
Laminar Burning Velocity and Ignition Delay Time of Oxygenated Biofuel



Abstract
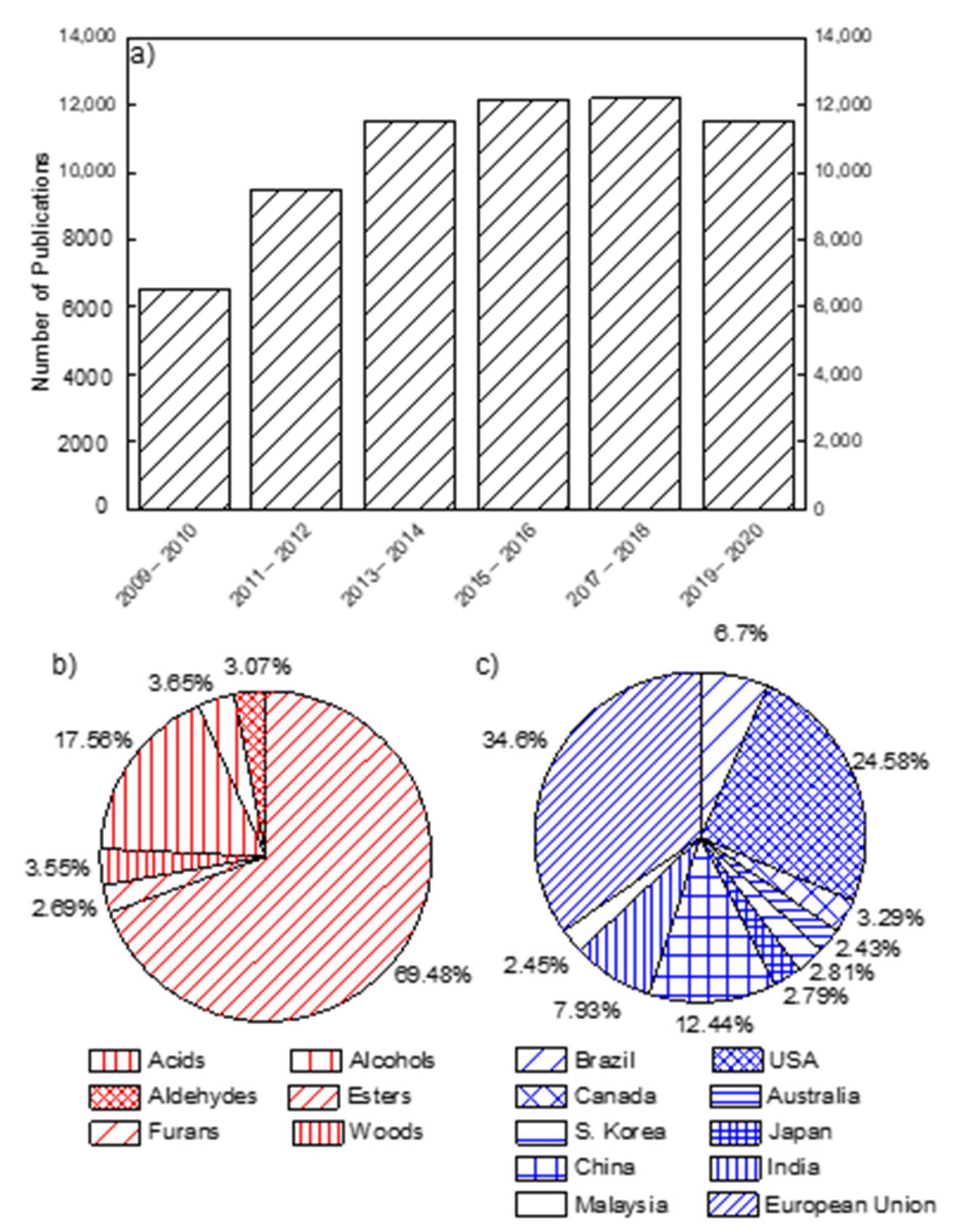
:1. Introduction
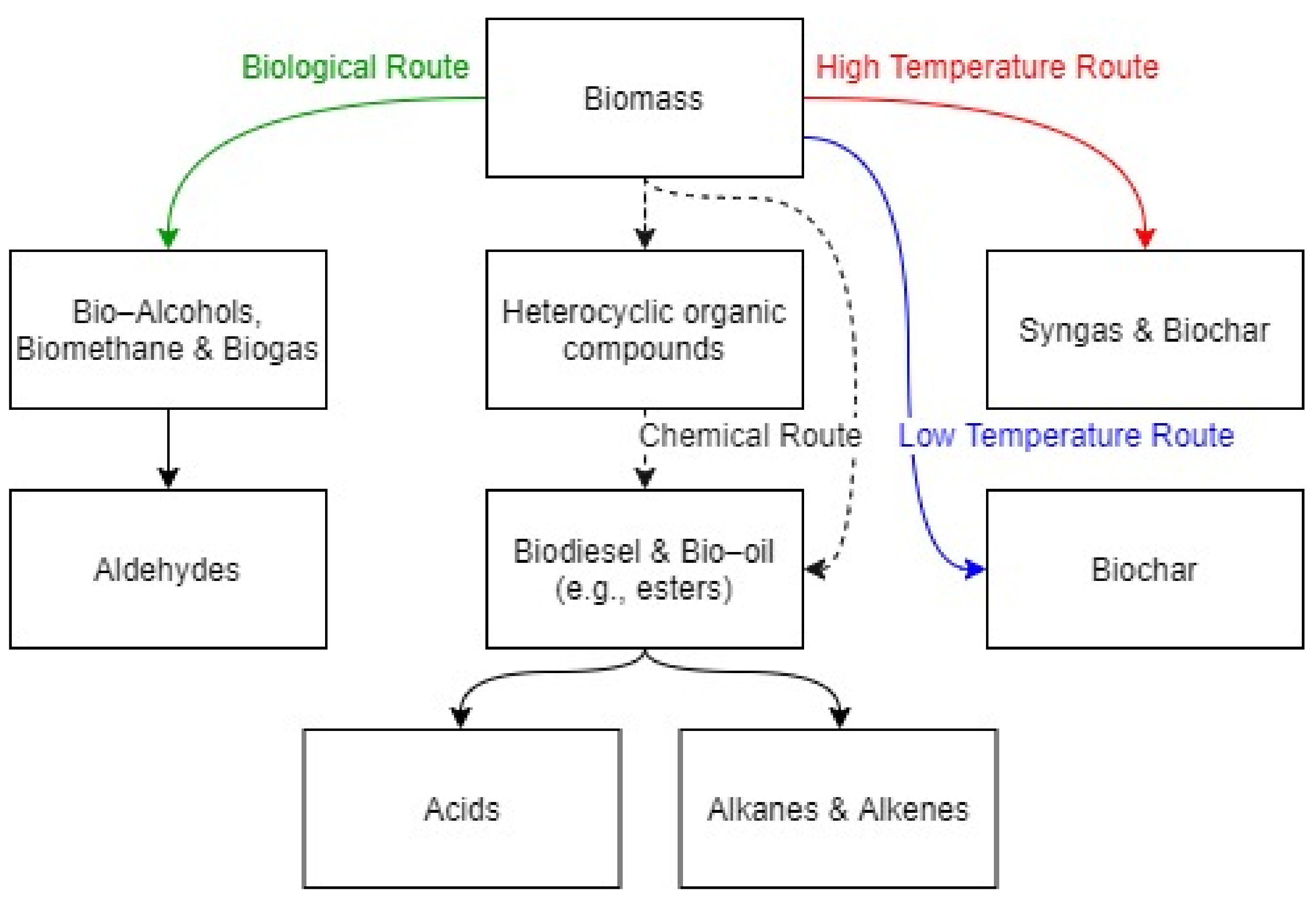
2. Research Metrics of Oxy-Biofuel
3. Combustion Chemistry of Oxy-Biofuels
3.1. Light Alcohols
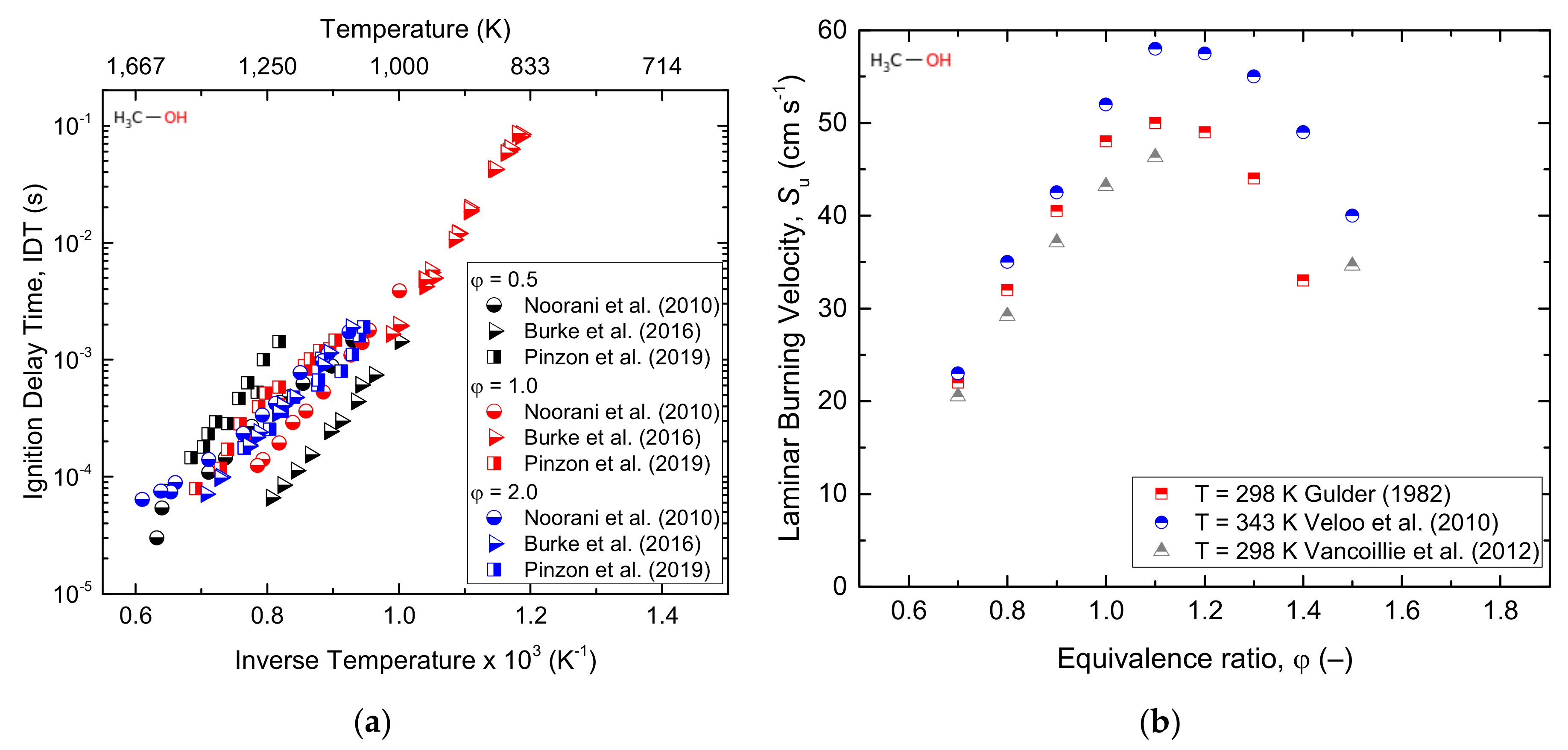
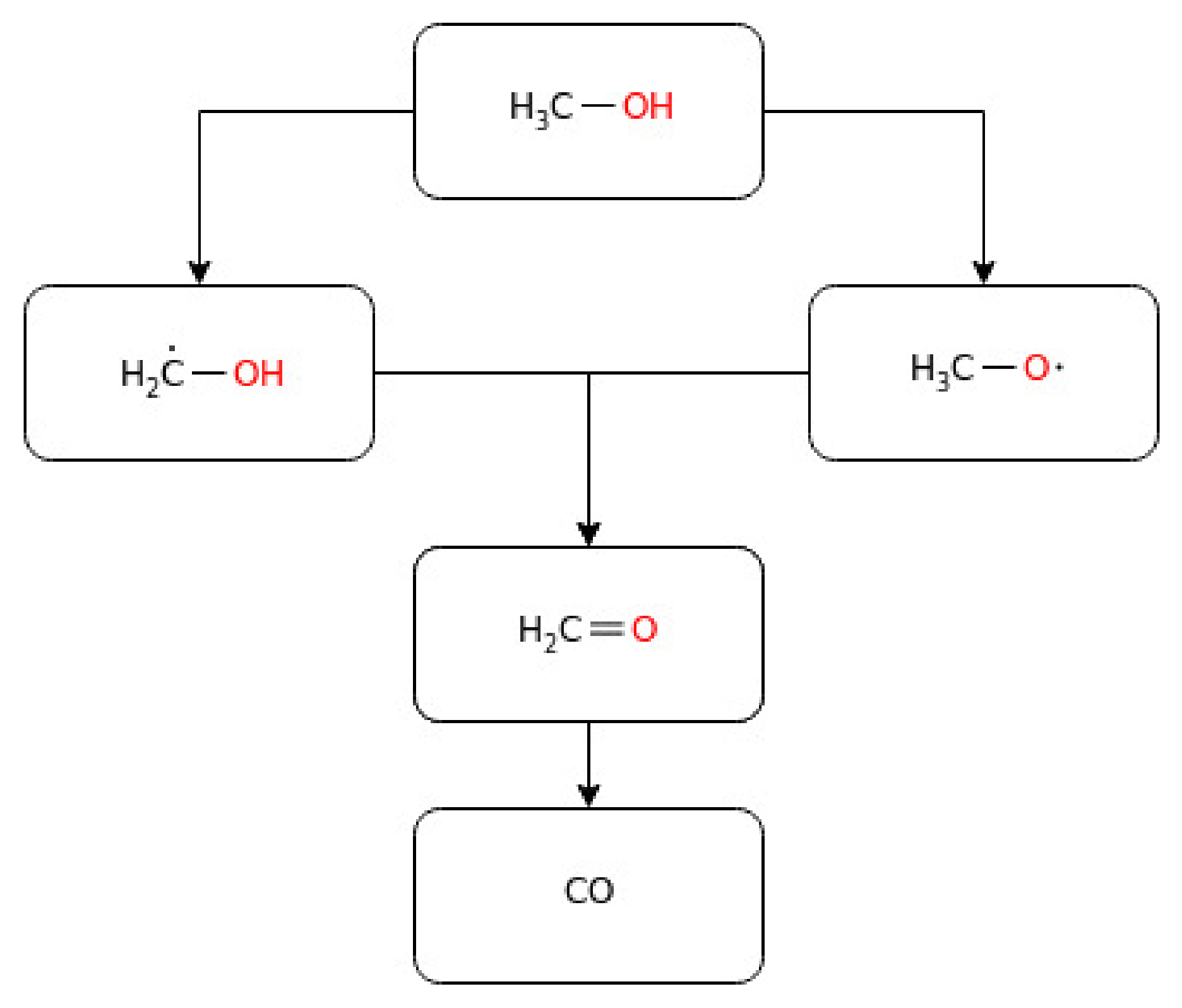
3.1.1. Methanol
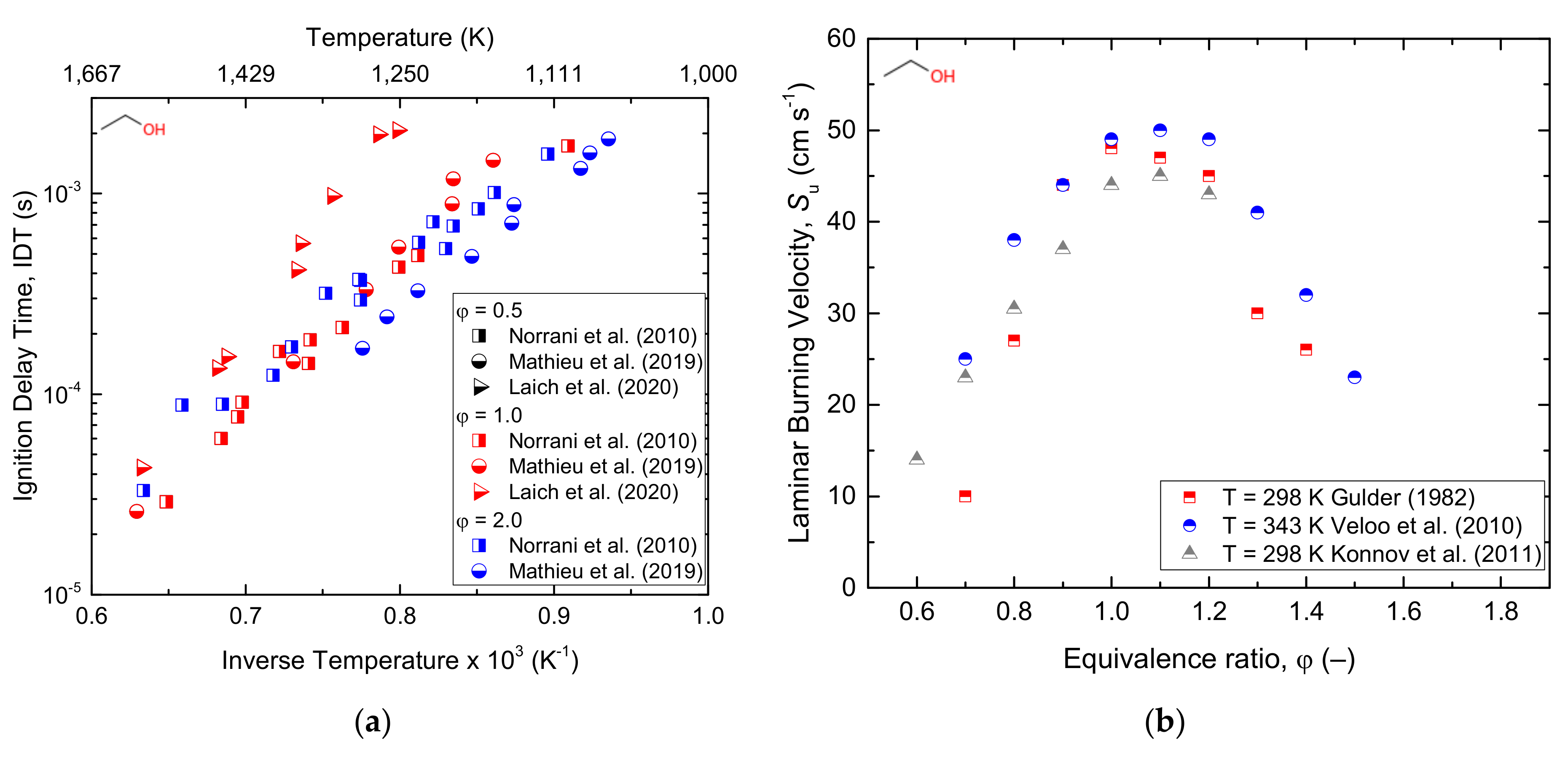
3.1.2. Ethanol
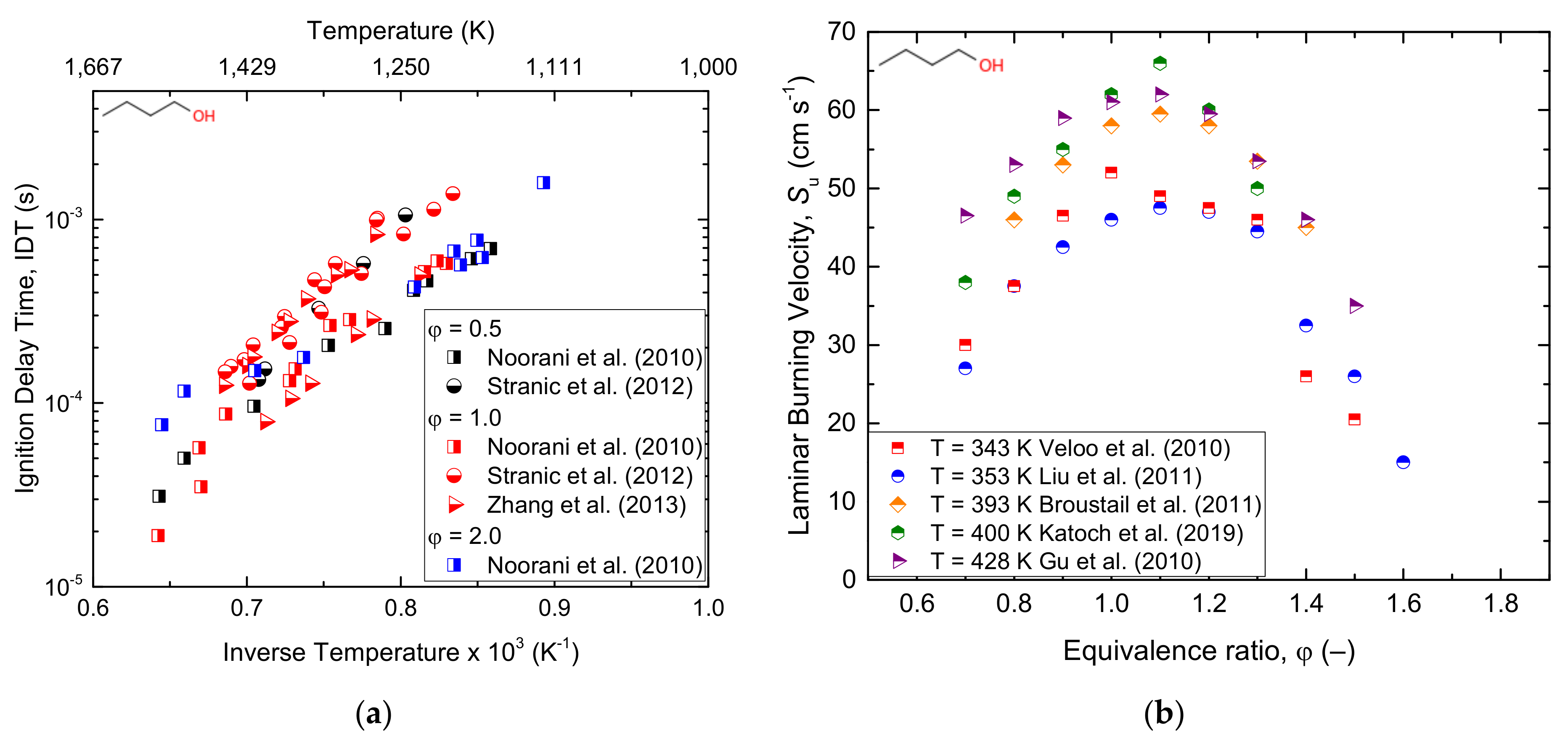
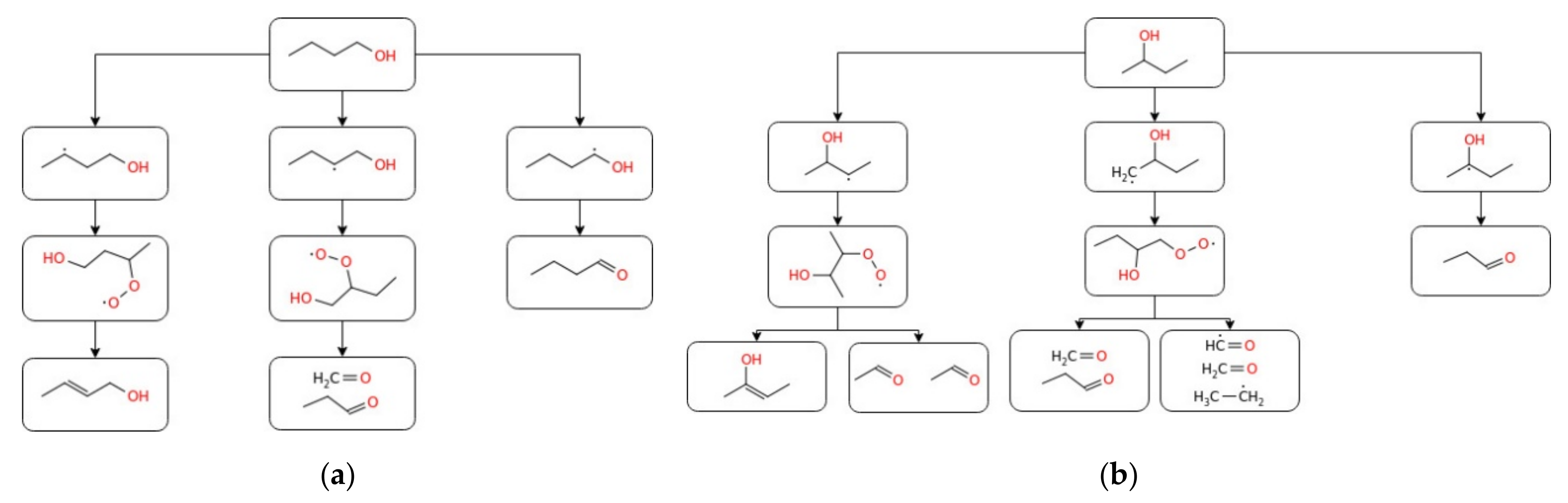
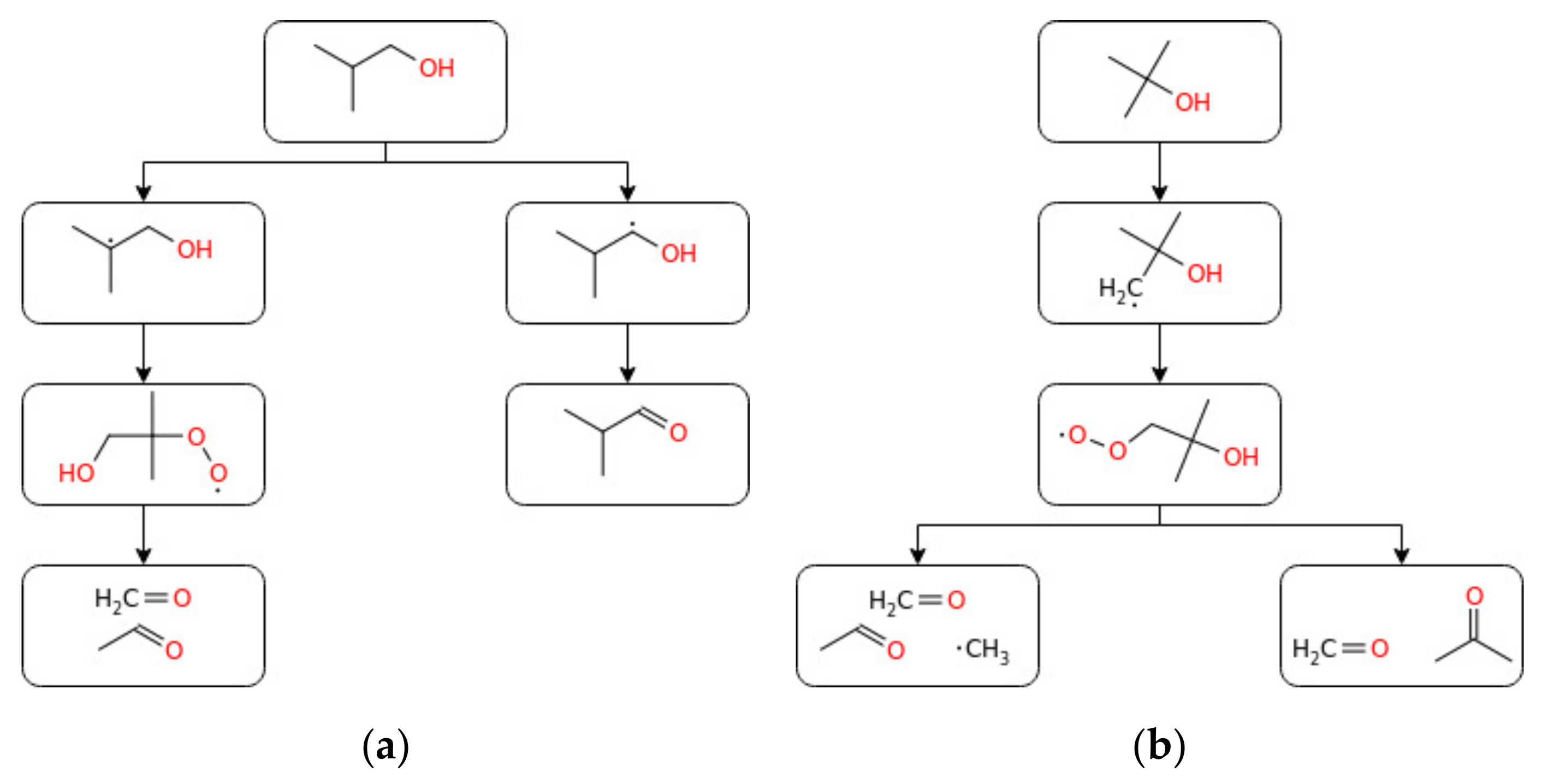
3.1.3. Butanol
3.2. Carboxylic Acids
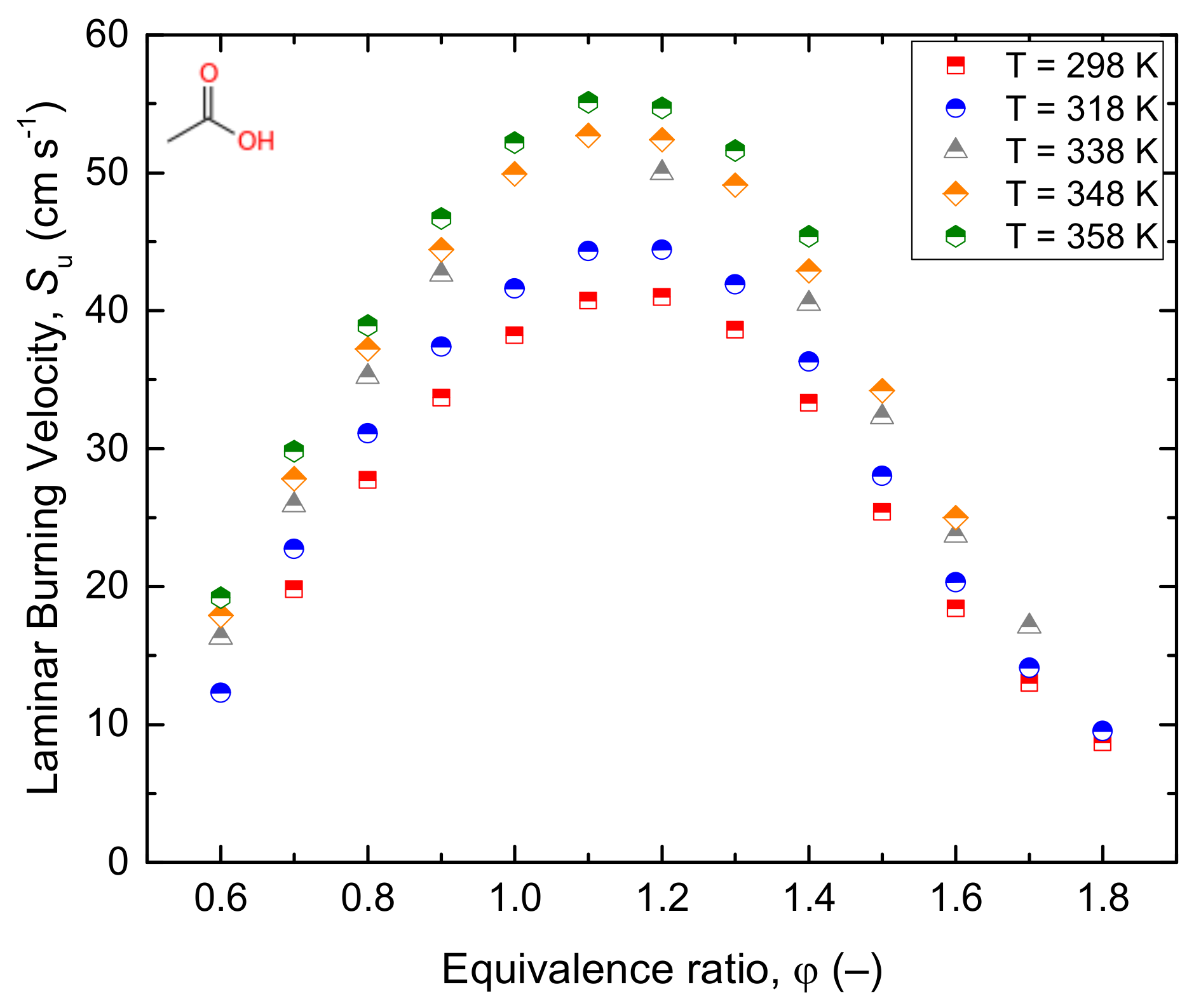
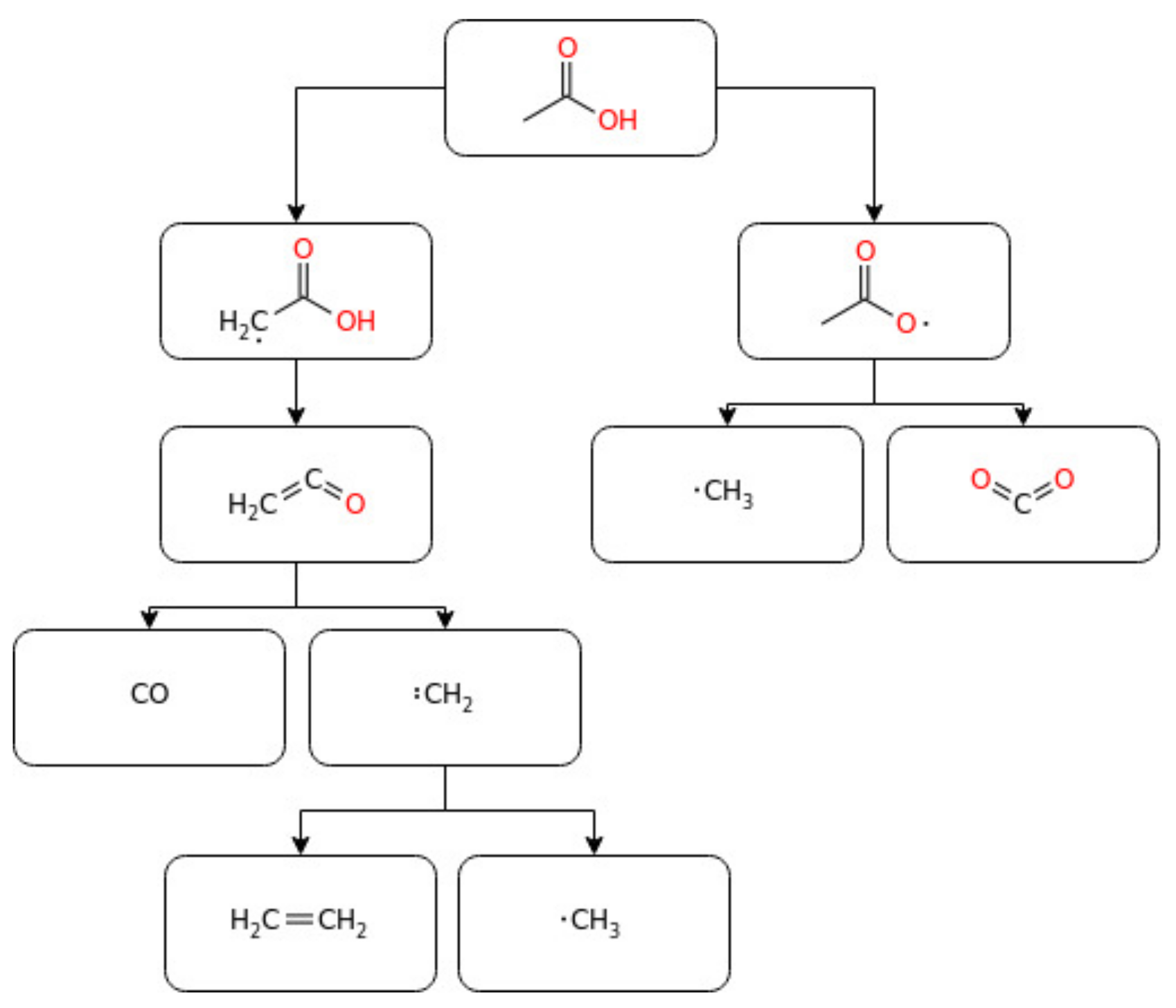
3.2.1. Acetic Acid
3.2.2. Crotonic Acid
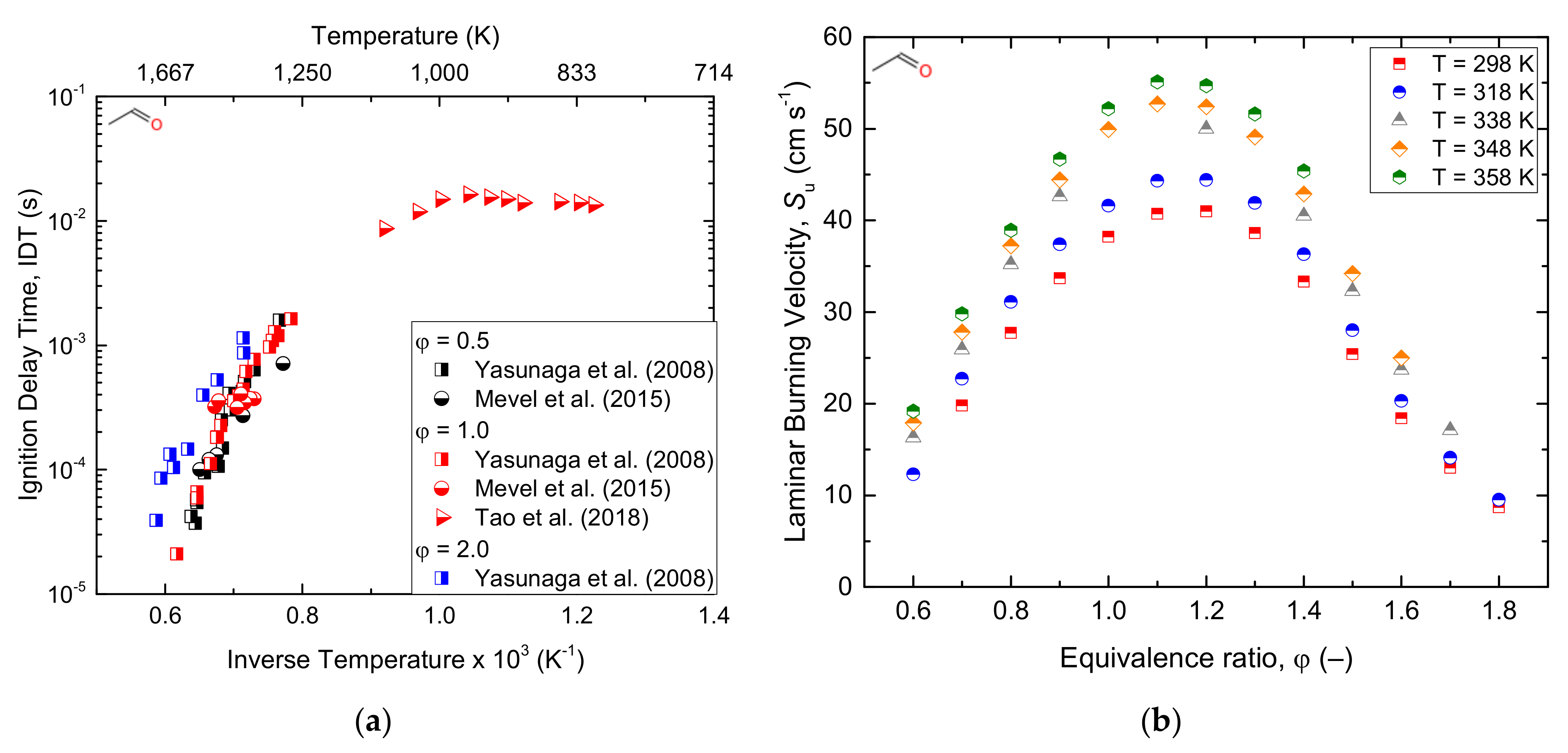
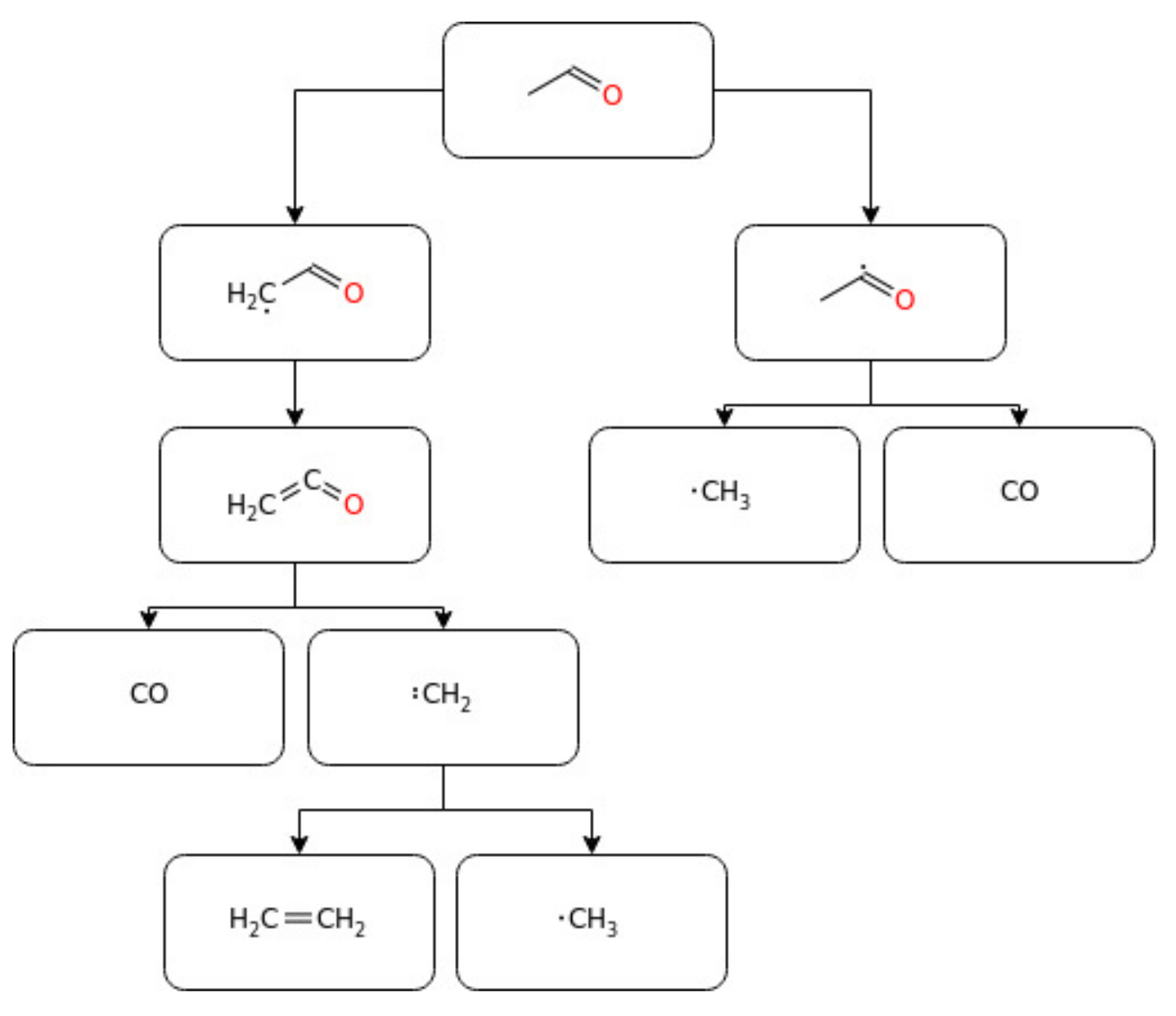
3.3. Light Aldehydes
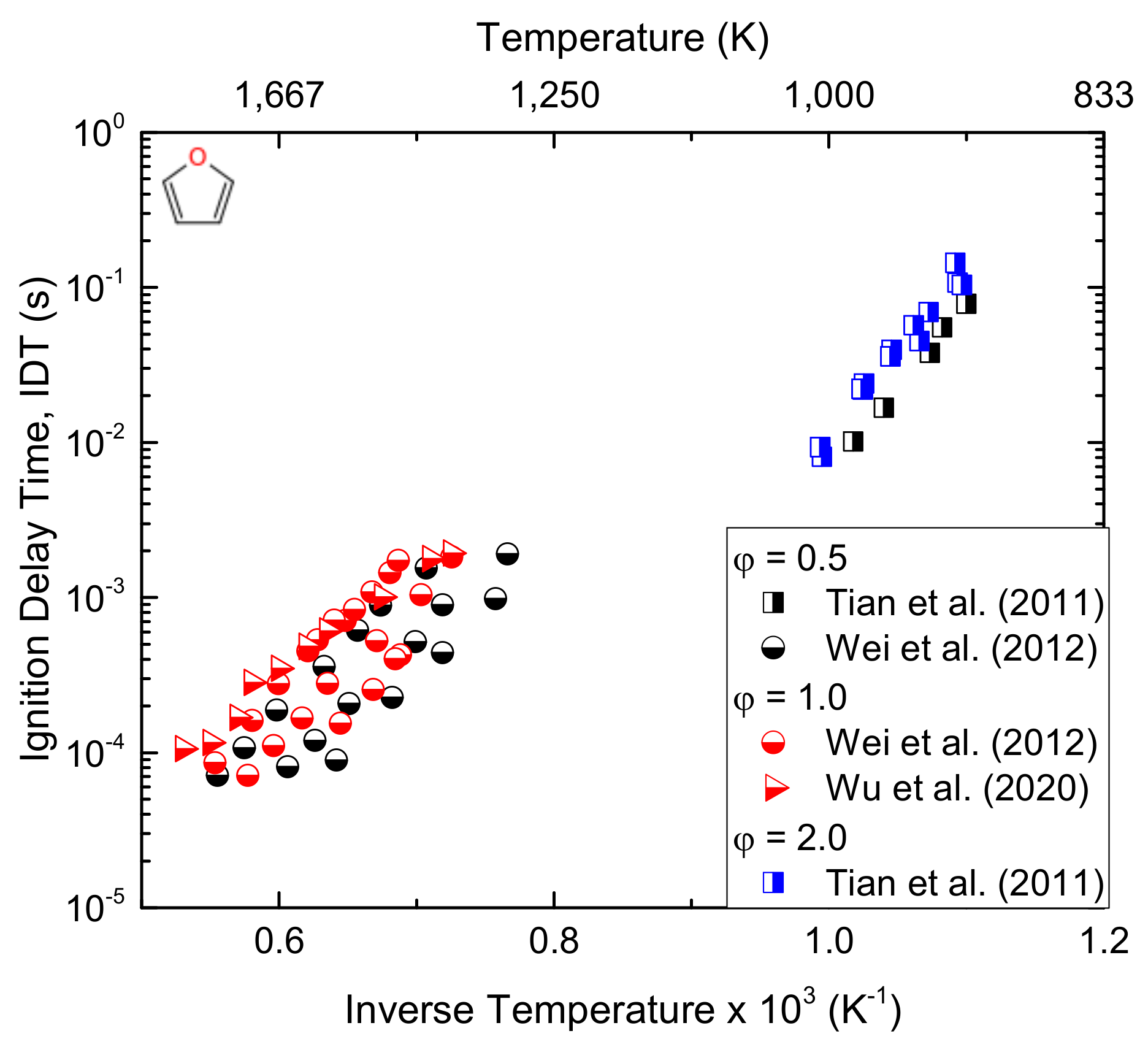
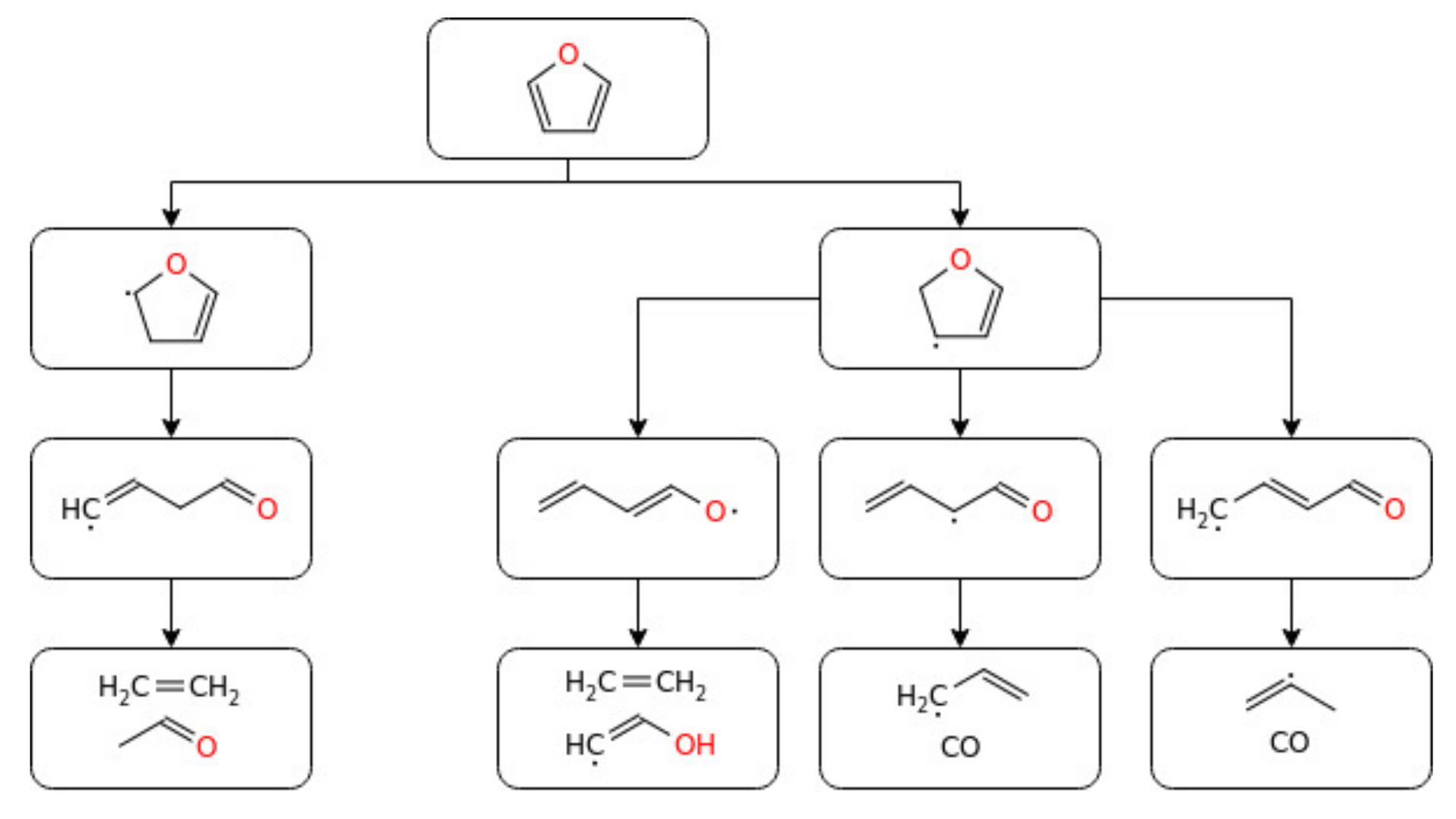
3.4. Heterocyclic Organic Compounds
4. Future Challenges
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Solomon, S.; Qin, D.; Manning, M.; Chen, Z.; Marquis, M.; Averyt, K.B.; Tignor, M.; Miller, H.L. Climate Change 2007—The Physical Science Basis: Working Group I Contribution to the Fourth Assessment Report of the IPCC; Cambridge University Press: Cambridge, MA, USA, 2007; Volume 4. [Google Scholar]
- Yasunaga, K.; Mikajiri, T.; Sarathy, S.M.; Koike, T.; Gillespie, F.; Nagy, T.; Simmie, J.M.; Curran, H.J. A shock tube and chemical kinetic modeling study of the pyrolysis and oxidation of butanols. Combust. Flame 2012, 159, 2009–2027. [Google Scholar] [CrossRef]
- Agarwal, A.K. Biofuels (alcohols and biodiesel) applications as fuels for internal combustion engines. Prog. Energy Combust. Sci. 2007, 33, 233–271. [Google Scholar] [CrossRef]
- Simmie, J.M. Detailed chemical kinetic models for the combustion of hydrocarbon fuels. Prog. Energy Combust. Sci. 2003, 29, 599–634. [Google Scholar] [CrossRef]
- Pelucchi, M.; Bissoli, M.; Rizzo, C.; Zhang, Y.; Somers, K.; Frassoldati, A.; Curran, H.; Faravelli, T. A Kinetic Modelling Study of Alcohols Operating Regimes in a HCCI Engine. SAE Int. J. Engines 2017, 10, 2354–2370. [Google Scholar] [CrossRef] [Green Version]
- Namysl, S.; Pelucchi, M.; Herbinet, O.; Frassoldati, A.; Faravelli, T.; Battin-Leclerc, F. A first evaluation of butanoic and pentanoic acid oxidation kinetics. Chem. Eng. J. 2019, 373, 973–984. [Google Scholar] [CrossRef]
- Pelucchi, M.; Namysl, S.; Ranzi, E.; Rodriguez, A.; Rizzo, C.; Somers, K.P.; Zhang, Y.; Herbinet, O.; Curran, H.J.; Battin-Leclerc, F.; et al. Combustion of n-C3–C6 Linear Alcohols: An Experimental and Kinetic Modeling Study. Part I: Reaction Classes, Rate Rules, Model Lumping, and Validation. Energy Fuels 2020, 34, 14688–14707. [Google Scholar] [CrossRef]
- Veloo, P.S.; Wang, Y.L.; Egolfopoulos, F.; Westbrook, C.K. A comparative experimental and computational study of methanol, ethanol, and n-butanol flames. Combust. Flame 2010, 157, 1989–2004. [Google Scholar] [CrossRef]
- Liu, D.; Togbé, C.; Tran, L.-S.; Felsmann, D.; Oßwald, P.; Nau, P.; Koppmann, J.; Lackner, A.; Glaude, P.-A.; Sirjean, B.; et al. Combustion chemistry and flame structure of furan group biofuels using molecular-beam mass spectrometry and gas chromatography—Part I: Furan. Combust. Flame 2014, 161, 748–765. [Google Scholar] [CrossRef] [Green Version]
- Ubando, A.T.; Africa, A.D.M.; Maniquiz-Redillas, M.C.; Culaba, A.B.; Chen, W.-H. Reduction of particulate matter and volatile organic compounds in biorefineries: A state-of-the-art review. J. Hazard. Mater. 2021, 403, 123955. [Google Scholar] [CrossRef]
- Nugroho, Y.K.; Zhu, L. Platforms planning and process optimization for biofuels supply chain. Renew. Energy 2019, 140, 563–579. [Google Scholar] [CrossRef]
- Huheey, J.; Cottrell, T. The Strengths of Chemical Bonds; Butterworths: London, UK, 1958. [Google Scholar]
- Zhang, X.; Mei, B.; Ma, S.; Zhang, Y.; Cao, C.; Li, W.; Ye, L.; Li, Y. Characterizing the fuel-specific combustion chemistry of acetic acid and propanoic acid: Laminar flame propagation and kinetic modeling studies. Proc. Combust. Inst. 2020, 38, 449–457. [Google Scholar] [CrossRef]
- Christensen, M.; Konnov, A.A. Laminar burning velocity of acetic acid + air flames. Combust. Flame 2016, 170, 12–29. [Google Scholar] [CrossRef]
- Marshall, P.; Glarborg, P. Ab initio and kinetic modeling studies of formic acid oxidation. Proc. Combust. Inst. 2015, 35, 153–160. [Google Scholar] [CrossRef]
- Li, Y.; Strathmann, T.J. Kinetics and mechanism for hydrothermal conversion of polyhydroxybutyrate (PHB) for wastewater valorization. Green Chem. 2019, 21, 5586–5597. [Google Scholar] [CrossRef]
- Fisher, E.; Pitz, W.; Curran, H.; Westbrook, C. Detailed chemical kinetic mechanisms for combustion of oxygenated fuels. Proc. Combust. Inst. 2000, 28, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Dafnomilis, I.; Hoefnagels, R.; Pratama, Y.W.; Schott, D.L.; Lodewijks, G.; Junginger, M. Review of solid and liquid biofuel demand and supply in Northwest Europe towards 2030—A comparison of national and regional projections. Renew. Sustain. Energy Rev. 2017, 78, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Griffin, W.; Saville, B.; MacLean, H. Ethanol Use in the United States: Status, Threats and the Potential Future. In Global Bioethanol; Elsevier BV: Amsterdam, The Netherlands, 2016; pp. 34–62. [Google Scholar]
