
Review of the Hydrogen Evolution Reaction—A Basic Approach




Abstract
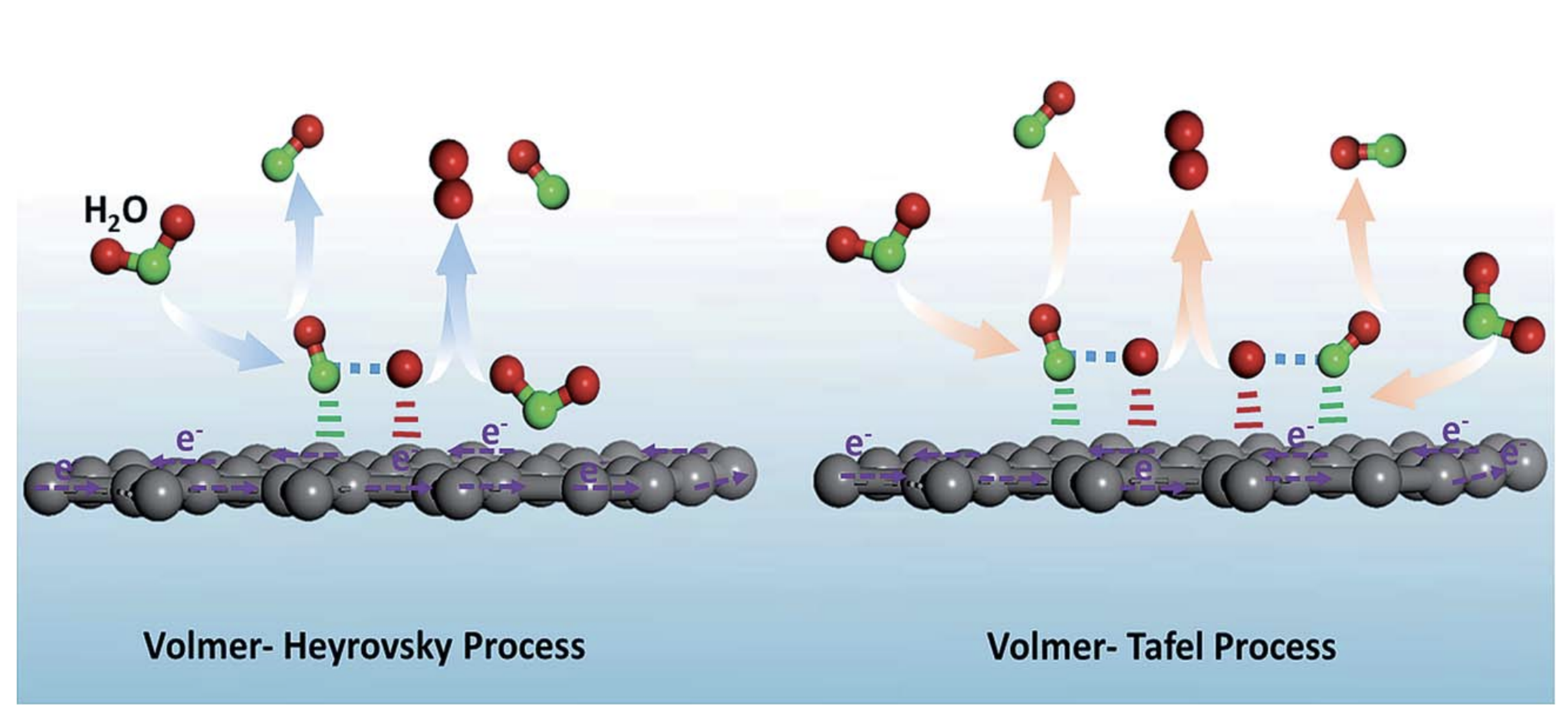
:1. Introduction
2. Modifying the Electronic Structure
2.1. Alloys and Oxides
2.2. Phosphides
2.3. Nitrides
2.4. Co-Doping
2.5. Chalcogenides
2.5.1. Sulfides
2.5.2. Selenides
2.5.3. Tellurides
2.6. Dichalcogenides
3. Creating Specific Sites for Hydroxide and Hydrogen Adsorption
4. Altering the Surface of a Catalyst
4.1. Strain
4.2. Additional Geometrical Modification Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Material | Overpotential | Tafel Slope | Stability | Stability From |
|---|---|---|---|---|
| at 10 mA cm | [mV dec] | from CA/CP [h] | CVs [% of Current] | |
| Electronic | ||||
| Structure | ||||
| Alloys and oxides | ||||
| Np-CuTi [23] | 50 mV * | 110 * | - | ∼98% (5000) |
| (@ 30 mA cm) | ||||
| Ni-Co-Ti-2 [24] | 125 mV * | 47 * | 150 (↓88.4%@ 10 mA cm) | - |
| (CP) | ||||
| NiO/Ni/CNT [25] | 80 mV * | 82 * | 2 (↑91.1%@ 20 mA cm) | - |
| (CP) | ||||
| CrNi [26] | 106 mV | 71 | 6 (↑99%@ 10 mA cm) | - |
| (CA) | ||||
| NiMo [27] | 185 mV * | 120 * | 20 (↑98.5%@ ∼14 mA cm) | - |
| (CA) | ||||
| Ni-LaCeO/C [33] | 114 mV * | 72.7 * | 24 (↑76.2%@ 10 mA cm) | ∼88% (500) |
| (CP) | (@ 25 mA cm) | |||
| 2%La-CoO-350 [34] | 98 mV | 72.8 | 3.3 (↓44.7%@ 10 mA cm) | ∼ 93% (3000) |
| (CP) | (@ 100 mA cm) | |||
| CoS NS/NF [35] | 93 mV | 55.1 | 220 (↑93%@ 10 mA cm) | - |
| (CP) | ||||
| mMoO [37] | 138 mV * | 56 * | 12 (↓96%@ ∼23 mA cm) | - |
| (CA) | ||||
| PBSCF [39] | 237 mV * | 45 * | 25 (↑99%@ 50 mA cm) | 100% (1000) |
| (CP) | (@ 100 mA cm) | |||
| SrNbCoFeO [40] | 262 mV * | 135 * | 10 (↑100%@ 10 mA cm) | ∼99% (1000) |
| (CP) | (@ 40 mA cm) | |||
| Phosphides | ||||
| NiP NP [41] | 291 mV * | 119 * | 16 (↑99%@ 10 mA cm) | - |
| (CP) | ||||
| (FeNi)P [43] | 103 mV | 77 | 20 (↑95.2%@ 100 mA cm) | ∼99% (1000) |
| (CA) | (@ 100 mA cm) | |||
| Pt-CoP [44] | 2.0 mV * | 44 * | 200 ** (↓@ 100 mA cm) | - |
| (CA) | ||||
| 10 wt.% Pt/C [44] | 20 mV * | 65 * | ∼55 ** (↓@ 100 mA cm) | - |
| (CA) | ||||
| RuP NF [45] | 9 mV * | 38 * | - | ∼88% (10,000) |
| (@ 100 mA cm) | ||||
| RhP-500 [46] | 4.3 mV * | 35.2 * | 16.7 (↓81.8%@ 10 mA cm) | ∼99% (2000) |
| (CA) | (@ 50 mA cm) | |||
| MoP/MoO [47] | 79 mV * | 41 * | 24 (↑98.2%@ 10 mA cm) | ∼84% (1000) |
