Prediction of Performance Variation Caused by Manufacturing Tolerances and Defects in Gas Diffusion Electrodes of Phosphoric Acid (PA)–Doped Polybenzimidazole (PBI)-Based High-Temperature Proton Exchange Membrane Fuel Cells



Abstract
:
1. Introduction
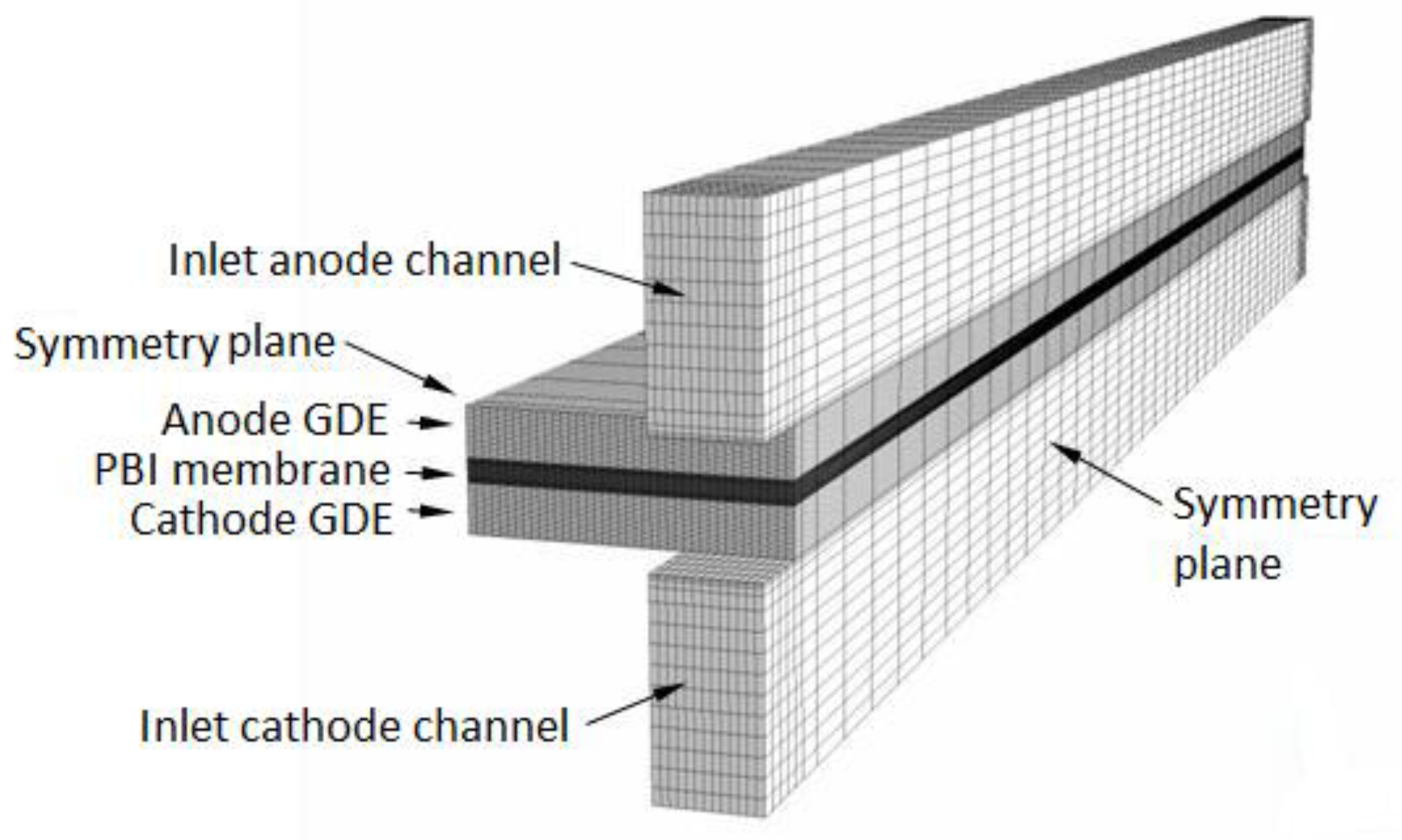
2. Mathematical Model
3. Results and Discussion
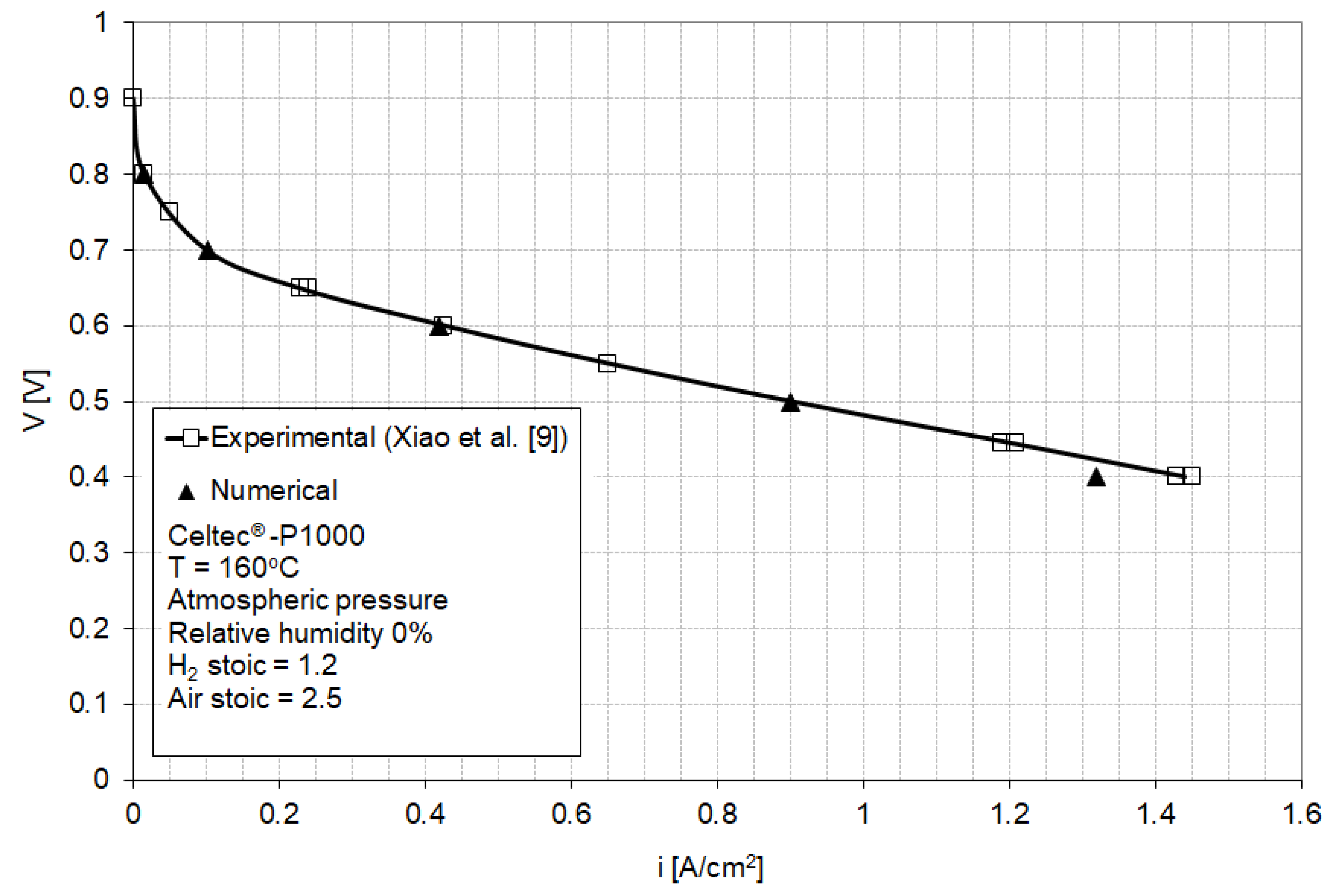
3.1. Baseline Performance of MEAs
3.2. Study of Agglomerate Distribution or Uncoated Regions in the Cathode Catalyst Layer That Would Cause a 10% Drop in Performance
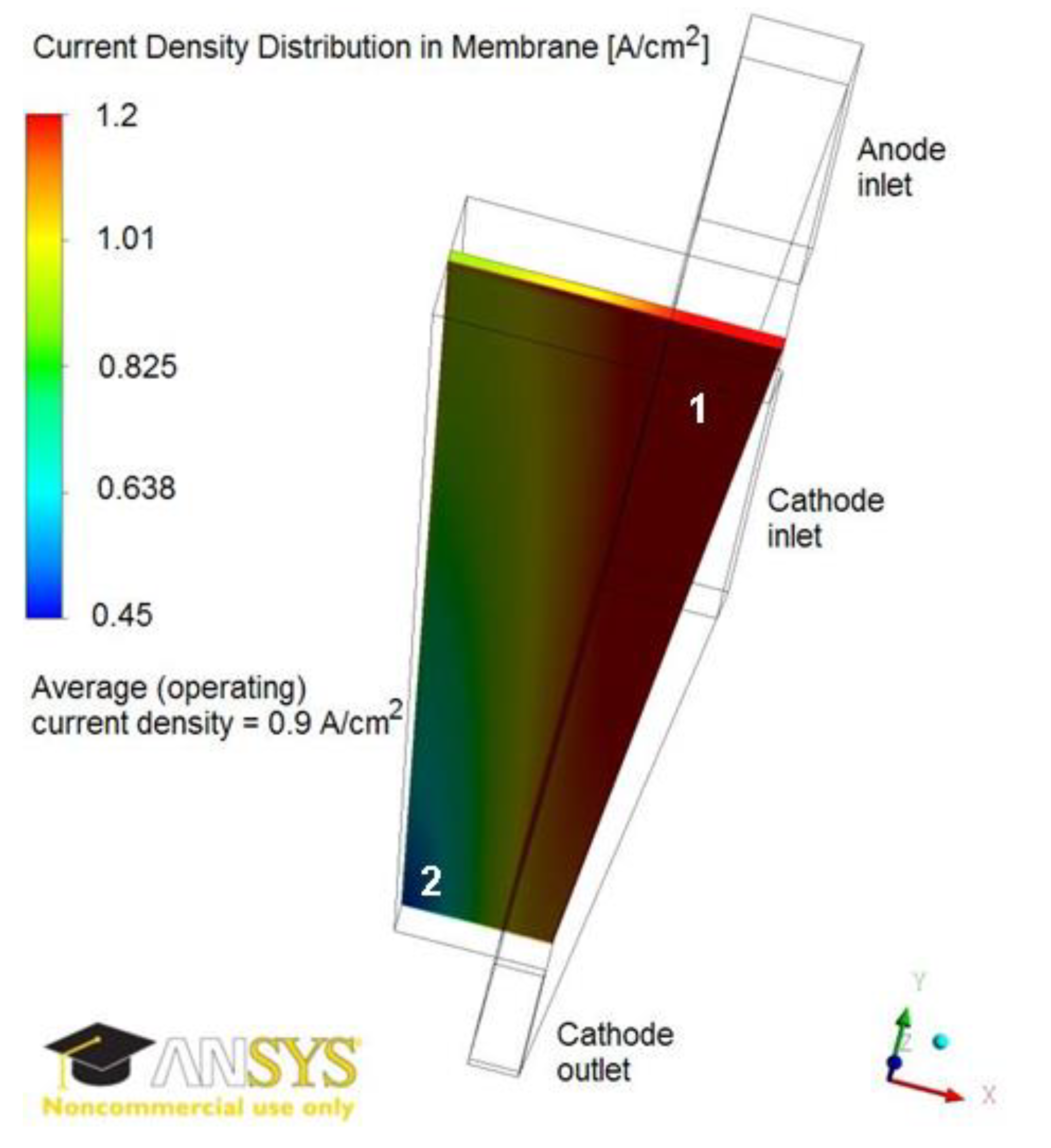
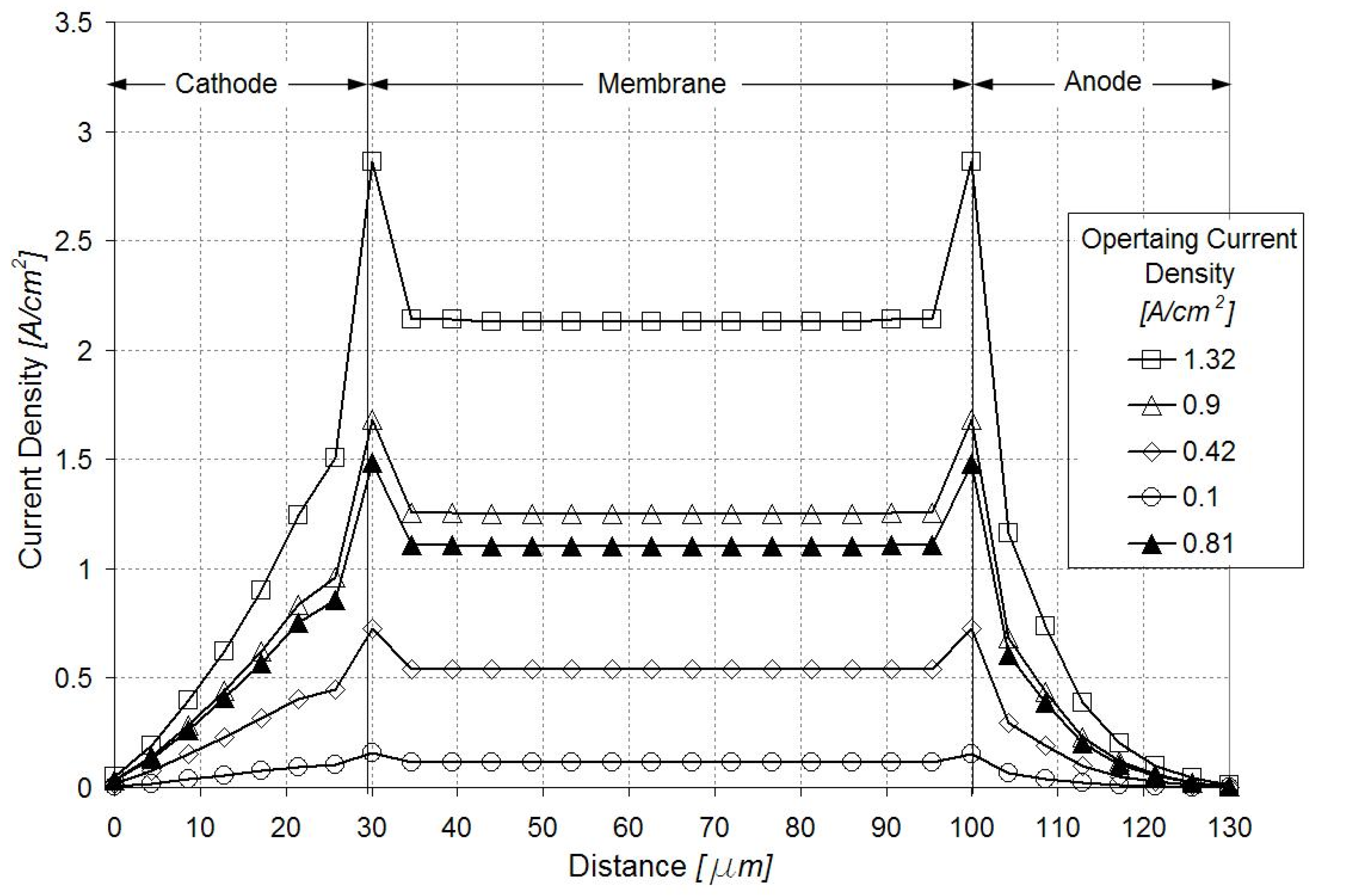
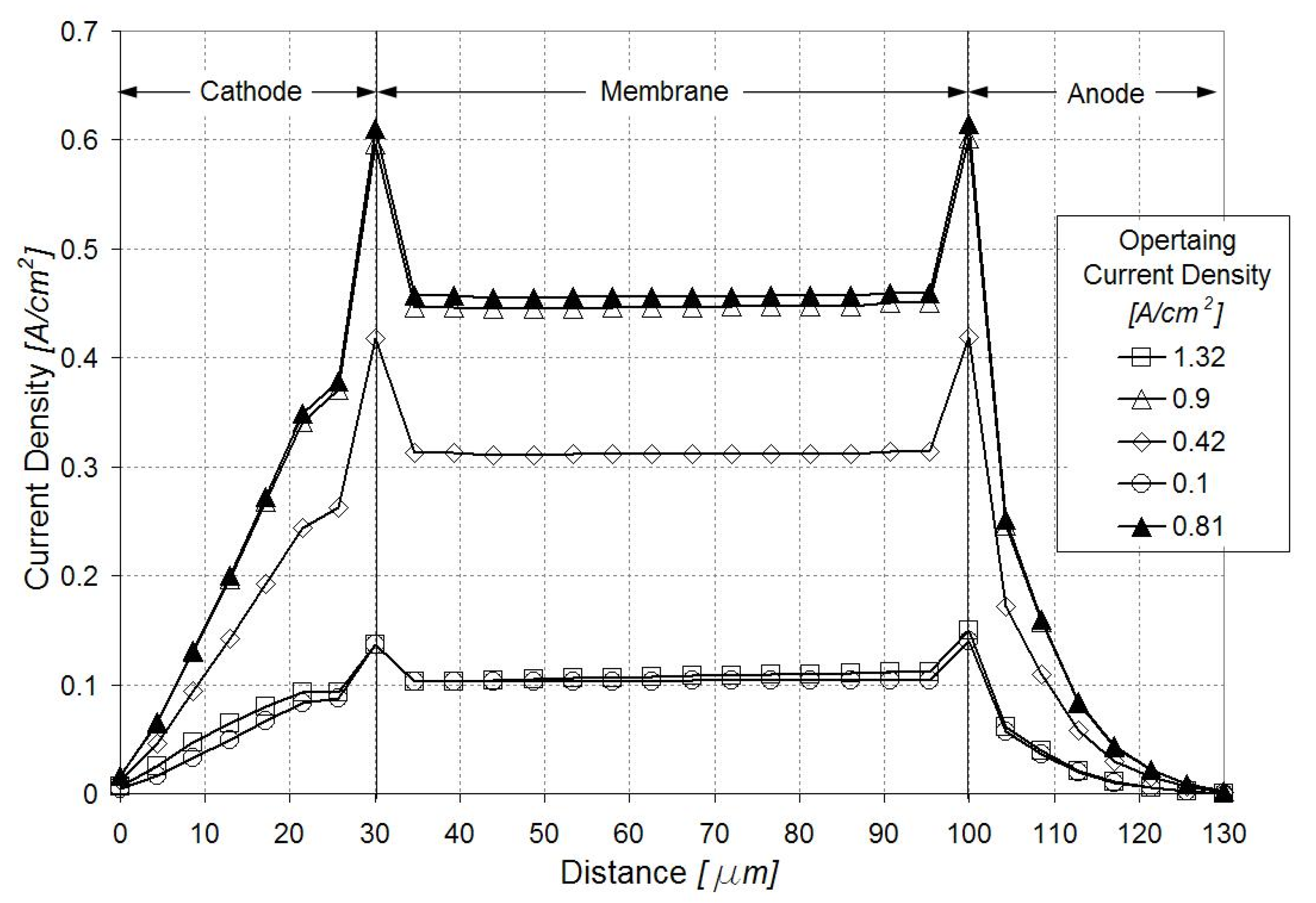
3.3. Sensitivity Analysis for the Defect Location
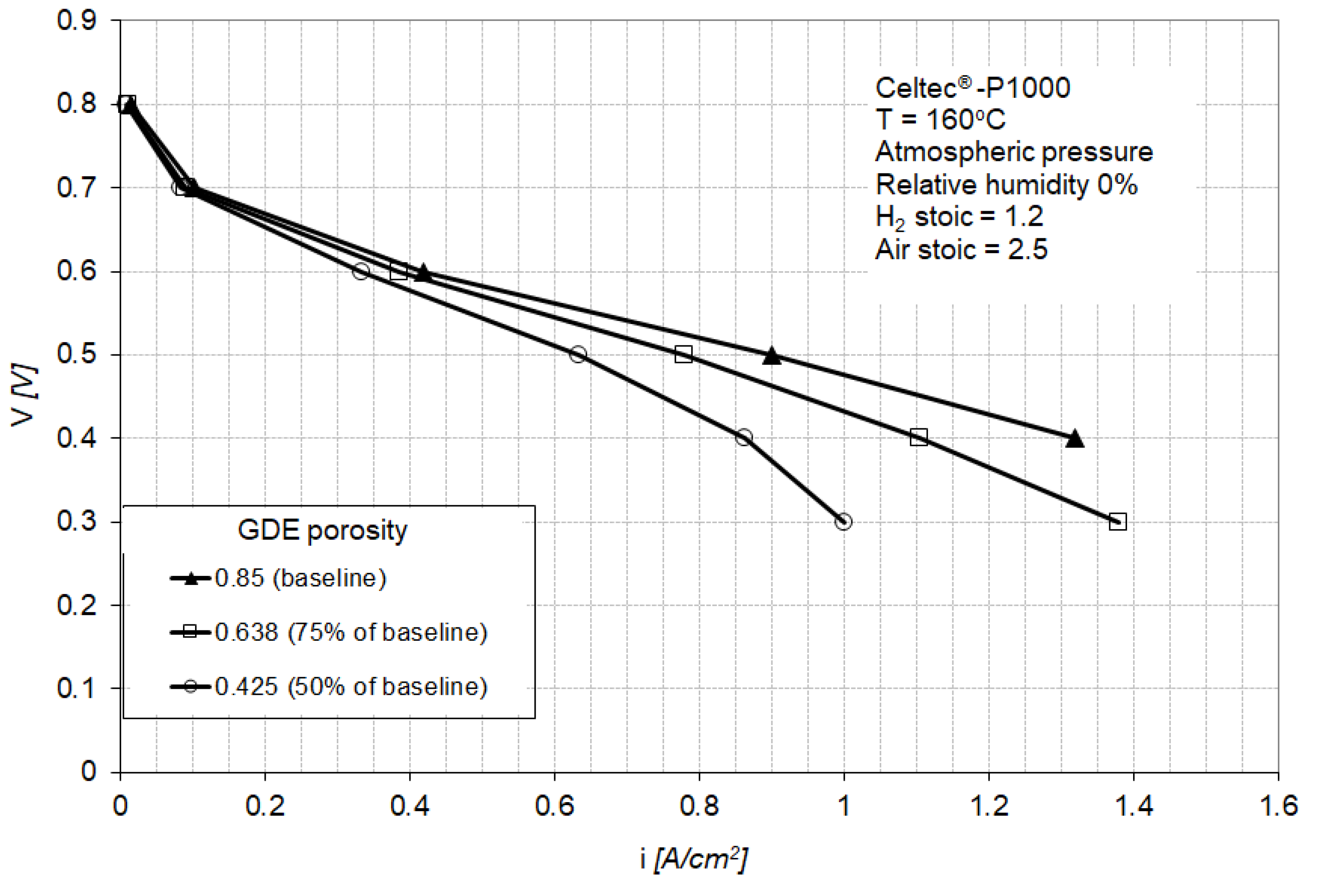
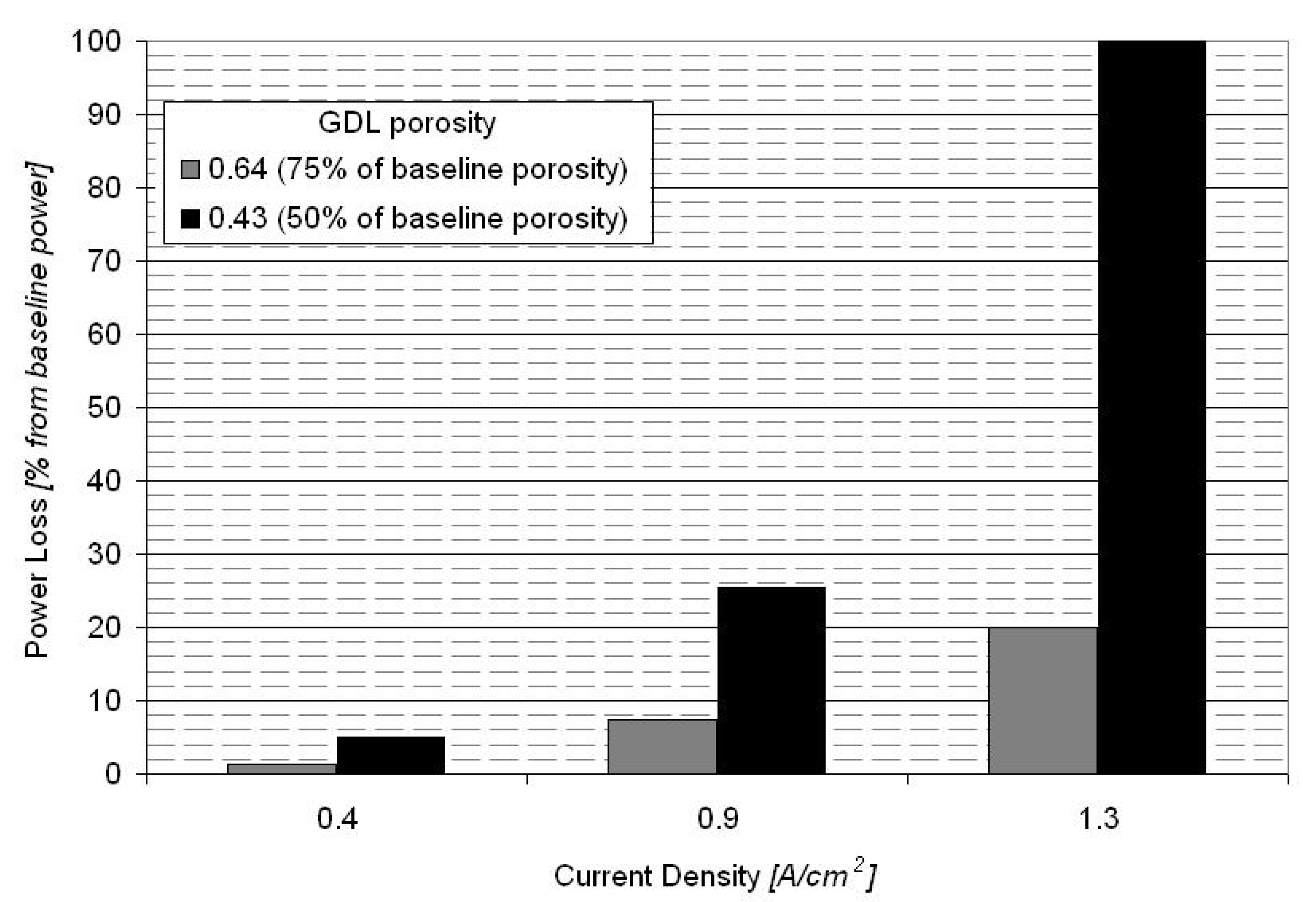
3.4. Sensitivity Analysis for GDE Porosity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
Appendix A.1. Transport Equations in the Flow Field
Appendix A.1.1. Mass Transport
Appendix A.1.2. Momentum Conservation
Appendix A.1.3. Conservation of Chemical Species
Appendix A.2. Transport Equations in the Ionomer (PA)-Phase
Conservation of Charge
Appendix A.3. Constitutive Relations
Appendix B
Appendix B.1. Nomenclature
| Aact | active area of catalyst layer (cm2) |
| effective catalyst surface area per unit volume (cm2/cm3) | |