- Kohse-Höinghaus, K.; Osswald, P.; Cool, T.A.; Kasper, T.; Hansen, N.; Qi, F.; Westbrook, C.K.; Westmoreland, P.R. Biofuel Combustion Chemistry: From Ethanol to Biodiesel. Angew. Chem. Int. Ed. 2010, 49, 3572–3597. [Google Scholar] [CrossRef]
- Saggese, C.; Frassoldati, A.; Cuoci, A.; Faravelli, T.; Ranzi, E. A wide range kinetic modeling study of pyrolysis and oxidation of benzene. Combust. Flame 2013, 160, 1168–1190. [Google Scholar] [CrossRef]
- Cossu, R.; Liu, J.; Pivato, A.; Ragazzi, M. Biomass to biofuels: Challenges and opportunities. Renew. Energy 2020, 158, 1–2. [Google Scholar] [CrossRef]
- Ruscic, B. Active Thermochemical Tables: Sequential Bond Dissociation Enthalpies of Methane, Ethane, and Methanol and the Related Thermochemistry. J. Phys. Chem. A 2015, 119, 7810–7837. [Google Scholar] [CrossRef] [Green Version]
- Pelucchi, M.; Cavallotti, C.; Ranzi, E.; Frassoldati, A.; Faravelli, T. Relative Reactivity of Oxygenated Fuels: Alcohols, Aldehydes, Ketones, and Methyl Esters. Energy Fuels 2016, 30, 8665–8679. [Google Scholar] [CrossRef] [Green Version]
- Nau, P.; Seipel, A.; Lucassen, A.; Brockhinke, A.; Kohse-Höinghaus, K. Intermediate species detection in a morpholine flame: Contributions to fuel-bound nitrogen conversion from a model biofuel. Exp. Fluids 2010, 49, 761–773. [Google Scholar] [CrossRef]
- Pelucchi, M.; Namysl, S.; Ranzi, E.; Frassoldati, A.; Herbinet, O.; Battin-Leclerc, F.; Faravelli, T. An experimental and kinetic modelling study of n-C4C6 aldehydes oxidation in a jet-stirred reactor. Proc. Combust. Inst. 2019, 37, 389–397. [Google Scholar] [CrossRef]
- Pelucchi, M.; Ranzi, E.; Frassoldati, A.; Faravelli, T. Alkyl radicals rule the low temperature oxidation of long chain aldehydes. Proc. Combust. Inst. 2017, 36, 393–401. [Google Scholar] [CrossRef]
- Pelucchi, M.; Somers, K.P.; Yasunaga, K.; Burke, U.; Frassoldati, A.; Ranzi, E.; Curran, H.J.; Faravelli, T. An experimental and kinetic modeling study of the pyrolysis and oxidation of n-C3C5 aldehydes in shock tubes. Combust. Flame 2015, 162, 265–286. [Google Scholar] [CrossRef] [Green Version]
- Veloo, P.; Dagaut, P.; Togbe, C.; Dayma, G.; Sarathy, S.; Westbrook, C.; Egolfopoulos, F. Jet-stirred reactor and flame studies of propanal oxidation. Proc. Combust. Inst. 2013, 34, 599–606. [Google Scholar] [CrossRef]
- Pelucchi, M.; Cavallotti, C.; Cuoci, A.; Faravelli, T.; Frassoldati, A.; Ranzi, E. Detailed kinetics of substituted phenolic species in pyrolysis bio-oils. React. Chem. Eng. 2019, 4, 490–506. [Google Scholar] [CrossRef] [Green Version]
- Namysl, S.; Pelucchi, M.; Maffei, L.P.; Herbinet, O.; Stagni, A.; Faravelli, T.; Battin-Leclerc, F. Experimental and modeling study of benzaldehyde oxidation. Combust. Flame 2020, 211, 124–132. [Google Scholar] [CrossRef]
- Maffei, L.P.; Pelucchi, M.; Faravelli, T.; Cavallotti, C. Theoretical study of sensitive reactions in phenol decomposition. React. Chem. Eng. 2020, 5, 452–472. [Google Scholar] [CrossRef]
- Coniglio, L.; Bennadji, H.; Glaude, P.-A.; Herbinet, O.; Billaud, F. Combustion chemical kinetics of biodiesel and related compounds (methyl and ethyl esters): Experiments and modeling—Advances and future refinements. Prog. Energy Combust. Sci. 2013, 39, 340–382. [Google Scholar] [CrossRef] [Green Version]
- Akrich, R.; Vovelle, C.; Delbourgo, R. Flame profiles and combustion mechanisms of methanol-air flames under reduced pressure. Combust. Flame 1978, 32, 171–179. [Google Scholar] [CrossRef]
- Chen, C.-C.; Liaw, H.-J.; Shu, C.-M.; Hsieh, Y.-C. Autoignition Temperature Data for Methanol, Ethanol, Propanol, 2-Butanol, 1-Butanol, and 2-Methyl-2,4-pentanediol. J. Chem. Eng. Data 2010, 55, 5059–5064. [Google Scholar] [CrossRef]
- Dagaut, P.; Sarathy, S.M.; Thomson, M. A chemical kinetic study of n-butanol oxidation at elevated pressure in a jet stirred reactor. Proc. Combust. Inst. 2009, 32, 229–237. [Google Scholar] [CrossRef]
- Cavallotti, C.; Pelucchi, M.; Frassoldati, A. Analysis of acetic acid gas phase reactivity: Rate constant estimation and kinetic sim-ulations. Proc. Combust. Inst. 2019, 37, 539–546. [Google Scholar] [CrossRef]
- Wilhoit, R.C.; Chao, J.; Hall, K.R. Thermodynamic Properties of Key Organic Oxygen Compounds in the Carbon Range C1 to C4. Part 1. Properties of Condensed Phases. J. Phys. Chem. Ref. Data 1985, 14, 1–175. [Google Scholar] [CrossRef]
- Li, J.; Kazakov, A.; Chaos, M.; Dryer, F.L. Chemical kinetics of ethanol oxidation. In Proceedings of the 5th US Combustion Meeting, San Diego, CA, USA, 25–28 March 2007. [Google Scholar]
- Zhang, X.; Ye, L.; Li, Y.; Zhang, Y.; Cao, C.; Yang, J.; Zhongyue, Z.; Zhen, H.; Fei, Q. Acetaldehyde oxidation at low and intermediate temperatures: An experimental and kinetic modeling investigation. Combust. Flame 2018, 191, 431–441. [Google Scholar] [CrossRef]
- Christensen, E.; Fioroni, G.M.; Kim, S.; Fouts, L.; Gjersing, E.; Paton, R.; McCormick, R. Experimental and theoretical study of oxidative stability of alkylated furans used as gasoline blend components. Fuel 2018, 212, 576–585. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Akih-Kumgeh, B. Recent Trends in the Production, Combustion and Modeling of Furan-Based Fuels. Energies 2018, 11, 512. [Google Scholar] [CrossRef] [Green Version]