| (CP) | (@ 100 mA cm) | |||
| 2D/3D NiCoP/NF [48] | 37 mV * | 50.8 * | 50 (↑81.8%@ 10 mA cm) | ∼97% (5000) |
| (CP) | (@ 200 mA cm) | |||
| Cr-CoP [49] | 36 mV * | 54 * | 514 (↓65.8%@ 10 mA cm) | ∼98% (3000) |
| (CP) | (@ 200 mA cm) | |||
| NiP/Ni(PO) HSs [51] | 114 mV * | 93.1 * | 10 (↑100%@ 10 mA cm) | ∼99% (1000) |
| (CP) | (@ 37.5 mA cm) | |||
| Nitrides | ||||
| (NiN/NF) [53] | 121 mV * | 109 * | 200 (↑92.6%@ 200 mA cm) | ∼97% (1000) |
| (CA) | (@ 360 mA cm) | |||
| MoN@NC [55] | 85 mV | 54 | 10 (↑89.7%@ 10 mA cm) | 100% (1000) |
| (CA) | (@ 80 mA cm) | |||
| N-CoS@NC/Ti [56] | 140 mV | 92 | 10 (↑98.1%@ 30 mA cm) | 96% (1000) |
| (CP) | (@ 100 mA cm) | |||
| Mo(CN) [59] | 80 mV * | 40 * | 24 (↓91.1%@ 18–19 mA cm) | ∼98% (5000) |
| (CA) | @ 300 mA cm) | |||
| NiN/NF [60] | 45 mV | 97 | 20 (↓78.2%@ ∼20 mA cm) | ∼91% (1000) |
| (CA) | (@ 80 mA cm) | |||
| Co-NPs/N-rGO [61] | 59 mV * | 105 * | 480 (↑100%@ 10 mA cm) | - |
| (CP) | ||||
| Co-NPs/N-rGO [61] | 200 (↑100%@ 500 mA cm) | - | ||
| (CP) | ||||
| Ni/V-N [63] | 43 mV * | 33 * | 20 (↓87.4%@ 20 mA cm) | - |
| (CP) | ||||
| Co-doping | ||||
| N-P-Ni/C [67] | 26 mV | 34 | 50 (↑96.8%@ 12 mA cm) | ∼85% (1000) |
| (CA) | @ 10 mA cm) | |||
| Zn,S-CoP | 49 mV * | 42 * | 20 (↑100%@ 10 mA cm) | ∼94% (1000) |
| NRCs/CP [68] | (CP) | (@ 100 mA cm) | ||
| O, Cu-CoP-2 [69] | 72 mV * | 58 * | 24 (↑100%@ 50 mA cm) | ∼98% (5000) |
| (CP) | (@ 200 mA cm) | |||
| V, N-CoP [70] | 57 mV | 51 | 50 (↑94.1%@ 10 mA cm) | ∼99% (1000) |
| (CA) | (@ 100 mA cm) | |||
| Ni(Cu)VO/NF [73] | 21 mV | 28 | 100 (↓55.5%@ 100 mA cm) | - |
| (CP) | ||||
| Ni(Cu)VO/NF [73] | 10 mV * | - | - | - |
| Ru-ReP/NPC [74] | 39 mV * | 29.9 * | 12 (↑100%@ 10 mA cm) | - |
| (CP) | ||||
| NiP@NC/NF [75] | 93 mV * | 77.8 * | 30 (↓83.3%@ 20 mA cm) | ∼97% (3000) |
| (CP) | (@ 120 mA cm) | |||
| (NiP-FeP)/C/CF [76] | 23.6 mV * | 52 * | 50 (↓62.9%@ 1000 mA cm) | - |
| (CP) | ||||
| (Ni, Zn)-CoO NR [77] | 53 mV | 47 | 24 ** (↑96%@ 10 mA cm) | - |
| (CP) (in 6.0M KOH) | ||||
| Chalcogenides | ||||
| MoSNiSMoO [81] | 91 mV * | 54 * | 20 (↑86.4%@ ∼10 mA cm) | 96.5% (1000) |
| (CP) | (@ 100 mA cm) | |||
| V-NiS [82] | 68 mV | 112 | 12 (↑100%@ 20 mA cm) | - |
| (CP) | ||||
| NiS/MoS/NF [83] | 71 mV | 79 | 20 (↑96.7%@ 30 mA cm) | - |