| ad | catalyst area with defects per unit volume (cm2/cm3) |
| constant in Slattery–Bird correlation (A25) | |
| b | Tafel slope (V/decade) |
| exponent in Slattery–Bird correlation (A25) | |
| D | diffusion coefficient (m2/s) |
| Dij | binary diffusion coefficient (m2/s) |
| equilibrium potential (V) | |
| F | Faraday constant 96487 (C/mols H+) |
| reference exchange current density (A/m2) | |
| current density (A/cm2) | |
| j | charge transfer current (A/cm3) |
| absolute viscous permeability (cm2) | |
| mass source (g/cm3s) | |
| n | number of defects |
| pressure (Pa) | |
| t | time (s) |
| tcl | thickness of catalyst area (mm) |
| temperature (K) | |
| velocity vector (cm/s) | |
| V | volume of catalyst layer (m3) |
| Vd | volume of defects in catalyst layer (m3) |
| V1d | volume of a single defect (mm) |
| molecular weight (g/mols) | |
| mass fraction of chemical species |
Appendix B.2. Greek Symbols
| surface porosity | |
| directional surface permeability of ionomer | |
| volumetric porosity | |
| activation polarization (V) | |
| electrochemical reaction rate at cathode (mols/cm3s) | |
| density (g/cm3) | |
| viscosity (Ns/cm2) | |
| stoichiometric coefficient | |
| ionomer conductivity | |
| potential (V) |
Appendix B.3. Subscripts
| a | anode |
| c | cathode |
| cr | critic |