- Bowman, C.T. A shock-tube investigation of the high-temperature oxidation of methanol. Combust. Flame 1975, 25, 343–354. [Google Scholar] [CrossRef]
- Kumar, K.; Sung, C.-J. Autoignition of methanol: Experiments and computations. Int. J. Chem. Kinet. 2011, 43, 175–184. [Google Scholar] [CrossRef]
- Noorani, K.E.; Akih-Kumgeh, B.; Bergthorson, J. Comparative High Temperature Shock Tube Ignition of C1−C4 Primary Alcohols. Energy Fuels 2010, 24, 5834–5843. [Google Scholar] [CrossRef]
- Pinzón, L.; Mathieu, O.; Mulvihill, C.; Schoegl, I.; Petersen, E. Ignition delay time and H2O measurements during methanol oxidation behind reflected shock waves. Combust. Flame 2019, 203, 143–156. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, Y.; Liu, W.; Wang, Z. Investigation of methanol ignition phenomena using a rapid compression machine. Combust. Flame 2020, 211, 147–157. [Google Scholar] [CrossRef]
- Gülder, Ö.L. Laminar burning velocities of methanol, ethanol and isooctane-air mixtures. Symp. Int. Combust. 1982, 19, 275–281. [Google Scholar] [CrossRef]
- Xiao, P.; Lee, C.-F.; Wu, H.; Liu, F. Effects of hydrogen addition on the laminar methanol-air flame under different initial temperatures. Renew. Energy 2020, 154, 209–222. [Google Scholar] [CrossRef]
- Botet, C.B.; Wagnon, S.; Wooldridge, M.S. Combustion Chemistry of Ethanol: Ignition and Speciation Studies in a Rapid Compression Facility. J. Phys. Chem. A 2016, 120, 7408–7418. [Google Scholar] [CrossRef]
- Laich, A.R.; Ninnemann, E.; Neupane, S.; Rahman, R.; Barak, S.; Pitz, W.J.; Vasu, S.S. High-pressure shock tube study of ethanol oxidation: Ignition delay time and CO time-history measurements. Combust. Flame 2020, 212, 486–499. [Google Scholar] [CrossRef]
- Mathieu, O.; Pinzón, L.T.; Atherley, T.M.; Mulvihill, C.R.; Schoel, I.; Petersen, E.L. Experimental study of ethanol oxidation behind reflected shock waves: Ignition delay time and H2O laser-absorption measurements. Combust. Flame 2019, 208, 313–326. [Google Scholar] [CrossRef]
- Wadkar, C.; Chinnathambi, P.; Toulson, E. Analysis of rapid compression machine facility effects on the auto-ignition of ethanol. Fuel 2020, 264, 116546. [Google Scholar] [CrossRef]
- Leplat, N.; Dagaut, P.; Togbé, C.; Vandooren, J. Numerical and experimental study of ethanol combustion and oxidation in laminar premixed flames and in jet-stirred reactor. Combust. Flame 2011, 158, 705–725. [Google Scholar] [CrossRef]
- Konnov, A.; Meuwissen, R.; de Goey, L. The temperature dependence of the laminar burning velocity of ethanol flames. Proc. Combust. Inst. 2011, 33, 1011–1019. [Google Scholar] [CrossRef]
- Van Treek, L.; Lavadera, M.L.; Seidel, L.; Mauss, F.; Konnov, A.A. Experimental and modelling study of laminar burning velocity of aqueous ethanol. Fuel 2019, 257, 116069. [Google Scholar] [CrossRef]
- Xu, C.; Wang, H.; Zhou, K.; Li, X.; Zhou, W.; Liu, W.; Wang, C. Laminar Burning Velocity of Premixed Ethanol–Air Mixtures with Laser-Induced Spark Ignition Using the Constant-Volume Method. Energy Fuels 2019, 33, 7749–7758. [Google Scholar] [CrossRef]
- Xu, C.; Hu, Y.; Li, X.; Zhou, X.; Zhong, A. Comparative experimental study of ethanol-air premixed laminar combustion characteristics by laser induced spark and electric spark ignition. Korean J. Chem. Eng. 2017, 34, 574–579. [Google Scholar] [CrossRef]
- Katoch, A.; Millán-Merino, A.; Kumar, S. Measurement of laminar burning velocity of ethanol-air mixtures at elevated temperatures. Fuel 2018, 231, 37–44. [Google Scholar] [CrossRef]
- Stranic, I.; Chase, D.P.; Harmon, J.T.; Yang, S.; Davidson, D.F.; Hanson, R.K. Shock tube measurements of ignition delay times for the butanol isomers. Combust. Flame 2012, 159, 516–527. [Google Scholar] [CrossRef]
- Hantouche, M.; Sarathy, S.M.; Knio, O.M. Global sensitivity analysis of n-butanol ignition delay times to thermodynamics class and rate rule parameters. Combust. Flame 2020, 222, 355–369. [Google Scholar] [CrossRef]
- Gu, X.; Huang, Z.; Wu, S.; Li, Q. Laminar burning velocities and flame instabilities of butanol isomers–air mixtures. Combust. Flame 2010, 157, 2318–2325. [Google Scholar] [CrossRef]
- Katoch, A.; Alfazazi, A.; Sarathy, S.M.; Kumar, S. Experimental and numerical investigations on the laminar burning velocity of n-butanol + air mixtures at elevated temperatures. Fuel 2019, 249, 36–44. [Google Scholar] [CrossRef]
- Talukder, N.; Lee, K.Y. Laminar flame speeds for n-butanol/air mixtures at elevated pressures and temperatures: An experimental and numerical study. J. Mech. Sci. Technol. 2018, 32, 1827–1834. [Google Scholar] [CrossRef]
- Broustail, G.; Seers, P.; Halter, F.; Moréac, G.; Mounaim-Rousselle, C. Experimental determination of laminar burning velocity for butanol and ethanol iso-octane blends. Fuel 2011, 90, 1–6. [Google Scholar] [CrossRef]
- Broustail, G.; Halter, F.; Seers, P.; Moréac, G.; Mounaïm-Rousselle, C. Experimental determination of laminar burning velocity for butanol/iso-octane and ethanol/iso-octane blends for different initial pressures. Fuel 2013, 106, 310–317. [Google Scholar] [CrossRef]