| (CA) | ||||
| CoNiSe/Ti [84] | 64 mV * | 63 * | 60 (↓66.4%@ 10–30 mA cm) | ∼95% (5000) |
| (CP) | (@ 90 mA cm) | |||
| Chalcogenides | ||||
| NiTe NW [85] | 113 mV * | 69 * | 16.5 (↑98.3%@ ∼50 mA cm) | 86.7% (1000) |
| (CP) | (@ 100 mA cm) | |||
| NiP/NiTe [86] | 63 mV * | 80 * | 50 (↑98%@ 50 mA cm) | ∼91% after 50 h CA |
| (CA) | (@ 100 mA cm) | |||
| Te@NiTe/NiS | 101 mV | 118 | 10 (↑100%@ 50 mA cm) | ∼99% (1000) |
| /AB [87] | (CA) | (@ 350 mA cm) | ||
| MoS [88] | 173 mV * | 110 * | 12 (↓89%@ 50 mA cm) | - |
| (CP) | ||||
| MoSe [88] | 208 mV * | 66 * | 12 (↓91.8%@ 50 mA cm) | - |
| (CP) | ||||
| MoTe [88] | 283 mV * | 102 * | 12 (↑100%@ 50 mA cm) | - |
| (CP) | ||||
| Mo SAs/MLMoS [89] | 209 mV | 35 | - | - |
| Creating | ||||
| sites | ||||
| NiW-WO/NF [97] | 48 mV | 33 | 14 (↑87.3%@ ∼20 mA cm) | - |
| (CA) | ||||
| 1T–MoS QS/Ni(OH) | 57 mV | 30 | 100 (↓85.8%@ 100 mA cm) | ∼99% (1000) |
| [98] | (CP) | (@ 100 mA cm) | ||
| Ni(OH)/MoS [99] | 80 mV * | 60 * | 16 (↓84.9%@ 10 mA cm) | - |
| (CP) | ||||
| PtNi/NiS NW [100] | 42 mV | - | 5 (↓63.5%@ 5 mA cm) | - |
| Co-WO/CoW | 25 mV * | 20 * | 100 (↑99%@ 10 mA cm) | ∼92% (5000) |
| /NF-1/3 [101] | (CP) | (@ 200 mA cm) | ||
| MoNi/MoO@NF [102] | 15 mV | 30 | 10 (↑96.3%@ 100 mA cm) | ∼99% (2000) |
| (CP) | (@ 600 mA cm) | |||
| WC–Ni(OH) [103] | 60 mV | 55.9 | 20+10 (↑100% and ↓94.7% | - |
| @ 10 and 100 mA cm)(CP) | ||||
| NiMoN/NiN [104] | 28 mV * | 49 * | 24 (↓87.5%@ 10 mA cm) | ∼99% (2000) |
| (CP) | (@ 200 mA cm) | |||
| CoO-Ag [105] | 51 mV | 49 | 100+100 (↑100% and ↓96.8% | - |
| @ 10 and 50 mA cm)(CP) | ||||
| NiFeN/NiFe [106] | 74 mV | 54 | 12 (↑100%@ 10 mA cm) | - |
| (CP) | ||||
| Ni/NiO-3.8 [107] | 90 mV * | 41 * | - | ∼98% (5000) |
| (@ 60 mA cm) | ||||
| Cu NDs/NiS | 128 mV | 76 | 30 (↑99%@ 87 mA cm) | - |
| NTs-CF [108] | (CP) | |||
| Altering | ||||
| surface | ||||
| NiSSe [113] | 70 mV | 78 | 300 (↓73.5%@ 100 mA cm) | ∼99% (10,000) |
| (CP) | (@ 300 mA cm) | |||
| 3.0% S-CoO NRs [117] | 73 mV * | 82 * | 28 (↓91.7%@ 9-10 mA cm) | ∼93% (1000) |
| (CA) | (@ 200 mA cm) | |||
| CoP NWs 40s/CC [118] | 65 mV * | 43 * | 35 (↓95.2%@ 110 mA cm) | ∼94% (5000) |
| (CA) | (@ 100 mA cm) | |||
| N-MoP/CC [120] | 70 mV | 55 | 36 (↑100%@ ∼15 mA cm) | - |
