopy. J. Phys. Chem. Solids 1994, 55, 305–312. [Google Scholar] [CrossRef]
- McIntyre, N.; Cook, M. X-ray photoelectron studies on some oxides and hydroxides of cobalt, nickel, and copper. Anal. Chem. 1975, 47, 2208–2213. [Google Scholar] [CrossRef]
- Schenck, C.; Dillard, J.; Murray, J. Surface analysis and the adsorption of Co (II) on goethite. J. Colloid Interface Sci. 1983, 95, 398–409. [Google Scholar] [CrossRef]
- Chuang, T.; Brundle, C.; Rice, D. Interpretation of the x-ray photoemission spectra of cobalt oxides and cobalt oxide surfaces. Surf. Sci. 1976, 59, 413–429. [Google Scholar] [CrossRef]
- Chen, R.; Yang, C.; Cai, W.; Wang, H.-Y.; Miao, J.; Zhang, L.; Chen, S.; Liu, B. Use of Platinum as the Counter Electrode to Study the Activity of Nonprecious Metal Catalysts for the Hydrogen Evolution Reaction. ACS Energy Lett. 2017, 2, 1070–1075. [Google Scholar] [CrossRef]
- Chen, J.G.; Jones, C.W.; Linic, S.; Stamenkovic, V.R. Best Practices in Pursuit of Topics in Heterogeneous Electrocatalysis. ACS Catal. 2017, 7, 6392–6393. [Google Scholar] [CrossRef] [Green Version]
- Campos-Roldán, C.; González-Huerta, R.; Alonso-Vante, N. Experimental protocol for HOR and ORR in alkaline electrochemical measurements. J. Electrochem. Soc. 2018, 165, J3001–J3007. [Google Scholar] [CrossRef]
- Davydova, E.S.; Speck, F.D.; Paul, M.T.Y.; Dekel, D.R.; Cherevko, S. Stability Limits of Ni-Based Hydrogen Oxidation Electrocatalysts for Anion Exchange Membrane Fuel Cells. ACS Catal. 2019, 9, 6837–6845. [Google Scholar] [CrossRef]
- Alsabet, M.; Grden, M.; Jerkiewicz, G. Electrochemical growth of surface oxides on nickel. Part 1: Formation of α-Ni(OH)2 in relation to the polarization potential, polarization time, and temperature. Electrocatalysis 2011, 2, 317–330. [Google Scholar] [CrossRef]
- Alsabet, M.; Grdeń, M.; Jerkiewicz, G. Electrochemical growth of surface oxides on nickel. Part 3: Formation of β-NiOOH in relation to the polarization potential, polarization time, and temperature. Electrocatalysis 2015, 6, 60–71. [Google Scholar] [CrossRef]
- Darab, M.; Barnett, A.O.; Lindbergh, G.; Thomassen, M.S.; Sunde, S. The influence of catalyst layer thickness on the performance and degradation of PEM fuel cell cathodes with constant catalyst loading. Electrochim. Acta 2017, 232, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-Q.; Xu, Q.-Z.; Wang, J.-Y.; Li, N.; Guo, S.-H.; Su, Y.-Z.; Wang, H.-J.; Zhang, J.-H.; Chen, S. Facile hydrothermal synthesis of urchin-like NiCo2O4 spheres as efficient electrocatalysts for oxygen reduction reaction. Int. J. Hydrogen Energy 2013, 38, 6657–6662. [Google Scholar] [CrossRef]
- Carenco, S.; Boissiere, C.; Nicole, L.; Sanchez, C.; Le Floch, P.; Mézailles, N. Controlled design of size-tunable monodisperse nickel nanoparticles. Chem. Mater. 2010, 22, 1340–1349. [Google Scholar] [CrossRef]