| i | chemical species i |
| i | ionomer (PA)-phase |
| s | solid (electrically-conductive)-phase |
Appendix B.4. Superscripts
| a | anodic |
| c | cathodic |
References
- Schmidt, T.J. High-Temperature Polymer Electrolyte Fuel Cells: Durability Insights. In Polymer Electrolyte Fuel Cell Durability; Buchi, F.N., Inaba, M., Schmidt, T.J., Eds.; Springer: New York, NY, USA, 2009; pp. 199–221. [Google Scholar]
- Wasmus, S.; Valeriu, A.; Mateescu, G.D.; Tryk, D.A.; Savinell, R.F. Characterization of H3P04-Equilibrated Nafion® 117 Membranes Using 1H and 31p NMR Spectroscopy. Solid State Ion. 1995, 80, 87–92. [Google Scholar] [CrossRef]
- Savinell, R.F.; Litt, M.H. Proton Conducting Polymers Used as Membranes. U.S. Patent 5,525,436, 11 June 1996. [Google Scholar]
- Wang, J.T.; Savinell, R.F.; Wainright, J.S.; Litt, M.H.; Yu, H. A H2/O2 Fuel Cell Using Acid Doped Polybenzimidazole as Polymer Electrolyte. Electrochim. Acta 1996, 41, 193–197. [Google Scholar] [CrossRef]
- Savinell, R.; Yeger, E.; Tryk, D.; Landau, U.; Wainright, J.; Weng, D.; Lux, K.; Litt, M.; Rogers, C. A Polymer Electrolyte for Operation at Temperatures up to 200 °C. J. Electrochem. Soc. 1994, 141, L46–L48. [Google Scholar] [CrossRef]
