Taxonomic Binning Approaches and Functional Characteristics of the Microbial Community during the Anaerobic Digestion of Hydrolyzed Corncob

 , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Substrate Sample and Characterization
2.2. Alkaline Organosolv Pre-Treatment
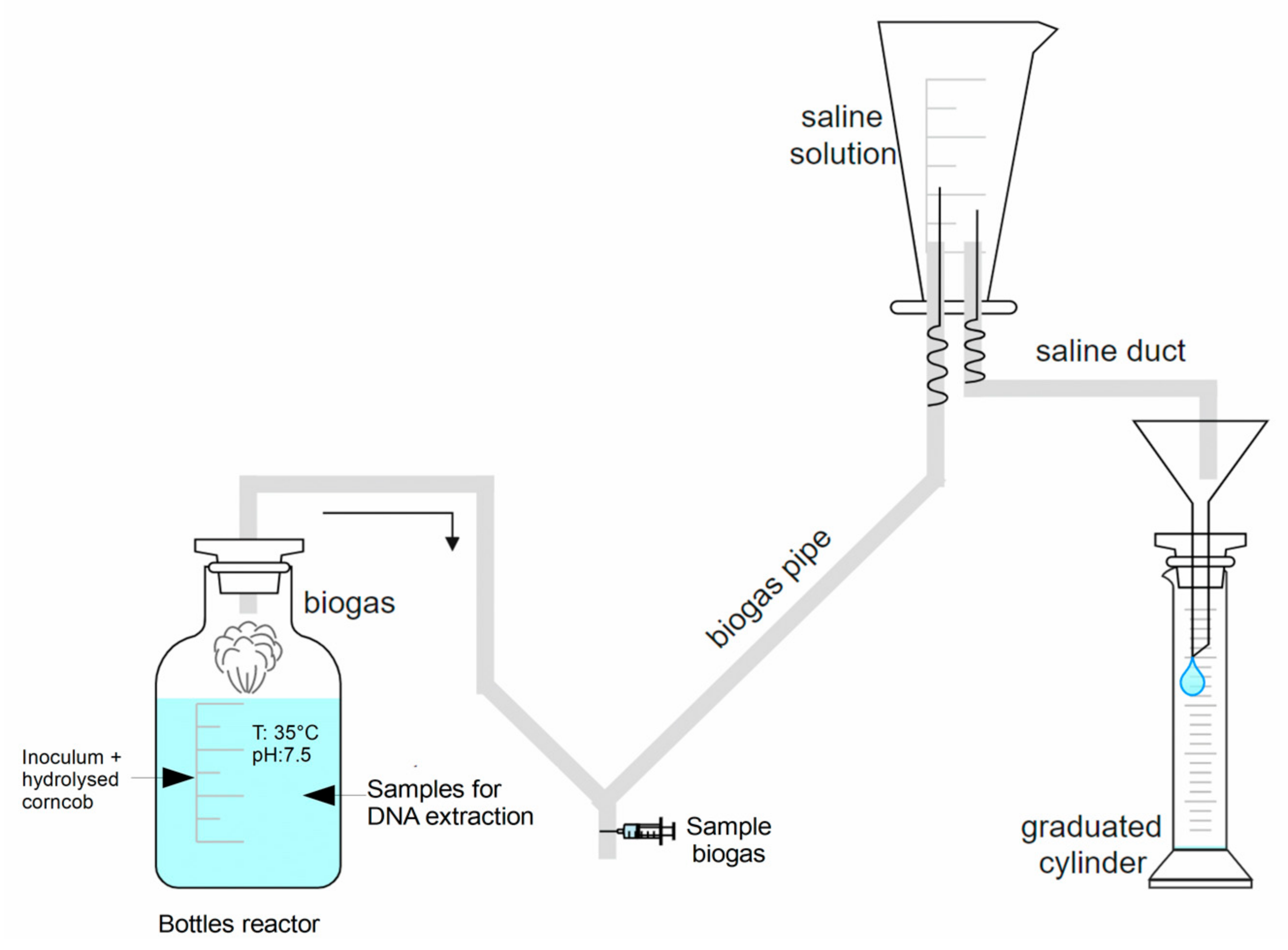
2.3. Batch Anaerobic Digestion Experiments
2.4. Hydrolysate and Digestate Characterization
2.5. DNA Extraction and Sequencing
2.6. Bioinformatics Analysis
2.7. Statistical Analysis
3. Results and Discussion
3.1. Anaerobic Digester Performance of Corncob
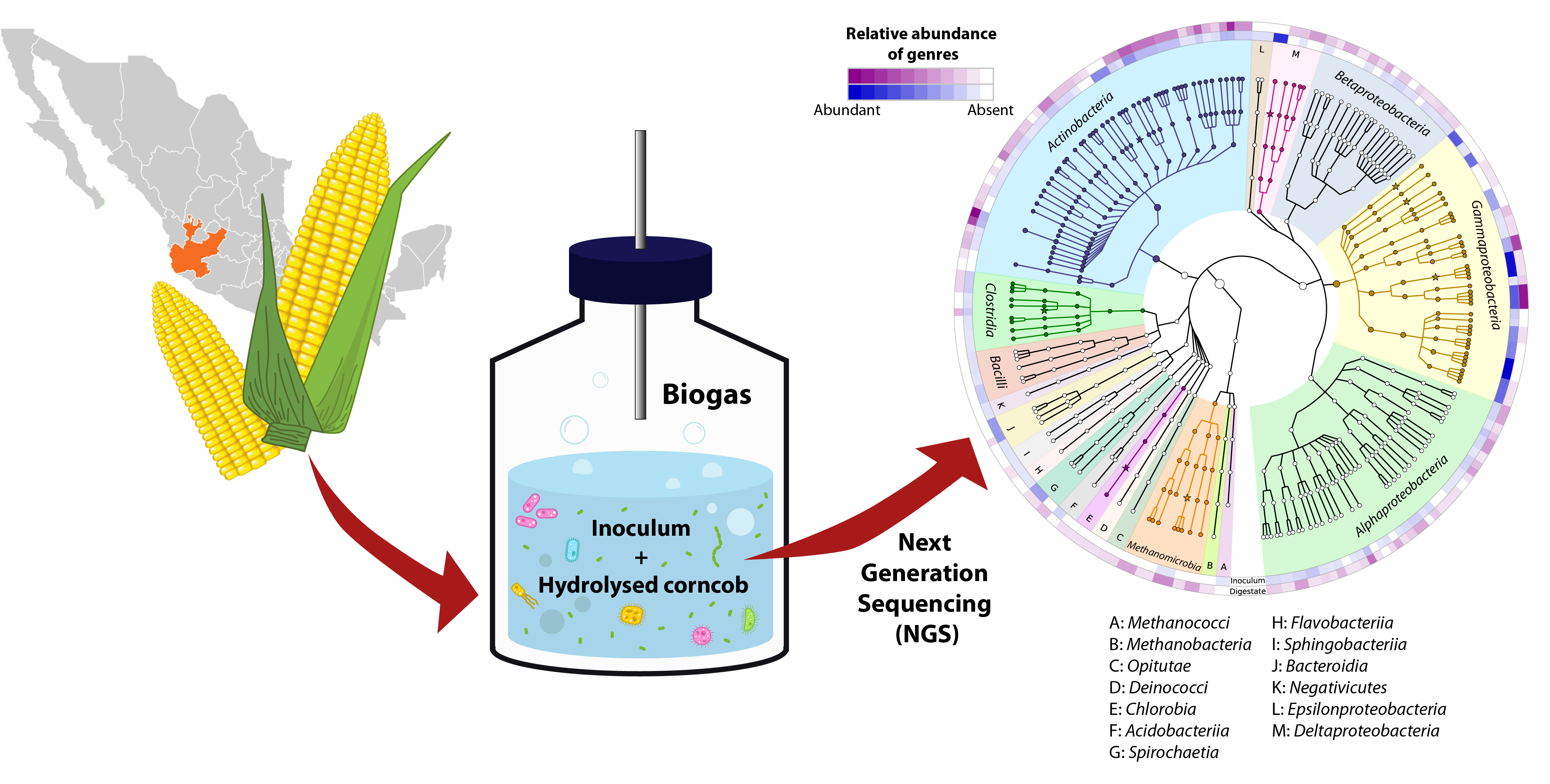
3.2. The Microbial Community at the Begging and after the Anaerobic Digestion of Corn Cob Hydrolysate
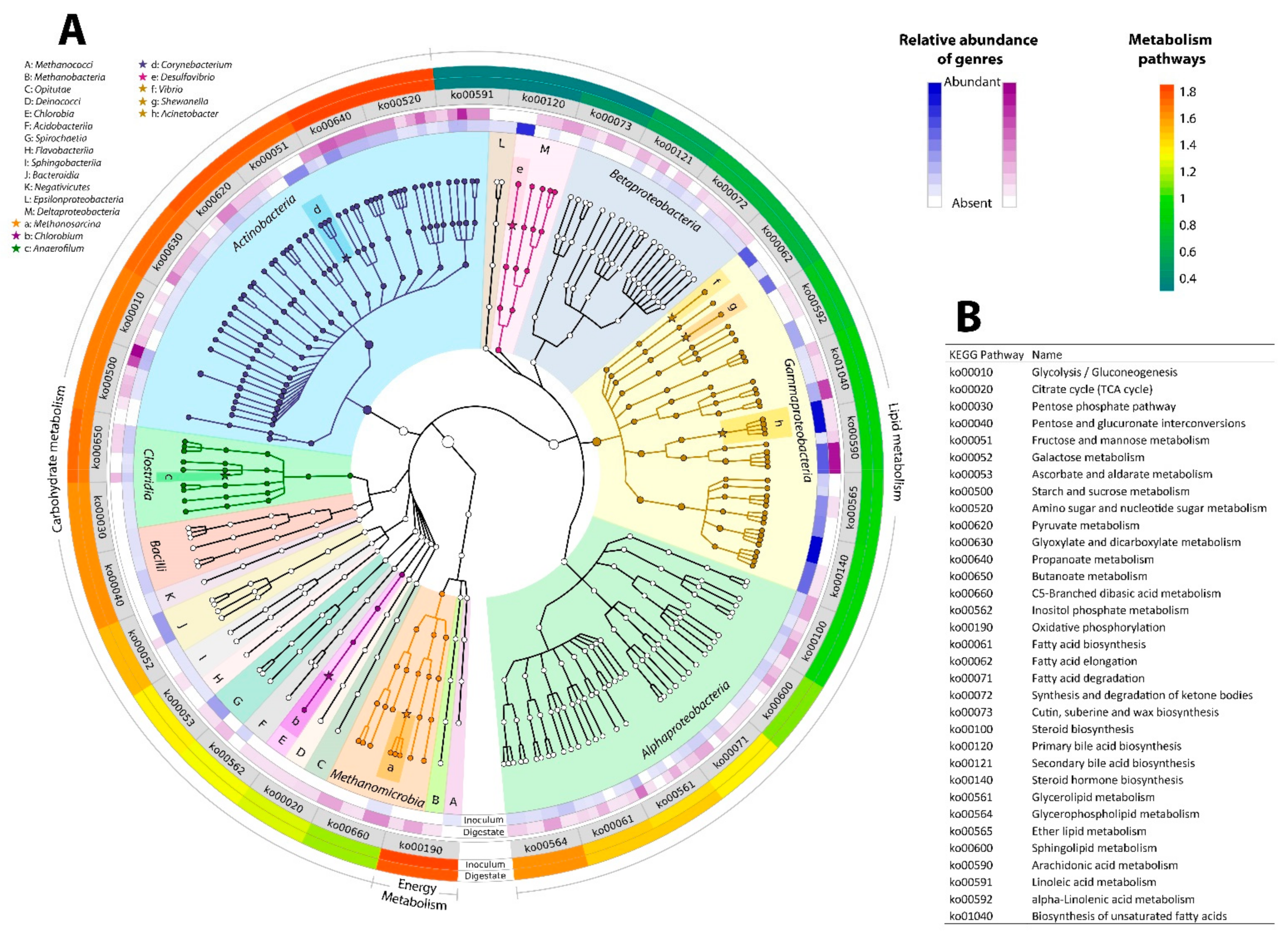
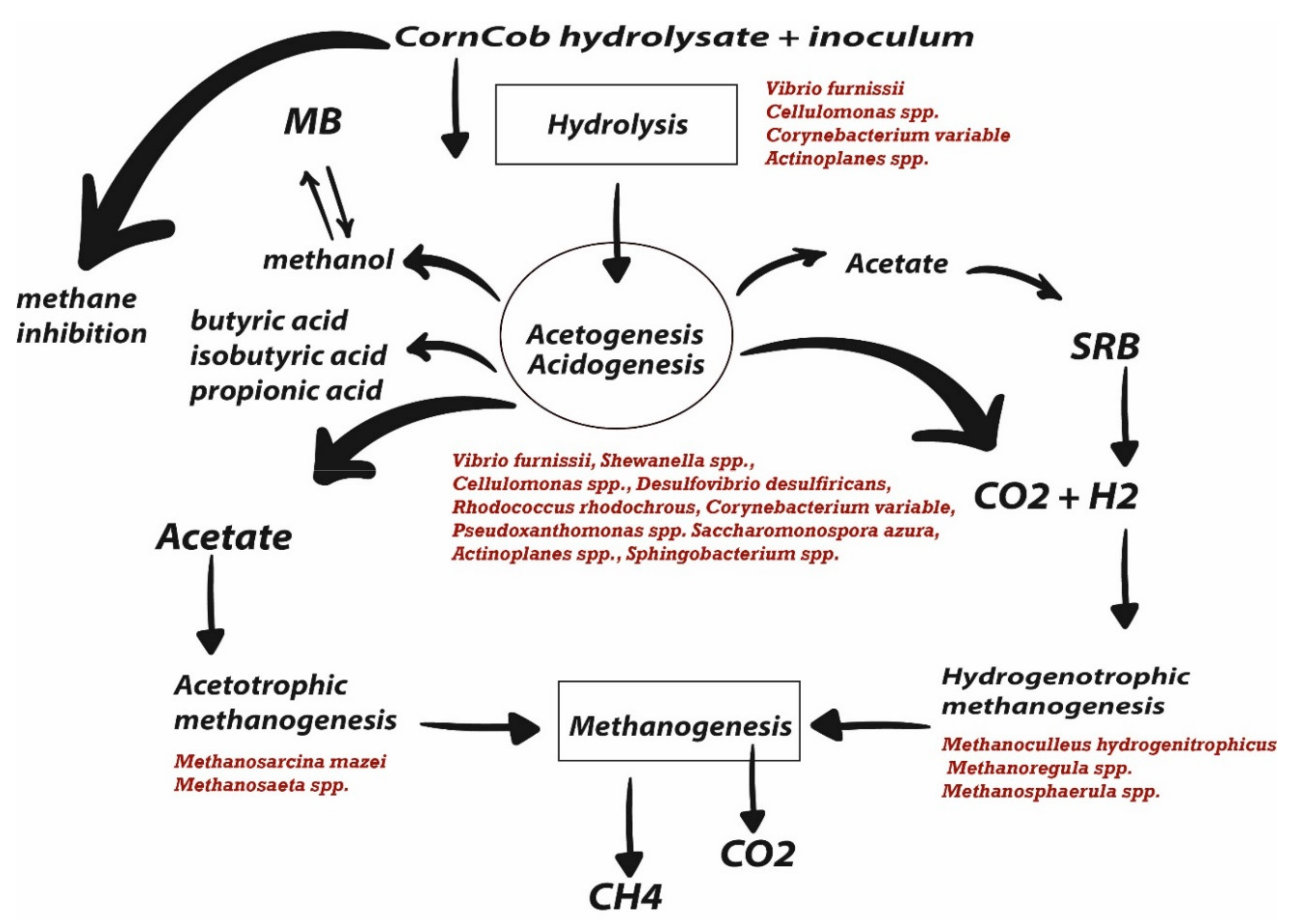
3.3. The Community Interaction Networks and Their Potential Functional Profile
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gielen, D.; Boshell, F.; Saygin, D.; Bazilian, M.D.; Wagner, N.; Gorini, R. The role of renewable energy in the global energy transformation. Energy Strat. Rev. 2019, 24, 38–50. [Google Scholar] [CrossRef]
- Meunier, F. The greenhouse effect: A new source of energy. Appl. Therm. Eng. 2007, 27, 658–664. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Broughton, K.; Osanai, Y.; Anderson, I.C.; Bange, M.P.; Tissue, D.T.; Singh, B.K. Effects of elevated temperature and elevated CO2 on soil nitrification and ammonia-oxidizing microbial communities in field-grown crop. Sci. Total. Environ. 2019, 675, 81–89. [Google Scholar] [CrossRef]
- Nüsser, M.; Dame, J.; Kraus, B.; Baghel, R.; Schmidt, S. Socio-hydrology of “artificial glaciers” in Ladakh, India: Assessing adaptive strategies in a changing cryosphere. Reg. Environ. Chang. 2018, 19, 1327–1337. [Google Scholar] [CrossRef] [Green Version]
- Carrillo-Reyes, J.; Buitrón, G. Biohydrogen and methane production via a two-step process using an acid pretreated native microalgae consortium. Bioresour. Technol. 2016, 221, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Moraes, B.; Triolo, J.; Lecona, V.; Zaiat, M.; Sommer, S. Biogas production within the bioethanol production chain: Use of co-substrates for anaerobic digestion of sugar beet vinasse. Bioresour. Technol. 2015, 190, 227–234. [Google Scholar] [CrossRef]
- Sivagurunathan, P.; Kumar, G.; Mudhoo, A.; Rene, E.R.; Saratale, G.D.; Kobayashi, T.; Xu, K.; Kim, S.-H.; Kim, D.-H. Fermentative hydrogen production using lignocellulose biomass: An overview of pre-treatment methods, inhibitor effects and detoxification experiences. Renew. Sustain. Energy Rev. 2017, 77, 28–42. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, N.; Garcia-Bernet, D.; Domínguez, J.M. Extrusion and enzymatic hydrolysis as pretreatments on corn cob for biogas production. Renew. Energy 2017, 107, 597–603. [Google Scholar] [CrossRef]