- Sarathy, S.M.; Thomson, M.; Togbé, C.; Dagaut, P.; Halter, F.; Mounaim-Rousselle, C. An experimental and kinetic modeling study of n-butanol combustion. Combust. Flame 2009, 156, 852–864. [Google Scholar] [CrossRef]
- Mackie, J.; Doolan, K.R. High-temperature kinetics of thermal decomposition of acetic acid and its products. Int. J. Chem. Kinet. 1984, 16, 525–541. [Google Scholar] [CrossRef]
- Wagner, H.G.; Zabel, F. Zum thermischen Zerfall von Keten in der Gasphase. Ber. Bunsenges. Phys. Chem. 1971, 75, 114–118. [Google Scholar] [CrossRef]
- Leplat, N.; Vandooren, J. Numerical and experimental study of the combustion of acetic acid in laminar premixed flames. Combust. Flame 2012, 159, 493–499. [Google Scholar] [CrossRef]
- Yasunaga, K.; Kubo, S.; Hoshikawa, H.; Kamesawa, T.; Hidaka, Y. Shock-tube and modeling study of acetaldehyde pyrolysis and oxidation. Int. J. Chem. Kinet. 2008, 40, 73–102. [Google Scholar] [CrossRef]
- Mével, R.; Chatelain, K.; Catoire, L.; Green, W.H.; Shepherd, J.E. Chemical Kinetics of Acetaldehyde Pyrolysis and Oxidation. In Proceedings of the 9th US National Combustion Meeting, Cincinnati, OH, USA, 17–20 May 2015. [Google Scholar]
- Tao, T.; Kang, S.; Sun, W.; Wang, J.; Liao, H.; Moshammer, K.; Hansen, N.; Law, C.K.; Yang, B. A further experimental and modeling study of acetaldehyde combustion kinetics. Combust. Flame 2018, 196, 337–350. [Google Scholar] [CrossRef]
- Wei, L.; Tang, C.; Man, X.; Jiang, X.; Huang, Z. High-Temperature Ignition Delay Times and Kinetic Study of Furan. Energy Fuels 2012, 26, 2075–2081. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, N.; Yang, M.; Liu, Y.; Tang, C.; Huang, Z. Ignition delay time measurement and kinetic modeling of furan, and comparative studies of 2,3-dihydrofuran and tetrahydrofuran at low to intermediate temperatures by using a rapid compression machine. Combust. Flame 2020, 213, 226–236. [Google Scholar] [CrossRef]
- Tian, Z.; Yuan, T.; Fournet, R.; Glaude, P.-A.; Sirjean, B.; Battin-Leclerc, F.; Zhang, K.; Qi, F. An experimental and kinetic investigation of premixed furan/oxygen/argon flames. Combust. Flame 2011, 158, 756–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, J.; Liu, H.; Zheng, Z. Chemical Kinetics Study on Combustion of Ethanol/biodiesel/n-heptane. Renew. Energy 2020, 148, 150–167. [Google Scholar] [CrossRef]
- Gainey, B.; Lawler, B. The role of alcohol biofuels in advanced combustion: An analysis. Fuel 2021, 283, 118915. [Google Scholar] [CrossRef]
- Kwon, H.; Lapointe, S.; Zhang, K.; Wagnon, S.W.; Pitz, W.J.; Zhu, J.; McEnally, C.S.; Pfefferle, L.D.; Xuan, Y. Sooting tendencies of 20 bio-derived fuels for advanced spark-ignition engines. Fuel 2020, 276, 118059. [Google Scholar] [CrossRef]
- Pelucchi, M.; Namysl, S.; Ranzi, E.; Rodriguez, A.; Rizzo, C.; Somers, K.P.; Zhang, Y.; Herbinet, O.; Curran, H.J.; Faravelli, T. Combustion of n-C3–C6 Linear Alcohols: An Experimental and Kinetic Modeling Study. Part II: Speciation Measurements in a Jet-Stirred Reactor, Ignition Delay Time Measurements in a Rapid Compression Machine, Model Validation, and Kinetic Analysis. Energy Fuels 2020, 34, 14708–14725. [Google Scholar] [CrossRef]
- Mack, J.H.; Schuler, D.; Butt, R.H.; Dibble, R. Experimental investigation of butanol isomer combustion in Homogeneous Charge Compression Ignition (HCCI) engines. Appl. Energy 2016, 165, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Svensson, E.; Tuner, M.; Verhelst, S. Influence of Injection Strategies on Engine Efficiency for a Methanol PPC Engine. SAE Tech. Pap. Ser. 2019, 2, 653–671. [Google Scholar] [CrossRef]
- Togbé, C.; Halter, F.; Foucher, F.; Mounaim-Rousselle, C.; Dagaut, P. Experimental and detailed kinetic modeling study of 1-pentanol oxidation in a JSR and combustion in a bomb. Proc. Combust. Inst. 2011, 33, 367–374. [Google Scholar] [CrossRef]
- Togbé, C.; Dagaut, P.; Mzé-Ahmed, A.; Diévart, P.; Halter, F.; Foucher, F. Experimental and Detailed Kinetic Modeling Study of 1-Hexanol Oxidation in a Pressurized Jet-Stirred Reactor and a Combustion Bomb. Energy Fuels 2010, 24, 5859–5875. [Google Scholar] [CrossRef]
- Fieweger, K.; Blumenthal, R.; Adomeit, G. Self-ignition of S.I. engine model fuels: A shock tube investigation at high pressure. Combust. Flame 1997, 109, 599–619. [Google Scholar] [CrossRef]
- Cathonnet, M.; Boettner, J.C.; James, H. Étude expérimentale et simulation de la pyrolyse du méthanol. J. Chim. Phys. Phys. Chim. Biol. 1979, 76, 183–189. [Google Scholar] [CrossRef]
- Aniolek, K.W.; Wilk, R.D. Preflame Oxidation Characteristics of Methanol. Energy Fuels 1995, 9, 395–405. [Google Scholar] [CrossRef]
- Egolfopoulos, F.N.; Du, D.; Law, C. A comprehensive study of methanol kinetics in freely-propagating and burner-stabilized flames, flow and static reactors, and shock tubes. Combust. Sci. Technol. 1992, 83, 33–75. [Google Scholar] [CrossRef]
- Liao, S.; Jiang, D.; Huang, Z.; Shen, W.; Yuan, C.; Cheng, Q. Laminar burning velocities for mixtures of methanol and air at elevated temperatures. Energy Convers. Manag. 2007, 48, 857–863. [Google Scholar] [CrossRef]
- Vancoillie, J.; Christensen, M.; Nilsson, E.J.K.; Verhelst, S.; Konnov, A.A. Temperature Dependence of the Laminar Burning Velocity of Methanol Flames. Energy Fuels 2012, 26, 1557–1564. [Google Scholar] [CrossRef] [Green Version]