| (CA) | ||||
| Ni-S-OH/NF [121] | 179 mV * | 120 * | 25+25 (↑100% and ↓96.9% | - |
| @ 100 and 200 mA cm)(CP) | ||||
| Fe-(NiS/NiP)@C/NF [123] | 129 mV | 66.8 | 25 (↑100%@ ∼10 mA cm) | ∼84% (3000) |
| (CP) | (@ 150 mA cm) | |||
| NF-NiS-A [125] | 67 mV * | 63 * | 40 (↓100%@ 20 mA cm) | - |
| (CP) |
5. Perspective
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AEM | Anion Exchange Membrane |
| AFM | Atomic Force Microscopy |
| CA | Chronoamperometry |
| CC | Carbon Cloth |
| CF | Copper Foam |
| CNT | Carbon Nanotubes |
| COHP | Crystal Orbital Hamilton Population |
| CP | Chronopotentiometry |
| CV | Cyclic Voltammetry |
| DFT | Density Functional Theory |
| DOS | Density Of States |
| ECD | Exchange Current Density |
| ECSA | Electrochemical Surface Area |
| EIS | Electrochemical Impedance Spectroscopy |
| ELF | Electron Localisation Function |
| EXAFS | Extended X-ray Adsorption Fine Structure |
| FCC | Face centerd Cubic |
| HAADF | High-Angle Annular Dark-Field |
| HBE | Hydrogen Binding Energy |
| HER | Hydrogen Evolution Reaction |
| HOR | Hydrogen Oxidation Reaction |
| HRTEM | High Resolution Transmission Electron Spectroscopy |
| ICPMS | Inductively Coupled Plasma Mass Spectrometry |
| ICP-OES | Inductively Coupled Plasma Optical Emission Spectroscopy |
| LSV | Linear Sweep Voltammetry |
| MEA | Membrane Electrode Assembly |
| NF | Nickel Foam |
| NP | Nano Particle |
| NR | Nanorod |
| NS | Nanosheet |
| NW | Nanowire |
| OER | Oxygen Evolution Reaction |
| OPEM | Oxygen Plasma Engraving Method |
| PGM | Platinum Group Metal |
| RHE | Reversible Hydrogen Electrode |
| SCAN | Strongly Constrained and Appropriately Normed |
| SEM | Scanning Electron Spectroscopy |
| SHE | Standard Hydrogen Electrode |
| TEM | Transmission Electron Spectroscopy |
| TMDC | Transition Metal Dichalcogenides |
| UPS | Ultraviolet Photoelectron Spectroscopy |
| XANES | X-ray Adsorption Near Edge Spectroscopy |
| XAS | X-ray Adsorption Spectroscopy |
| XPS | X-ray Photoelectron Spectroscopy |
| XRD | X-ray Diffraction |
References
- Tilak, B.; Ramamurthy, A.; Conway, B. High performance electrode materials for the hydrogen evolution reaction from alkaline media. In Proceedings of the Indian Academy of Sciences-Chemical Sciences; Springer: Berlin/Heidelberg, Germany, 1986; Volume 97, pp. 359–393. [Google Scholar]