- Aslanzadeh, S.; Rajendran, K.; Jeihanipour, A.; Taherzadeh, M.J. The Effect of Effluent Recirculation in a Semi-Continuous Two-Stage Anaerobic Digestion System. Energies 2013, 6, 2966–2981. [Google Scholar] [CrossRef] [Green Version]
- Kavitha, S.; Banu, J.R.; Kumar, J.V.; Rajkumar, M. Improving the biogas production performance of municipal waste activated sludge via disperser induced microwave disintegration. Bioresour. Technol. 2016, 217, 21–27. [Google Scholar] [CrossRef]
- Amnuaycheewa, P.; Hengaroonprasan, R.; Rattanaporn, K.; Kirdponpattara, S.; Cheenkachorn, K.; Sriariyanun, M. Enhancing enzymatic hydrolysis and biogas production from rice straw by pretreatment with organic acids. Ind. Crop. Prod. 2016, 87, 247–254. [Google Scholar] [CrossRef]
- Bolado, S.; Toquero, C.; Martín-Juárez, J.; Travaini, R.; García-Encina, P.A. Effect of thermal, acid, alkaline and alkaline-peroxide pretreatments on the biochemical methane potential and kinetics of the anaerobic digestion of wheat straw and sugarcane bagasse. Bioresour. Technol. 2016, 201, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, C.; Ai, S.; Wang, H.; Gao, Y.; Yan, L.; Mei, Z.; Wang, W. Biological pretreatment enhances the activity of functional microorganisms and the ability of methanogenesis during anaerobic digestion. Bioresour. Technol. 2019, 290, 121660. [Google Scholar] [CrossRef] [PubMed]
- Romaní, A.; Larramendi, A.; Yáñez, R.; Cancela, Á.; Sánchez, Á.; Teixeira, J.A.; Domingues, L. Valorization of Eucalyptus nitens bark by organosolv pretreatment for the production of advanced biofuels. Ind. Crop. Prod. 2019, 132, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Sulbarán-Rangel, B.; Aguirre, J.S.A.; Breton-Deval, L.; Del Real-Olvera, J.; Gurubel-Tun, K.J. Improvement of Anaerobic Digestion of Hydrolysed Corncob Waste by Organosolv Pretreatment for Biogas Production. Appl. Sci. 2020, 10, 2785. [Google Scholar] [CrossRef] [Green Version]
- Enzmann, F.; Mayer, F.; Rother, M.; Holtmann, D. Methanogens: Biochemical background and biotechnological applications. AMB Express 2018, 8, 1. [Google Scholar] [CrossRef]
- Anderson, K.; Sallis, P.J.; Uyanik, S. Anaerobic Treatment Processes. In Handbook of Water and Waste Water Microbiology; Mara, D., Horan, N., Eds.; Academic Press: London, UK, 2003; Chapter 24; pp. 391–426. [Google Scholar] [CrossRef]
- Wojcieszak, M.; Pyzik, A.; Poszytek, K.; Krawczyk, P.S.; Sobczak, A.; Lipinski, L.; Roubinek, O.; Palige, J.; Sklodowska, A.; Drewniak, L. Adaptation of Methanogenic Inocula to Anaerobic Digestion of Maize Silage. Front. Microbiol. 2017, 8, 1881. [Google Scholar] [CrossRef]