- Burke, U.; Metcalfe, W.K.; Burke, S.M.; Heufer, K.A.; Dagaut, P.; Curran, H.J. A detailed chemical kinetic modeling, ignition delay time and jet-stirred reactor study of methanol oxidation. Combust. Flame 2016, 165, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Westbrook, C.K.; Dryer, F.L. Comprehensive Mechanism for Methanol Oxidation. Combust. Sci. Technol. 1979, 20, 125–140. [Google Scholar] [CrossRef]
- Norton, T.S.; Dryer, F.L. Some New Observations on Methanol Oxidation Chemistry. Combust. Sci. Technol. 1989, 63, 107–129. [Google Scholar] [CrossRef]
- Grotheer, H.; Kelm, S. Elementary Reactions in the Methanol Oxidation System. Implications on the Modeling of Laminar Burning Velocities. In Proceedings of the III International Seminar on Flame Structure, Alma-Ata, Kazakhstan, 18–22 September 1989. [Google Scholar]
- Grotheer, H.-H.; Kelm, S.; Driver, H.S.T.; Hutcheon, R.J.; Lockett, R.D.; Robertson, G.N. Elementary Reactions in the Methanol Oxidation System. Part I: Establishment of the Mechanism and Modelling of Laminar Burning Velocities. Ber. Bunsenges. Phys. Chem. 1992, 96, 1360–1376. [Google Scholar] [CrossRef]
- Aranda, V.; Christensen, J.M.; Alzueta, M.U.; Glarborg, P.; Gersen, S.; Gao, Y.; Marshall, P. Experimental and Kinetic Modeling Study of Methanol Ignition and Oxidation at High Pressure. Int. J. Chem. Kinet. 2013, 45, 283–294. [Google Scholar] [CrossRef]
- Cancino, L.R.; Oliveira, A.A.M.; Friki, M.; Schulz, C. Thermal Oxidation of Ethanol: Experimental and Numerical Analysis of Ignition Chemistry of Ethanol-Air Mixtures in Shock Heated Gases. In Proceedings of the Twenty Seventh International Symposium in Shock Waves, St. Petersburg, Russia, 19–24 July 2009. [Google Scholar]
- Cancino, L.; Fikri, M.; Oliveira, A.; Schulz, C. Ignition delay times of ethanol-containing multi-component gasoline surrogates: Shock-tube experiments and detailed modeling. Fuel 2011, 90, 1238–1244. [Google Scholar] [CrossRef]
- Aghsaee, M.; Nativel, D.; Bozkurt, M.; Fikri, M.; Chaumeix, N.; Schulz, C. Experimental study of the kinetics of ethanol pyrolysis and oxidation behind reflected shock waves and in laminar flames. Proc. Combust. Inst. 2015, 35, 393–400. [Google Scholar] [CrossRef]
- Lee, C.; Vranckx, S.; Heufer, K.A.; Khomik, S.V.; Uygun, Y.; Olivier, H.; Fernandez, R.X. On the Chemical Kinetics of Ethanol Oxidation: Shock Tube, Rapid Compression Machine and Detailed Modeling Study. Z. Phys. Chem. 2012, 226, 1–28. [Google Scholar] [CrossRef]
- Mittal, G.; Burke, S.M.; Davies, V.A.; Parajuli, B.; Metcalfe, W.K.; Curran, H.J. Autoignition of ethanol in a rapid compression machine. Combust. Flame 2014, 161, 1164–1171. [Google Scholar] [CrossRef] [Green Version]
- Van Lipzig, J.; Nilsson, E.; de Goey, L.; Konnov, A. Laminar burning velocities of n-heptane, iso-octane, ethanol and their binary and tertiary mixtures. Fuel 2011, 90, 2773–2781. [Google Scholar] [CrossRef]
- Dirrenberger, P.; Glaude, P.; Bounaceur, R.; Le Gall, H.; da Cruz, A.P.; Konnov, A.; Battin-Leclerc, F. Laminar burning velocity of gasolines with addition of ethanol. Fuel 2014, 115, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Vagelopoulos, C.; Egolfopoulos, F.; Law, C. Further considerations on the determination of laminar flame speeds with the counterflow twin-flame technique. Symp. Int. Combust. 1994, 25, 1341–1347. [Google Scholar] [CrossRef]
- Fiala, T.; Sattelmayer, T. Nonpremixed Counterflow Flames: Scaling Rules for Batch Simulations. J. Combust. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, H.; Zheng, Z.; Liu, J.; Reitz, R.D.; Yao, M. Development of a combined reduced primary reference fuel-alcohols (methanol/ethanol/propanols/butanols/n-pentanol) mechanism for engine applications. Energy 2016, 114, 542–558. [Google Scholar] [CrossRef]
- Hinton, N.; Stone, R.; Cracknell, R.; Olm, C. Aqueous ethanol laminar burning velocity measurements using constant volume bomb methods. Fuel 2018, 214, 127–134. [Google Scholar] [CrossRef]
- Dunphy, M.P.; Simmie, J.M. High-temperature oxidation of ethanol. Part 1. Ignition delays in shock waves. J. Chem. Soc. Faraday Trans. 1991, 87, 1691–1696. [Google Scholar] [CrossRef]
- Marinov, N.M. A detailed chemical kinetic model for high temperature ethanol oxidation. Int. J. Chem. Kinet. 1999, 31, 183–220. [Google Scholar] [CrossRef]
- Norton, T.S.; Dryer, F.L. An experimental and modeling study of ethanol oxidation kinetics in an atmospheric pressure flow reactor. Int. J. Chem. Kinet. 1992, 24, 319–344. [Google Scholar] [CrossRef]
- Natarajan, K.; Bhaskaran, K. An Experimental and Analytical Investigation of High Temperature Ignition of Ethanol; Indian Institute of Tech Madras Department of Mechanical Engineering: Tamil Nadu, India, 1981. [Google Scholar]
- Li, J.; Kazakov, A.; Dryer, F.L. Ethanol pyrolysis experiments in a variable pressure flow reactor. Int. J. Chem. Kinet. 2001, 33, 859–867. [Google Scholar] [CrossRef]
- Li, J.; Kazakov, A.; Dryer, F.L. Experimental and Numerical Studies of Ethanol Decomposition Reactions. J. Phys. Chem. A 2004, 108, 7671–7680. [Google Scholar] [CrossRef]
- Metcalfe, W.K.; Burke, S.M.; Ahmed, S.S.; Curran, H.J. A Hierarchical and Comparative Kinetic Modeling Study of C1−C2 Hydrocarbon and Oxygenated Fuels. Int. J. Chem. Kinet. 2013, 45, 638–675. [Google Scholar] [CrossRef]