- Li, S.; Kang, Q.; Baeyens, J.; Zhang, H.; Deng, Y. Hydrogen production: State of technology. In Proceedings of the IOP Conference Series: Earth and Environmental Science, Vienna, Austria, 18–21 May 2020; Volume 544, p. 012011. [Google Scholar]
- Wang, X.; Zheng, Y.; Sheng, W.; Xu, Z.; Jaroniec, M.; Qiao, S.Z. Strategies for design of electrocatalysts for hydrogen evolution under alkaline conditions. Mater. Today 2020, 36, 125–138. [Google Scholar] [CrossRef]
- Gerischer, H.; Mehl, W. Zum mechanismus der kathodischen wasserstoffabscheidung an quecksilber, silber und kupfer. Z. Elektrochem. Berichte Bunsenges. Phys. Chem. 1955, 59, 1049–1059. [Google Scholar]
- Chen, Z.; Duan, X.; Wei, W.; Wang, S.; Ni, B.J. Recent advances in transition metal-based electrocatalysts for alkaline hydrogen evolution. J. Mater. Chem. A 2019, 7, 14971–15005. [Google Scholar] [CrossRef]
- Firouzjaie, H.; Mustain, W. Catalytic Advantages, Challenges, and Priorities in Alkaline Membrane Fuel Cells; American Chemical Society: Washington, DC, USA, 2019. [Google Scholar]
- Rossmeisl, J.; Chan, K.; Skulason, E.; Björketun, M.; Tripkovic, V. On the pH dependence of electrochemical proton transfer barriers. Catal. Today 2016, 262, 36–40. [Google Scholar] [CrossRef]
- Zheng, J.; Sheng, W.; Zhuang, Z.; Xu, B.; Yan, Y. Universal dependence of hydrogen oxidation and evolution reaction activity of platinum-group metals on pH and hydrogen binding energy. Sci. Adv. 2016, 2, e1501602. [Google Scholar] [CrossRef] [Green Version]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.C.; Uchimura, M.; Paulikas, A.; Stamenkovic, V.; Markovic, N. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Subbaraman, R.; Tripkovic, D.; Chang, K.C.; Strmcnik, D.; Paulikas, A.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N. Trends in activity for the water electrolyzer reactions on 3d-M(Ni, Co, Fe, Mn)-hydr(oxy)oxide catalysts. Nat. Mater. 2012, 11, 550–557. [Google Scholar] [CrossRef]
- Mustain, W. Understanding how high-performance anion exchange membrane fuel cells were achieved: Component, interfacial, and cell-level factors. Curr. Opin. Electrochem. 2018, 12, 233–239. [Google Scholar] [CrossRef]