- Techtmann, S.M.; Hazen, T.C. Metagenomic applications in environmental monitoring and bioremediation. J. Ind. Microbiol. Biotechnol. 2016, 43, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef] [Green Version]
- Treu, L.; Campanaro, S.; Kougias, P.G.; Sartori, C.; Bassani, I.; Angelidaki, I. Hydrogen-Fueled Microbial Pathways in Biogas Upgrading Systems Revealed by Genome-Centric Metagenomics. Front. Microbiol. 2018, 9, 1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Toril, E.; Aguilera, Á. Chapter 14—Microbial Ecology in Extreme Acidic Environments: Use of Molecular Tools. In Microbial Diversity in the Genomic Era; Surajit, D., Dash, H.R., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 227–238. [Google Scholar]
- Breton-Deval, L.; Méndez-Acosta, H.O.; González-Álvarez, V.; Snell-Castro, R.; Gutiérrez-Sánchez, D.; Arreola-Vargas, J. Agave tequilana bagasse for methane production in batch and sequencing batch reactors: Acid catalyst effect, batch optimization and stability of the semi-continuous process. J. Environ. Manag. 2018, 224, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Tsapekos, P.; Kougias, P.; Treu, L.; Campanaro, S.; Angelidaki, I. Process performance and comparative metagenomic analysis during co-digestion of manure and lignocellulosic biomass for biogas production. Appl. Energy 2017, 185, 126–135. [Google Scholar] [CrossRef]
- Abendroth, C.; Pérez, A.L.; Porcar, M.; Simeonov, C.; Luschnig, O.; Vilanova, C.; Pascual, J. Shedding light on biogas: Phototrophic biofilms in anaerobic digesters hold potential for improved biogas production. Syst. Appl. Microbiol. 2020, 43, 126024. [Google Scholar] [CrossRef] [PubMed]
- Elnaker, N.A.; Elektorowicz, M.; Naddeo, V.; Hasan, S.W.; Yousef, A.F. Assessment of Microbial Community Structure and Function in Serially Passaged Wastewater Electro-Bioreactor Sludge: An Approach to Enhance Sludge Settleability. Sci. Rep. 2018, 8, 7013. [Google Scholar] [CrossRef] [PubMed]
- Liguori, R.; Ventorino, V.; Pepe, O.; Faraco, V. Bioreactors for lignocellulose conversion into fermentable sugars for production of high added value products. Appl. Microbiol. Biotechnol. 2016, 100, 597–611. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.G.; Han, G.; Lee, J.; Shin, J.; Hwang, S. A snapshot of microbial community structures in 20 different field-scale anaerobic bioreactors treating food waste. J. Environ. Manag. 2019, 248, 109297. [Google Scholar] [CrossRef]
- Guo, J.; Peng, Y.; Ni, B.-J.; Han, X.; Fan, L.; Yuan, Z. Dissecting microbial community structure and methane-producing pathways of a full-scale anaerobic reactor digesting activated sludge from wastewater treatment by metagenomic sequencing. Microb. Cell Factories 2015, 14, 33. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Ren, J.; Sun, F. Reads Binning Improves Alignment-Free Metagenome Comparison. Front. Genet. 2019, 10, 1156. [Google Scholar] [CrossRef] [Green Version]