- Weng, D.; Wainright, J.S.; Landau, U.; Savinell, R.F. Electro-osmotic Drag Coefficient of Water and Methanol in Polymer Electrolytes at Elevated Temperatures. J. Electrochem. Soc. 1996, 143, 1260–1263. [Google Scholar] [CrossRef]
- Sammes, S.R.; Wasmus, S.; Savinell, R.F. Thermal Stability of Proton Conducting Acid Doped Polybenzimidazole in Simulated Fuel Cell Environments. J. Electrochem. Soc. 1996, 143, 1225–1232. [Google Scholar] [CrossRef]
- Wainright, J.S.; Litt, M.H.; Savinell, R.F. High-Temperature Membranes. In Handbook of Fuel Cells. Fundamentals, Technology and Applications; Vielstich, W., Lamm, A., Gasteiger, H.A., Eds.; John Wiley & Sons: New York, NY, USA, 2003; Volume 3, p. 436. [Google Scholar]
- Xiao, L.; Zhang, H.; Scanlon, E.; Ramanathan, L.S.; Choe, E.-W.; Rogers, D.; Apple, T.; Beniecewicz, B.C. High-Temperature Polybenzimidazole Fuel Cell Membranes via a Sol-Gel Process. Chem. Mater. 2005, 17, 5328–5333. [Google Scholar] [CrossRef]
- Xiao, L.; Zhang, H.; Scanlon, E.; Chen, R.; Choe, E.-W.; Ramanathan, L.S.; Yu, S.; Beniecewicz, B.C. Synthesis and Characterization of Pyridine-Based Polybenzimidazoles for High Temperature Polymer Electrolyte Membrane Fuel Cell Applications. Fuel Cells 2005, 5, 287–295. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Baurmeister, J. Properties of High-Temperature PEFC Celtec®-P 1000 MEAs in Start/Stop Operation Mode. J. Power Sourcce 2008, 176, 428–434. [Google Scholar] [CrossRef]