- Ferriday, T.; Middleton, P. Alkaline fuel cell technology—A review. Int. J. Hydrogen Energy 2021, 46, 18489–18510. [Google Scholar] [CrossRef]
- Gutić, S.; Dobrota, A.; Fako, E.; Skorodumova, N.; López, N.; Pašti, I. Hydrogen evolution reaction-from single crystal to single atom catalysts. Catalysts 2020, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Qi, J.; Lei, F.; Xie, Y. Modulation of electronic structures in two-dimensional electrocatalysts for the hydrogen evolution reaction. Chem. Commun. 2020, 56, 11910–11930. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.; Bligaard, T.; Logadottir, A.; Kitchin, J.; Chen, J.; Pandelov, S.; Stimming, U. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef] [Green Version]
- Ge, Z.; Fu, B.; Zhao, J.; Li, X.; Ma, B.; Chen, Y. A review of the electrocatalysts on hydrogen evolution reaction with an emphasis on Fe, Co and Ni-based phosphides. J. Mater. Sci. 2020, 55, 14081–14104. [Google Scholar] [CrossRef]
- Anantharaj, S.; Kundu, S.; Noda, S. Progress in nickel chalcogenide electrocatalyzed hydrogen evolution reaction. J. Mater. Chem. A 2020, 8, 4174–4192. [Google Scholar] [CrossRef]
- Xu, M.; Liang, T.; Shi, M.; Chen, H. Graphene-Like Two-Dimensional Materials. Chem. Rev. 2013, 113, 3766–3798. [Google Scholar] [CrossRef] [PubMed]
- Chia, X.; Sofer, Z.; Luxa, J.; Pumera, M. Layered Noble Metal Dichalcogenides: Tailoring Electrochemical and Catalytic Properties. ACS Appl. Mater. Interfaces 2017, 9, 25587–25599. [Google Scholar] [CrossRef]
- Nørskov, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhu, Y.; Vasileff, A.; Jiao, Y.; Chen, S.; Song, L.; Zheng, B.; Zheng, Y.; Qiao, S.Z. Strain effect in bimetallic electrocatalysts in the hydrogen evolution reaction. ACS Energy Lett. 2018, 3, 1198–1204. [Google Scholar] [CrossRef]
- Jin, H.; Ruqia, B.; Park, Y.; Kim, H.; Oh, H.S.; Choi, S.I.; Lee, K. Nanocatalyst Design for Long-Term Operation of Proton/Anion Exchange Membrane Water Electrolysis. Adv. Energy Mater. 2021, 11, 2003188. [Google Scholar] [CrossRef]