- Qing, Q.; Zhou, L.; Guo, Q.; Gao, X.; Zhang, Y.; He, Y.; Zhang, Y. Mild alkaline presoaking and organosolv pretreatment of corn stover and their impacts on corn stover composition, structure, and digestibility. Bioresour. Technol. 2017, 233, 284–290. [Google Scholar] [CrossRef]
- Van Soest, P.V.; Robertson, J.B.; Lewis, B.A. Methods for Dietary Fiber, Neutral Detergent Fiber, and Nonstarch Polysaccharides in Relation to Animal Nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef]
- Dubois, M.Y.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.A.; Smith, F.G. A Colorimetric Method for the Determination of Sugars. Nat. Cell Biol. 2006, 168, 167. [Google Scholar] [CrossRef] [PubMed]
- Lins, P.; Malin, C.; Wagner, A.O.; Illmer, P. Reduction of accumulated volatile fatty acids by an acetate-degrading enrichment culture. FEMS Microbiol. Ecol. 2010, 71, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Baird, R.; Eaton, A.; Rice, E. Standard Methods for the Examination of Water and Wastewater, 23 ed.; American Public Health Association: Washington, DC, USA, 2017. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Asnicar, F.; Weingart, G.; Tickle, T.L.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Ecai, M.; Ewilkins, D.; Echen, J.; Eng, S.K.; Elu, H.; Ejia, Y.; Lee, P.K.H. Metagenomic Reconstruction of Key Anaerobic Digestion Pathways in Municipal Sludge and Industrial Wastewater Biogas-Producing Systems. Front. Microbiol. 2016, 7, 778. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [Green Version]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Velázquez-Valadez, U.; Farías-Sánchez, J.C.; Vargas-Santillán, A.; Castro-Montoya, A.J. Tequilana weber Agave Bagasse Enzymatic Hydrolysis for the Production of Fermentable Sugars: Oxidative-Alkaline Pretreatment and Kinetic Modeling. BioEnergy Res. 2016, 9, 998–1004. [Google Scholar] [CrossRef]
- Siegert, I.; Banks, C. The effect of volatile fatty acid additions on the anaerobic digestion of cellulose and glucose in batch reactors. Process Biochem. 2005, 40, 3412–3418. [Google Scholar] [CrossRef]
- Naraian, R.; Gautam, R.L. Chapter 6—Penicillium Enzymes for the Saccharification of Lignocellulosic Feedstocks. In New and Future Developments in Microbial Biotechnology and Bioengineering; Gupta, V.K., Rodriguez-Couto, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 121–136. [Google Scholar]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhao, D.; Huang, R.; Cao, X.; Zeng, J.; Yu, Z.; Hooker, K.V.; Hambright, K.D.; Wu, Q. Contrasting Network Features between Free-Living and Particle-Attached Bacterial Communities in Taihu Lake. Microb. Ecol. 2018, 76, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Guo, B.; Zhang, L.; Zhang, Y.; Liu, Y. Microbial community dynamics in anaerobic digesters treating conventional and vacuum toilet flushed blackwater. Water Res. 2019, 160, 249–258. [Google Scholar] [CrossRef]