- Uchida, M.; Aoyama, Y.; Eda, N.; Ohta, A. Investigation of the Microstructure in the Catalyst Layer and Effects of Both Perfluorosulfonate Ionomer and PTFE-Loaded Carbon on the Catalyst Layer of Polymer Electrolyte Fuel Cells. J. Electrochem. Soc. 1995, 142, 4143–4149. [Google Scholar] [CrossRef]
- Watanabe, M.; Igarashi, H.; Yosioka, K. An experimental prediction of the preparation conditions of Nafion-coated catalyst layers for PEFCs. Electrochim. Acta 1995, 40, 329–334. [Google Scholar] [CrossRef]
- Scharifker, B.R.; Zelenay, P.; Bockris, J.M. The Kinetics of Oxygen Reduction in Molten Phosphoric Acid at High Temperatures. J. Electrochem. Soc. 1987, 134, 2714–2725. [Google Scholar] [CrossRef]
- Haug, A.T.; White, R.E. Oxygen Diffusion Coefficient and Solubility in a New Proton Exchange Membrane. J. Electrochem. Soc. 2000, 147, 980–983. [Google Scholar] [CrossRef] [Green Version]
- Cheddie, D.F.; Munroe, N.D. A Two-Phase Model of an Intermediate Temperature PEM Fuel Cell. Int. J. Hydrogen Energy 2007, 32, 832–841. [Google Scholar] [CrossRef]
- Siegel, C.; Bandlamudi, G.; Heinzel, A. Numerical Simulation of a High-Temperature PEM (HTPEM) Fuel Cell. In Proceedings of the COMSOL Users Conference 2007, Grenoble, France, 23–24 October 2007. [Google Scholar]