- Pan, J.; Chen, C.; Li, Y.; Wang, L.; Tan, L.; Li, G.; Tang, X.; Xiao, L.; Lu, J.; Zhuang, L. Constructing ionic highway in alkaline polymer electrolytes. Energy Environ. Sci. 2014, 7, 354–360. [Google Scholar] [CrossRef]
- Lindström, R.W.; Kortsdottir, K.; Wesselmark, M.; Oyarce, A.; Lagergren, C.; Lindbergh, G. Active Area Determination of Porous Pt Electrodes Used in Polymer Electrolyte Fuel Cells: Temperature and Humidity Effects. J. Electrochem. Soc. 2010, 157, B1795–B1801. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pd | 10 wt% Pd/C-CeO2 | FSP 18%Pd/CeO2 | Ref [31,32] |
|---|---|---|---|
| Pd(0) | 335.2 | N/A | 335.1–335.4 |
| Pd(II) | 335.9 | 336.7 | 336.7–336.8 |
| Pd(II) | 337.9 | 337.7 | see main text |
| Thickness (mm) | PTFE (wt%) | Air Permeability (sec) | Through Plane Resistance (mΩ cm2) | Mean Pore Diameter (µm) | Porosity (%) | Contact Angle (°) | ||
|---|---|---|---|---|---|---|---|---|
| MPL | Back | MPL | Back | |||||
| 0.31 | 30 | 30 | 99.5 | 11.9 | 36.69 | 64.06 | 146.2 | 147.5 |
| Tittle | Anodes | Cathodes | ||||||
|---|---|---|---|---|---|---|---|---|
| Cell | Catalyst | Pt, Pd or NiCo Loading (mg cm−2) | Carbon/Ceria Loading (mg cm−2) | Ionomer Loading (mg cm−2) | Catalyst | Pt or Co3O4 Loading (mg cm−2) | Carbon Loading (mg cm−2) | Ionomer Loading (mg cm−2) |
| 1 | 40% Pt/C | 0.5 | 0.75 | 0.25 | 40% Pt/C | 0.5 | 0.75 | 0.25 |
| 2 | 40% Pt/C | 0.5 | 0.75 | 0.25 | Co3O4 + C | 3 | 3 | 1.2 |
| 3 | 10% Pd/C CeO2 | 0.5 | 2.25/2.25 | 1 | Co3O4 + C | 3 | 3 | 1.2 |
| 4 | 18% Pd/CeO2 + C | 0.5 | 2.27/2.27 | 1 | Co3O4 + C | 3 | 3 | 1.2 |
| 5 | 50% NiCo/C | 5.0 | 5 | 5 | Co3O4 + C | 3 | 3 | 1.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Men Truong, V.; Richard Tolchard, J.; Svendby, J.; Manikandan, M.; A. Miller, H.; Sunde, S.; Yang, H.; R. Dekel, D.; Oyarce Barnett, A. Platinum and Platinum Group Metal-Free Catalysts for Anion Exchange Membrane Fuel Cells. Energies 2020, 13, 582. https://doi.org/10.3390/en13030582
Men Truong V, Richard Tolchard J, Svendby J, Manikandan M, A. Miller H, Sunde S, Yang H, R. Dekel D, Oyarce Barnett A. Platinum and Platinum Group Metal-Free Catalysts for Anion Exchange Membrane Fuel Cells. Energies. 2020; 13(3):582. https://doi.org/10.3390/en13030582
Chicago/Turabian StyleMen Truong, Van, Julian Richard Tolchard, Jørgen Svendby, Maidhily Manikandan, Hamish A. Miller, Svein Sunde, Hsiharng Yang, Dario R. Dekel, and Alejandro Oyarce Barnett. 2020. "Platinum and Platinum Group Metal-Free Catalysts for Anion Exchange Membrane Fuel Cells" Energies 13, no. 3: 582. https://doi.org/10.3390/en13030582
APA StyleMen Truong, V., Richard Tolchard, J., Svendby, J., Manikandan, M., A. Miller, H., Sunde, S., Yang, H., R. Dekel, D., & Oyarce Barnett, A. (2020). Platinum and Platinum Group Metal-Free Catalysts for Anion Exchange Membrane Fuel Cells. Energies, 13(3), 582. https://doi.org/10.3390/en13030582