- Tapadia-Maheshwari, S.; Pore, S.; Engineer, A.; Shetty, D.; Dagar, S.S.; Dhakephalkar, P.K. Illustration of the microbial community selected by optimized process and nutritional parameters resulting in enhanced biomethanation of rice straw without thermo-chemical pretreatment. Bioresour. Technol. 2019, 289, 121639. [Google Scholar] [CrossRef]
- Liu, C.; Li, H.; Zhang, Y.; Liu, C. Improve biogas production from low-organic-content sludge through high-solids anaerobic co-digestion with food waste. Bioresour. Technol. 2016, 219, 252–260. [Google Scholar] [CrossRef]
- Abendroth, C.; Vilanova, C.; Günther, T.; Luschnig, O.; Porcar, M. Eubacteria and archaea communities in seven mesophile anaerobic digester plants in Germany. Biotechnol. Biofuels 2015, 8, 87. [Google Scholar] [CrossRef] [Green Version]
- Azman, S.; Khadem, A.F.; Van Lier, J.B.; Zeeman, G.; Plugge, C.M. Presence and Role of Anaerobic Hydrolytic Microbes in Conversion of Lignocellulosic Biomass for Biogas Production. Crit. Rev. Environ. Sci. Technol. 2015, 45, 2523–2564. [Google Scholar] [CrossRef]
- Bae, H.-S.; Moe, W.M.; Yan, J.; Tiago, I.; Da Costa, M.S.; Rainey, F.A. Brooklawnia cerclae gen. nov., sp. nov., a propionate-forming bacterium isolated from chlorosolvent-contaminated groundwater. Int. J. Syst. Evol. Microbiol. 2006, 56, 1977–1983. [Google Scholar] [CrossRef] [Green Version]
- Ziels, R.M.; Karlsson, A.; Beck, D.A.; Ejlertsson, J.; Yekta, S.S.; Bjorn, A.; Stensel, H.D.; Svensson, B. Microbial community adaptation influences long-chain fatty acid conversion during anaerobic codigestion of fats, oils, and grease with municipal sludge. Water Res. 2016, 103, 372–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, D.; Shayegan, S.S.; Ng, W.J.; He, J. Development and characteristics of rapidly formed hydrogen-producing granules in an acidic anaerobic sequencing batch reactor (AnSBR). Biochem. Eng. J. 2010, 49, 119–125. [Google Scholar] [CrossRef]
- Alvarez, A.; Saez, J.M.; Costa, J.S.D.; Colin, V.L.; Fuentes, M.S.; Cuozzo, S.A.; Benimeli, C.S.; Polti, M.A.; Amoroso, M.J. Actinobacteria: Current research and perspectives for bioremediation of pesticides and heavy metals. Chemosphere 2017, 166, 41–62. [Google Scholar] [CrossRef]
- Anandan, R.; Dharumadurai, D.; Manogaran, G.P. An Introduction to Actinobacteria. In Actinobacteria—Basics and Biotechnological Applications; IntechOpen: Tamil Nadu, India, 2016. [Google Scholar]