- Siegel, C.; Bandlamudi, G.; Heinzel, A. Modeling Polybenzimidazole/Phosphoric Acid Membrane Behavior in a HTPEM Fuel Cell. In Proceedings of the COMSOL Conference 2008, Hannover, Germany, 4–6 November 2008. [Google Scholar]
- Hu, J.; Zhang, H.; Gang, L. Diffusion-Convection/Electrochemical Model studies on Polybenzimidazole (PBI) Fuel Cell Based on AC Impedance Technique. Energy Convers. Manag. 2008, 49, 1019–1027. [Google Scholar] [CrossRef]
- Gurau, V.; Zawodzinski, T.A.; Mann, J.A. Two-Phase Transport In PEM Fuel Cell Cathodes. J. Fuel Cell Sci. Technol. 2008, 5, 021009. [Google Scholar] [CrossRef]
- Gurau, V.; Edwards, R.V.; Mann, J.A.; Zawodzinski, T.A. A Look at the Multiphase Mixture Model for PEM Fuel Cell Simulations. Electrochem. Solid-State Lett. 2008, 11, B132–B135. [Google Scholar] [CrossRef]
- Gurau, V.; Zawodzinski, T.A.; Mann, J.A. Spatiotemporal Behavior of Water and Two-Phase Transport in the Porous Electrodes for PEM Fuel Cells. Characterization of Porous Materials 2. ECS Trans. 2009, 19, 29–38. [Google Scholar]
- Gurau, V.; Mann, J.A. A Critical Overview of Computational Fluid Dynamics Multiphase Models for Proton Exchange Membrane Fuel Cells. SIAM J. Appl. Math. 2009, 70, 410–454. [Google Scholar] [CrossRef]
- Gurau, V.; Mann, J.A. A Continuum Model for Water Transport in the Ionomer-Phase of Catalyst Coated Membranes for PEMFCs. Adv. Mech. Eng. 2010, 2010, 372795. [Google Scholar] [CrossRef]