- Lutz, A.E.; Kee, R.J.; Miller, J.A. SENKIN: A FORTRAN Program for Predicting Homogeneous Gas Phase Chemical Kinetics with Sensitivity Analysis; Sandia National Labs.: Livermore, CA, USA, 1988. [Google Scholar]
- Millán-Merino, A.; Fernández-Tarrazo, E.; Sánchez-Sanz, M.; Williams, F.A. A multipurpose reduced mechanism for ethanol combustion. Combust. Flame 2018, 193, 112–122. [Google Scholar] [CrossRef]
- Hashemi, H.; Christensen, J.M.; Gersen, S.; Levinsky, H.; Klippenstein, S.; Glarborg, P. High-pressure oxidation of methane. Combust. Flame 2016, 172, 349–364. [Google Scholar] [CrossRef] [Green Version]
- Zyada, A.; Samimi-Abianeh, O. Ethanol Kinetic Model Development and Validation at Wide Ranges of Mixture Temperatures, Pressures, and Equivalence Ratios. Energy Fuels 2019, 33, 7791–7804. [Google Scholar] [CrossRef]
- Roy, S.; Askari, O. A New Detailed Ethanol Kinetic Mechanism at Engine-Relevant Conditions. Energy Fuels 2020, 34, 3691–3708. [Google Scholar] [CrossRef]
- Geng, Z.; Xu, L.; Li, H.; Wang, J.; Huang, Z.; Lu, X. Shock tube measurements and modeling study on the ignition delay times of n-butanol/dimethyl ether mixtures. Energy Fuels 2014, 28, 4206–4215. [Google Scholar] [CrossRef]
- Feng, P.; Gu, X.; Huang, Z. Measurement of laminar burning velocities of iso-butanol-air mixtures. Chin. Sci. Bull. 2010, 55, 2046–2056. [Google Scholar] [CrossRef]
- Gu, X.; Li, Q.; Huang, Z.; Zhang, N. Measurement of laminar flame speeds and flame stability analysis of tert-butanol–air mixtures at elevated pressures. Energy Convers. Manag. 2011, 52, 3137–3146. [Google Scholar] [CrossRef]
- Veloo, P.S.; Egolfopoulos, F. Flame propagation of butanol isomers/air mixtures. Proc. Combust. Inst. 2011, 33, 987–993. [Google Scholar] [CrossRef]
- Wu, F.; Law, C.K. Laminar Flame Speed, Markstein Length and Flame Chemistry of the Butanol Isomers From 1 atm to 5 atm; Western States Section/Combustion Institute: Park City, UT, USA, 2013. [Google Scholar]
- Moss, J.T.; Berkowitz, A.M.; Oehlschlaeger, M.A.; Biet, J.; Warth, V.; Glaude, P.-A.; Battin-Leclerc, F. An Experimental and Kinetic Modeling Study of the Oxidation of the Four Isomers of Butanol. J. Phys. Chem. A 2008, 112, 10843–10855. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, T.K.; Lna, M.C.; Lin, C.-Y.; Sanders, W.A. Thermal Decomposition of t-Butyl Alcohol in Shock Waves. Combust. Sci. Technol. 1990, 71, 219–232. [Google Scholar] [CrossRef]
- Norton, T.S.; Dryer, F.L. The flow reactor oxidation of C1−C4 alcohols and MTBE. Symp. Int. Combust. 1991, 23, 179–185. [Google Scholar] [CrossRef]
- Liu, W.; Kelley, A.P.; Law, C.K. Non-premixed ignition, laminar flame propagation, and mechanism reduction of n-butanol, iso-butanol, and methyl butanoate. Proc. Combust. Inst. 2011, 33, 995–1002. [Google Scholar] [CrossRef]
- Zhang, J.; Niu, S.; Zhang, Y.; Tang, C.; Jiang, X.; Hu, E.; Huang, Z. Experimental and modeling study of the auto-ignition of n-heptane/n-butanol mixtures. Combust. Flame 2013, 160, 31–39. [Google Scholar] [CrossRef]
- Kohse-Höinghaus, K. Combustion in the future: The importance of chemistry. Proc. Combust. Inst. 2020, 38, 1–56. [Google Scholar] [CrossRef]
- Grana, R.; Frassoldati, A.; Faravelli, T.; Niemann, U.; Ranzi, E.; Seiser, R.; Cattolica, R.; Seshadri, K. An experimental and kinetic modeling study of combustion of isomers of butanol. Combust. Flame 2010, 157, 2137–2154. [Google Scholar] [CrossRef]
- Sarathy, S.M.; Vranckx, S.; Yasunaga, K.; Mehl, M.; Oßwald, P.; Metcalfe, W.K.; Westbrook, C.K.; Pitz, W.J.; Kohse-Höinghaus, K.; Fernandes, R.X.; et al. A comprehensive chemical kinetic combustion model for the four butanol isomers. Combust. Flame 2012, 159, 2028–2055. [Google Scholar] [CrossRef]
- Oasmaa, A.; Solantausta, Y.; Arpiainen, V.; Kuoppala, E.; Sipilä, K. Fast Pyrolysis Bio-Oils from Wood and Agricultural Residues. Energy Fuels 2010, 24, 1380–1388. [Google Scholar] [CrossRef]
- Bertero, M.; de la Puente, G.; Sedran, U. Fuels from bio-oils: Bio-oil production from different residual sources, characterization and thermal conditioning. Fuel 2012, 95, 263–271. [Google Scholar] [CrossRef]
- Onarheim, K.; Solantausta, Y.; Lehto, J. Process Simulation Development of Fast Pyrolysis of Wood Using Aspen Plus. Energy Fuels 2014, 29, 205–217. [Google Scholar] [CrossRef]
- Doolan, K.R.; Mackie, J.; Reid, C.R. High temperature kinetics of the thermal decomposition of the lower alkanoic acids. Int. J. Chem. Kinet. 1986, 18, 575–596. [Google Scholar] [CrossRef]
- Elwardany, A.; Nasir, E.F.; Es-Sebbar, E.; Farooq, A. Unimolecular decomposition of formic and acetic acids: A shock tube/laser absorption study. Proc. Combust. Inst. 2015, 35, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Zervas, E.; Montagne, X.; Lahaye, J. Influence of Fuel Composition on the Emission of Oxygenated Pollutants (Organic Acids, Alcohols and Carbonyl Compounds) from a SI Engine. Tech. Chron. Sci. J. 2004, 1, 49–58. [Google Scholar]
- Zervas, E.; Montagne, X.; Lahaye, J. Emission of Alcohols and Carbonyl Compounds from a Spark Ignition Engine. Influence of Fuel and Air/Fuel Equivalence Ratio. Environ. Sci. Technol. 2002, 36, 2414–2421. [Google Scholar] [CrossRef]