- Welte, C.U.; Deppenmeier, U. Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1837, 1130–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bräuer, S.L.; Cadillo-Quiroz, H.; Ward, R.J.; Yavitt, J.B.; Zinder, S.H. Methanoregula boonei gen. nov., sp. nov., an acidiphilic methanogen isolated from an acidic peat bog. Int. J. Syst. Evol. Microbiol. 2011, 61, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Kougias, P.G.; Campanaro, S.; Treu, L.; Zhu, X.; Angelidaki, I. A novel archaeal species belonging to Methanoculleus genus identified via de-novo assembly and metagenomic binning process in biogas reactors. Anaerobe 2017, 46, 23–32. [Google Scholar] [CrossRef]
- Lv, L.; Mbadinga, S.M.; Wang, L.-Y.; Liu, J.-F.; Gu, J.-D.; Mu, B.-Z.; Yang, S.-Z. Acetoclastic methanogenesis is likely the dominant biochemical pathway of palmitate degradation in the presence of sulfate. Appl. Microbiol. Biotechnol. 2015, 99, 7757–7769. [Google Scholar] [CrossRef]
- Tian, G.; Xi, J.; Yeung, M.; Ren, G. Characteristics and mechanisms of H2S production in anaerobic digestion of food waste. Sci. Total. Environ. 2020, 724, 137977. [Google Scholar] [CrossRef]
- Hassa, J.; Maus, I.; Off, S.; Pühler, A.; Scherer, P.; Klocke, M.; Schlüter, A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl. Microbiol. Biotechnol. 2018, 102, 5045–5063. [Google Scholar] [CrossRef] [Green Version]
- Joo, J.-Y.; Park, C.-H.; Han, G.-B. Optimization of two-phased anaerobic sludge digestion using the pressurized ultra filtration membrane with a mesh screen (MS-PUFM). Chem. Eng. J. 2016, 300, 20–28. [Google Scholar] [CrossRef]
- Doi, Y. Glycerol metabolism and its regulation in lactic acid bacteria. Appl. Microbiol. Biotechnol. 2019, 103, 5079–5093. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, B.; Khanna, N.; Das, D. Chapter 4—Dark-Fermentative Biohydrogen Production, in Biohydrogen, 2nd ed.; Ashok Pandey, S., Mohan, V., Chang, J., Patrick, C., Larroche, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 79–122. [Google Scholar]
- Esquivel-Elizondo, S.; Ilhan, Z.E.; Garcia-Peña, E.I.; Krajmalnik-Brown, R. Insights into Butyrate Production in a Controlled Fermentation System via Gene Predictions. mSystems 2017, 2, e00051-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.W.; Trottier, T.M. Effect of sulfur-containing compounds on anaerobic degradation of cellulose to methane by mixed cultures obtained from sewage sludge. Appl. Environ. Microbiol. 1978, 35, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomvong, K.; Kordić, V.; Li, X.; Bauer, S.; Gillespie, A.E.; Ha, S.-J.; Oh, E.J.; Galazka, J.; Jin, Y.-S.; Cate, J.H.D. Overcoming inefficient cellobiose fermentation by cellobiose phosphorylase in the presence of xylose. Biotechnol. Biofuels 2014, 7, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liaquat, R.; Jamal, A.; Tauseef, I.; Qureshi, Z.; Farooq, U.; Imran, M.; Ali, M. Characterizing Bacterial Consortia from an Anaerobic Digester Treating Organic Waste for Biogas Production. Pol. J. Environ. Stud. 2017, 26, 709–716. [Google Scholar] [CrossRef]