- Gurau, V.; Mann, J.A. Effect of Interfacial Phenomena at the Gas Diffusion Layer-Channel Interface on the Water Evolution in a PEM Fuel Cell. J. Electrochem. Soc. 2010, 157, B512–B521. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Model | Measured |
|---|---|---|
| Tafel slope for oxygen reduction reaction, b | 86 mV/decade | 90 mV/decade (Reference [11]) |
| Equilibrium potential, Eeq | 1.15 V | - |
| Reference exchange current density x effective catalyst area/unit volume, | 5.5 × 10−4 A/cm2 | - |
| Membrane conductivity, | 0.135 S/cm | 0.225 S/cm (Reference [9]) |
| Reference Exchange Current Density X Effective Catalyst Area/Unit Volume, (A/cm2) × 10−4 | Volume of Defects, Vd % of Total Catalyst Volume | Number of Defects/cm2 in Catalyst Active Area | |
|---|---|---|---|
| Pristine catalyst layer | 5.5 | 0% | 0 |
| Catalyst layer causing 10% performance reduction | 3.35 | 39% | 46.6 × 106 |
| Defect Location from Membrane/Catalyst Layer Interface, % of Catalyst Layer Thickness | Change in Local Current Density, % of Current in Pristine GDE | |
|---|---|---|
| Region 1 in Figure 4 | Region 2 in Figure 4 | |
| 7 | 1.6 | 1.6 |
| 36 | 0.6 | 1.1 |
| 64 | 0.4 | 0.1 |