- Fontaras, G.; Karavalakis, G.; Kousoulidou, M.; Ntziachristos, L.; Bakeas, E.; Stournas, S.; Samaras, Z. Effects of low concentration biodiesel blends application on modern passenger cars. Part 2: Impact on carbonyl compound emissions. Environ. Pollut. 2010, 158, 2496–2503. [Google Scholar] [CrossRef]
- Sarathy, S.M.; Oßwald, P.; Hansen, N.; Kohse-Höinghaus, K. Alcohol combustion chemistry. Prog. Energy Combust. Sci. 2014, 44, 40–102. [Google Scholar] [CrossRef]
- Jacobson, M.Z. Effects of Ethanol (E85) versus Gasoline Vehicles on Cancer and Mortality in the United States. Environ. Sci. Technol. 2007, 41, 4150–4157. [Google Scholar] [CrossRef]
- Gaffney, J.S.; Marley, N.A. The impacts of combustion emissions on air quality and climate—From coal to biofuels and beyond. Atmos. Environ. 2009, 43, 23–36. [Google Scholar] [CrossRef]
- Sivaramakrishnan, R.; Michael, J.V.; Harding, L.B.; Klippenstein, S. Resolving Some Paradoxes in the Thermal Decomposition Mechanism of Acetaldehyde. J. Phys. Chem. A 2015, 119, 7724–7733. [Google Scholar] [CrossRef]
- Tao, T.; Sun, W.; Yang, B.; Hansen, N.; Moshammer, K.; Law, C.K. Investigation of the chemical structures of laminar premixed flames fueled by acetaldehyde. Proc. Combust. Inst. 2017, 36, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Christensen, M.; Abebe, M.T.; Nilsson, E.J.; Konnov, A.A. Kinetics of premixed acetaldehyde + air flames. Proc. Combust. Inst. 2015, 35, 499–506. [Google Scholar] [CrossRef]
- Christensen, M.; Konnov, A.A. Laminar burning velocity of diacetyl + air flames. Further assessment of combustion chemistry of ketene. Combust. Flame 2017, 178, 97–110. [Google Scholar] [CrossRef]
- Halstead, M.P.; Prothero, A.; Quinn, C.P. A mathematical model of the cool-flame oxidation of acetaldehyde. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1971, 322, 377–403. [Google Scholar] [CrossRef]
- Felton, P.; Gray, B.; Shank, N. Low temperature oxidation in a stirred flow reactor—II. Acetaldehyde (theory). Combust. Flame 1976, 27, 363–376. [Google Scholar] [CrossRef]
- Cavanagh, J.; Cox, R.; Olson, G. Computer modeling of cool flames and ignition of acetaldehyde. Combust. Flame 1990, 82, 15–39. [Google Scholar] [CrossRef]
- Gibson, C.; Gray, P.; Griffiths, J.; Hasko, S. Spontaneous ignition of hydrocarbon and related fuels: A fundamental study of thermokinetic interactions. Symp. Int. Combust. 1985, 20, 101–109. [Google Scholar] [CrossRef]
- Kaiser, E.W.; Westbrook, C.K.; Pitz, W.J. Acetaldehyde oxidation in the negative temperature coefficient regime: Experimental and modeling results. Int. J. Chem. Kinet. 1986, 18, 655–688. [Google Scholar] [CrossRef]
- Pelucchi, M. Kinetic modeling of the low temperature cool flames of acetaldehyde in a well stirred reactor. In Proceedings of the Meeting of the Italian Section of the Combustion Institute, Lecce, Italy, 20–23 September 2015. [Google Scholar]
- Bentz, T.; Striebel, F.; Olzmann, M. Shock-Tube Study of the Thermal Decomposition of CH 3CHO and CH 3CHO + H Reaction. J. Phys. Chem. A 2008, 112, 6120–6124. [Google Scholar] [CrossRef]
- Hidaka, Y.; Kubo, S.; Hoshikawa, T.; Wakamatsu, H. Shock-tube study of acetaldehyde pyrolysis. In Shock Waves; Springer: Berlin/Heidelberg, Germany, 2005; pp. 603–608. [Google Scholar]
- Ernst, J.; Spindler, K.; Wagner, H.G. Untersuchungen zum thermischen Zerfall von Acetaldehyd und Aceton. Ber. Bunsenges. Phys. Chem. 1976, 80, 645–650. [Google Scholar] [CrossRef]
- Wei, L.; Tang, C.; Man, X.; Huang, Z. Shock-Tube Experiments and Kinetic Modeling of 2-Methylfuran Ignition at Elevated Pressure. Energy Fuels 2013, 27, 7809–7816. [Google Scholar] [CrossRef]
- Graedel, T.E.; Hawkins, D.T.; Claxton, L.D. Atmospheric Chemical Compounds: Sources, Occurrence and Bioassay; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Fulle, D.; Dib, A.A.; Kiefer, J.H.; Zhang, Q.; Yao, A.J.; Kern, R.D. Pyrolysis of Furan at Low Pressures: Vibrational Relaxation, Unimolecular Dissociation, and Incubation Times. J. Phys. Chem. A 1998, 102, 7480–7486. [Google Scholar] [CrossRef]
- Ohtomo, M.; Nishikawa, K.; Suzuoki, T.; Miyagawa, H.; Koike, M. Auto-ignition Characteristics of Biofuel Blends for SI Engines. SAE Tech. Pap. Ser. 2011. [Google Scholar] [CrossRef]
- Wang, C.; Xu, H.; Daniel, R.; Ghafourian, A.; Herreros, J.; Shuai, S.; Ma, X. Combustion characteristics and emissions of 2-methylfuran compared to 2,5-dimethylfuran, gasoline and ethanol in a DISI engine. Fuel 2013, 103, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Huang, Z.; Yuan, T.; Zhang, K.; Wei, L. Identification of combustion intermediates in a low-pressure premixed laminar 2,5-dimethylfuran/oxygen/argon flame with tunable synchrotron photoionization. Combust. Flame 2009, 156, 1365–1376. [Google Scholar] [CrossRef]
- Wu, X.; Huang, Z.; Wang, X.; Jin, C.; Tang, C.; Wei, L.; Law, C.K. Laminar burning velocities and flame instabilities of 2,5-dimethylfuran–air mixtures at elevated pressures. Combust. Flame 2011, 158, 539–546. [Google Scholar] [CrossRef]
- Organ, P.P.; Mackie, J. Kinetics of pyrolysis of furan. J. Chem. Soc. Faraday Trans. 1991, 87, 815–823. [Google Scholar] [CrossRef]
- Grela, M.A.; Amorebieta, V.T.; Colussi, A.J. Very low pressure pyrolysis of furan, 2-methylfuran and 2,5-dimethylfuran. The stability of the furan ring. J. Phys. Chem. 1985, 89, 38–41. [Google Scholar] [CrossRef]