- Schnürer, A.; Zellner, G.; Svensson, B.H. Mesophilic syntrophic acetate oxidation during methane formation in biogas reactors. FEMS Microbiol. Ecol. 1999, 29, 249–261. [Google Scholar] [CrossRef]
- Goenrich, M.; Thauer, R.K.; Yurimoto, H.; Kato, N. Formaldehyde activating enzyme (Fae) and hexulose-6-phosphate synthase (Hps) in Methanosarcina barkeri: A possible function in ribose-5-phosphate biosynthesis. Arch. Microbiol. 2005, 184, 41–48. [Google Scholar] [CrossRef]
- Nölling, J.; Pihl, T.D.; Reeve, J.N. Cloning, sequencing, and growth phase-dependent transcription of the coenzyme F420-dependent N5, N10-methylenetetrahydromethanopterin reductase-encoding genes from Methanobacterium thermoautotrophicum delta H and Methanopyrus kandleri. J. Bacteriol. 1995, 177, 7238–7244. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, E.; Henne, A.; Cramm, R.; Eitinger, T.; Friedrich, B.; Gottschalk, G. Complete Nucleotide Sequence of pHG1: A Ralstonia eutropha H16 Megaplasmid Encoding Key Enzymes of H2-based Lithoautotrophy and Anaerobiosis. J. Mol. Biol. 2003, 332, 369–383. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.; Hu, C.; Liu, H.; Bai, Y.; Qu, J. Sulfur-based mixotrophic denitrification corresponding to different electron donors and microbial profiling in anoxic fluidized-bed membrane bioreactors. Water Res. 2015, 85, 422–431. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters (g/L) | Hydrolysate | Digestate |
|---|---|---|
| Total sugars | 157.70 ± 6.29 | 1.35 ± 0.22 |
| Chemical oxygen demand (COD) | 219.85 ± 8.76 | 80.81 ± 10.42 |
| Glucose | 6.23 ± 0.34 | 1.66 ± 0.45 |
| Xylose | 4.12 ± 0.34 | 1.12 ± 0.19 |
| Galactose | 2.42 ± 0.17 | 0.41 ± 0.10 |
| Mannose | 1.66 ± 0.41 | 0.38 ± 0.051 |
| Acetic | 1.28 ± 0.37 | 2.47 ± 0.17 |
| Propionic | 0.29 ± 0.06 | 0.51 ± 0.05 |
| Butyric | 0.41 ± 0.6 | 1.37 ± 0.44 |
| Furfural | 0.02 ± 0.003 | 0.01 ± 0.006 |
| Genera | Completeness (%) | Genome Size (bp) | GC Content (%) |
|---|---|---|---|
| Acinetobacter.041 | 99.31 | 3,736,607 | 39.13 |
| Shewanella.040 | 98.07 | 4,617,115 | 53.42 |
| Corynebacterium.027 | 96.88 | 3,736,091 | 68.11 |
| Vibrio.029 | 96.64 | 4,889,876 | 50.24 |
| Chlorobium.022 | 96.63 | 3,730,513 | 36.61 |
| Desulfovibrio.039 | 96.47 | 3,218,158 | 58.09 |
| Anaerofilum.030 | 85.96 | 5,873,292 | 66.97 |
| Hydrolytic Enzyme | Catalyzed Reaction | Stage | EC Number |
|---|---|---|---|
| Cellobiose phosphorylase (CBP) | cellobiose: phosphate alpha-D-glucosyltransferase | Hydrolysis | EC:2.4.1.20 |