| 93 | 0.3 | 0.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurau, V.; De Castro, E.S. Prediction of Performance Variation Caused by Manufacturing Tolerances and Defects in Gas Diffusion Electrodes of Phosphoric Acid (PA)–Doped Polybenzimidazole (PBI)-Based High-Temperature Proton Exchange Membrane Fuel Cells. Energies 2020, 13, 1345. https://doi.org/10.3390/en13061345
Gurau V, De Castro ES. Prediction of Performance Variation Caused by Manufacturing Tolerances and Defects in Gas Diffusion Electrodes of Phosphoric Acid (PA)–Doped Polybenzimidazole (PBI)-Based High-Temperature Proton Exchange Membrane Fuel Cells. Energies. 2020; 13(6):1345. https://doi.org/10.3390/en13061345
Chicago/Turabian StyleGurau, Vladimir, and Emory S. De Castro. 2020. "Prediction of Performance Variation Caused by Manufacturing Tolerances and Defects in Gas Diffusion Electrodes of Phosphoric Acid (PA)–Doped Polybenzimidazole (PBI)-Based High-Temperature Proton Exchange Membrane Fuel Cells" Energies 13, no. 6: 1345. https://doi.org/10.3390/en13061345
APA StyleGurau, V., & De Castro, E. S. (2020). Prediction of Performance Variation Caused by Manufacturing Tolerances and Defects in Gas Diffusion Electrodes of Phosphoric Acid (PA)–Doped Polybenzimidazole (PBI)-Based High-Temperature Proton Exchange Membrane Fuel Cells. Energies, 13(6), 1345. https://doi.org/10.3390/en13061345