- Cullis, C.; Norris, A. The pyrolysis of organic compounds under conditions of carbon formation. Carbon 1972, 10, 525–537. [Google Scholar] [CrossRef]
- Lifshitz, A.; Bidani, M.; Bidani, S. Thermal reactions of cyclic ethers at high temperatures. III. Pyrolysis of furan behind reflected shocks. J. Phys. Chem. 1986, 90, 5373–5377. [Google Scholar] [CrossRef]
- Bruinsma, O.S.; Tromp, P.J.; Nolting, H.J.D.S.; Moulijn, J.A. Gas phase pyrolysis of coal-related aromatic compounds in a coiled tube flow reactor. Fuel 1988, 67, 334–340. [Google Scholar] [CrossRef]
- Sendt, K.; Bacskay, G.B.; Mackie, J. Pyrolysis of Furan: Ab Initio Quantum Chemical and Kinetic Modeling Studies. J. Phys. Chem. A 2000, 104, 1861–1875. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, X.; Zuo, T. The pyrolysis mechanism of furan revisited. Chem. Phys. Lett. 2000, 325, 457–464. [Google Scholar] [CrossRef]
- Holzer, G.; Oró, J.; Bertsch, W. Gas chromatographic-mass spectrometric evaluation of exhaled tobacco smoke. J. Chromatogr. A 1976, 126, 771–785. [Google Scholar] [CrossRef]
- Guarneri, F.; Ikeda, E.; Mackie, J. A Study of Furan as a Model Oxygenated Reburn Fuel for Nitric Oxide Reduction. Energy Fuels 2001, 15, 743–750. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, X.; Zhai, L. Theoretical investigation of unimolecular decomposition channels of furan4. J. Comput. Chem. 1998, 19, 240–249. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fuel | Structure | LHV (MJ L−1) | λ (kJ kg−1) | AIT (°C) | Abstracting Agents | Ref. |
|---|---|---|---|---|---|---|
| Methanol |  | 15.8 | 1103 | 465 | H, OH, HO2 | [34,35] |
| Ethanol |  | 21.4 | 904 | 420 | H, OH, O2, HO2 | [20,35] |
| Butanol |  | 26.9 | 582 | 343 | CH3, OH, HO2 | [35,36] |
| Acetic acid |  | 10.1 | 700 | 485 | H, OH, H O2, O2, CH3 | [30,37,38,39] |
| Crotonic acid |  | No data | 649 | 396 | No data | [38] |
| Acetaldehyde |  | 18.8 | 569 | 155 | H, OH, HO2, CH3, O2 | [40] |
| Furan |  | 23.2 | 406 | 390 | H, OH, CH3 | [41,42] |
| Parameter | Approach | T (K) | P (atm) | φ (–) | Ref. |
|---|---|---|---|---|---|
| Methanol | |||||
| IDT | Exp/Mod | 1545–2180 | 1.5–4.2 | 1 | [43] |
| IDT | Exp/Mo | 850–1100 | 7–30 | 0.25–1 | [44] |
| IDT | Exp/Mod | 1070–1760 | 2, 10, 12 | 0.5, 1, 2 | [45] |
| IDT | Exp/Mod | 940–1540 | 1–14.9 | 0.5, 1, 2 | [46] |
| IDT | Exp | 840–1000 | 12–24 | 1.0 | [47] |
| Su | Exp | 300 | 1 | 0.7–1.4 | [48] |
| Su | Exp/Mod | 340–450 | 0.02 | 0.8, 1, 1.2 | [49] |
| Ethanol | |||||
| IDT | Exp/Mod | 1070–1760 | 2, 10, 12 | 0.5, 1, 2 | [45] |
| IDT | Exp/Mod | 880–1150 | 3–10 | 1 | [50] |
| IDT | Exp/Mod | 960–1580 | 17.8–23.9 | 0.5, 1 | [51] |
| IDT | Exp/Mod | 944–1589 | 1.3–53 | 0.5, 1, 2 | [52] |
| IDT | Exp/Mod | 800–875 | 20 | 1.0 | [53] |
| Su | Exp/Mod | 300–453 | 1 | 0.8–1.4 | [54] |
| Su | Exp/Mod | 298–358 | 1 | 0.6–1.55 | [55] |
| Su | Exp | 358 | 1 | 0.7–1.4 | [56] |
| Su | Exp | 358 | 1–3 | 0.7–1.4 | [57] |
| Su | Exp/Mod | 358 | 1 | 0.6–1.6 | [58] |
| Su | Exp/Mod | 350–620 | 1 | 0.7–1.3 | [59] |
| Su | Exp | 300 | 1 | 0.7–1.4 | [48] |
| Butanol | |||||
| IDT | Exp/Mod | 1070–1760 | 2, 10, 12 | 0.5, 1, 2 | [45] |
| IDT | Exp/Mod | 1050–1600 | 1.5–43 | 0.5, 1 | [60] |
| IDT | Exp/Mod | 1000–1800 | 1–4 | 0.5, 1 | [2] |
| IDT | Exp/Mod | 700–1000 | 10–80 | 1.0 | [61] |
| Su | Exp/Mod | 428 | 1, 2.5, 7.5 | 0.7–1.5 | [62] |
| Su | Exp/Mod | 350–600 | 1 | 0.7–1.3 | [63] |
| Su | Exp | 400 | 5 | 0.7–1.5 | [64] |
| Su | Exp/Mod | 393 | 1 | 0.8–1.4 | [65] |
| Su | Exp/Mod | 423 | 1 | 0.7–1.4 | [66] |
| Su | Exp/Mod | 350 | 1 | 0.8–1.2 | [67] |
| Acetic acid | |||||
| IDT | Exp/Mod | 1300–1950 | 0.3–0.7 | - | [68] |
| IDT | Exp/Mod | 700–2100 | 0.1–100 | No data | [37] |
| IDT | Exp | 1300–2000 | No data | No data | [69] |
| Su | Exp/Mod | 423 | 1 | 0.7–1.4 | [13] |
| Su | Exp/Mod | 673–973 | 0.05 | 0.77–1.05 | [70] |
| Su | Exp/Mod | 298–358 | 1 | 0.6–1.8 | [14] |
| Crotonic acid | |||||
| IDT, Su | No data | No data | No data | No data | |
| Acetaldehyde | |||||
| IDT | Exp/Mod | 1000–1700 | 1.2–2.8 | 1 | [71] |
| IDT | Exp/Mod | 1295–1580 | 3–3.98 | 0.5, 1, 1.5 | [72] |
| IDT | Exp/Mod | 1000–1100 | 10 | 1 | [73] |
| Su | Exp/Mod | 298–358 | 1 | 0.6–1.8 | [14] |
| Su | Exp/Mod | 460–900 | 0.7–0.71 | 0.5–4 | [40] |
| Furan | |||||
| IDT | Exp/Mod | 1320–1880 | 1.2–10.4 | 0.5, 1, 2 | [74] |
| IDT | Exp/Mod | 850–1050 | 18–33 | 0.5, 1, 2 | [75] |
| Su | Exp/Mod | 300–400 | 0.046 | 1.4, 1.8, 2.2 | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosisa Wako, F.; Pio, G.; Salzano, E. Laminar Burning Velocity and Ignition Delay Time of Oxygenated Biofuel. Energies 2021, 14, 3562. https://doi.org/10.3390/en14123562
Mosisa Wako F, Pio G, Salzano E. Laminar Burning Velocity and Ignition Delay Time of Oxygenated Biofuel. Energies. 2021; 14(12):3562. https://doi.org/10.3390/en14123562
Chicago/Turabian StyleMosisa Wako, Fekadu, Gianmaria Pio, and Ernesto Salzano. 2021. "Laminar Burning Velocity and Ignition Delay Time of Oxygenated Biofuel" Energies 14, no. 12: 3562. https://doi.org/10.3390/en14123562
APA StyleMosisa Wako, F., Pio, G., & Salzano, E. (2021). Laminar Burning Velocity and Ignition Delay Time of Oxygenated Biofuel. Energies, 14(12), 3562. https://doi.org/10.3390/en14123562