| Pentose phosphate | beta-D-Glucose:NAD + 1-oxoreductase | Hydrolysis | EC:1.1.1.47 |
| Mannose-6-phosphate isomerase | D-mannose-6-phosphate aldose-ketose-isomerase | Hydrolysis | EC:5.3.1.8 |
| GDP-mannose 6-dehydrogenase | GDP-D-mannose: NAD + 6-oxidoreductase | Hydrolysis | EC:1.1.1.132 |
| Fructokinase | ATP + D-Fructose <=> ADP + D-Fructose 6-phosphate | Hydrolysis | EC:2.7.1.4 |
| Glucokinase | ATP: D-glucose 6-phosphotransferase | Hydrolysis | EC:2.7.1.2 |
| Pyruvate kinase | ATP: pyruvate 2-O-phosphotransferase | Acidogenesis | EC:2.7.1.40 |
| Malate dehydrogenase | (S)-malate: NAD + oxidoreductase | Acidogenesis | EC: 1.1.1.37 |
| Succinyl-CoA:acetate CoA-transferase | Succinyl-CoA + Acetate <=> Acetyl-CoA + Succinate | Acetogenesis | EC:2.8.3.18 |
| Formyltetrahydrofolate synthetase | formate—tetrahydrofolate ligase | Acetogenesis dependent on syntrophic relations | EC:6.3.4.3 |
| Tetrahydromethanopterin hydro-lyase | 5,6,7,8-tetrahydromethanopterin hydro-lyase | Methanogenesis | EC:4.2.1.147 |
| Anaerobic carbon monoxide dehydrogenase | CO + H2O + 2 Oxidized ferredoxin <=> CO2 + 2 Reduced ferredoxin + 2H+ | Methanogenesis | EC:1.2.7.4 |
| Formylmethanofuran—tetrahydromethanopterin N-formyltransferase | Formylmethanofuran:5,6,7,8-tetrahydromethanopterin 5-formyltransferase | Methanogenesis | EC:2.3.1.101 |
| NAD-reducing hydrogenase | Hydrogen: Coenzyme F420 oxidoreductase | Methanogenesis | EC:1.12.98.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breton-Deval, L.; Salinas-Peralta, I.; Alarcón Aguirre, J.S.; Sulbarán-Rangel, B.; Gurubel Tun, K.J. Taxonomic Binning Approaches and Functional Characteristics of the Microbial Community during the Anaerobic Digestion of Hydrolyzed Corncob. Energies 2021, 14, 66. https://doi.org/10.3390/en14010066
Breton-Deval L, Salinas-Peralta I, Alarcón Aguirre JS, Sulbarán-Rangel B, Gurubel Tun KJ. Taxonomic Binning Approaches and Functional Characteristics of the Microbial Community during the Anaerobic Digestion of Hydrolyzed Corncob. Energies. 2021; 14(1):66. https://doi.org/10.3390/en14010066
Chicago/Turabian StyleBreton-Deval, Luz, Ilse Salinas-Peralta, Jaime Santiago Alarcón Aguirre, Belkis Sulbarán-Rangel, and Kelly Joel Gurubel Tun. 2021. "Taxonomic Binning Approaches and Functional Characteristics of the Microbial Community during the Anaerobic Digestion of Hydrolyzed Corncob" Energies 14, no. 1: 66. https://doi.org/10.3390/en14010066
APA StyleBreton-Deval, L., Salinas-Peralta, I., Alarcón Aguirre, J. S., Sulbarán-Rangel, B., & Gurubel Tun, K. J. (2021). Taxonomic Binning Approaches and Functional Characteristics of the Microbial Community during the Anaerobic Digestion of Hydrolyzed Corncob. Energies, 14(1), 66. https://doi.org/10.3390/en14010066