- Lu, Q.; Hutchings, G.; Yu, W.; Zhou, Y.; Forest, R.; Tao, R.; Rosen, J.; Yonemoto, B.; Cao, Z.; Zheng, H.; et al. Highly porous non-precious bimetallic electrocatalysts for efficient hydrogen evolution. Nat. Commun. 2015, 6, 6567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, P.; Sivanantham, A.; Shanmugam, S. Nanostructured Nickel–Cobalt–Titanium Alloy Grown on Titanium Substrate as Efficient Electrocatalyst for Alkaline Water Electrolysis. ACS Appl. Mater. Interfaces 2017, 9, 12416–12426. [Google Scholar] [CrossRef]
- Gong, M.; Zhou, W.; Tsai, M.C.; Zhou, J.; Guan, M.; Lin, M.C.; Zhang, B.; Hu, Y.; Wang, D.Y.; Yang, J.; et al. Nanoscale nickel oxide/nickel heterostructures for active hydrogen evolution electrocatalysis. Nat. Commun. 2014, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Ji, S.J.; Xue, H.G.; Suen, N.T. HER activity of MxNi1-x (M = Cr, Mo and W; x ≈ 0.2) alloy in acid and alkaline media. Int. J. Hydrogen Energy 2020, 45, 17533–17539. [Google Scholar] [CrossRef]
- Faid, A.; Oyarce Barnett, A.; Seland, F.; Sunde, S. Highly active nickel-based catalyst for hydrogen evolution in anion exchange membrane electrolysis. Catalysts 2018, 8, 614. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zheng, Y.R.; Montoya, J.; Hochfilzer, D.; Cao, A.; Kibsgaard, J.; Chorkendorff, I.; Nørskov, J. Origins of the Instability of Nonprecious Hydrogen Evolution Reaction Catalysts at Open-Circuit Potential. ACS Energy Lett. 2021, 6, 2268–2274. [Google Scholar] [CrossRef]
- Schalenbach, M.; Speck, F.; Ledendecker, M.; Kasian, O.; Goehl, D.; Mingers, A.; Breitbach, B.; Springer, H.; Cherevko, S.; Mayrhofer, K. Nickel-molybdenum alloy catalysts for the hydrogen evolution reaction: Activity and stability revised. Electrochim. Acta 2018, 259, 1154–1161. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, X.; Montoya, J.; Nørskov, J. Predicting aqueous stability of solid with computed Pourbaix diagram using SCAN functional. Npj Comput. Mater. 2020, 6, 1–7. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.; Hautier, G.; Chen, W.; Richards, W.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gao, Y.; Kong, H.; Kim, J.; Choi, S.; Ciucci, F.; Hao, Y.; Yang, S.; Shao, Z.; Lim, J. Non-precious-metal catalysts for alkaline water electrolysis: Operando characterizations, theoretical calculations, and recent advances. Chem. Soc. Rev. 2020, 49, 9154–9196. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Yang, J.; Jeong, J.; Kim, G.; Kwon, C.; Myung, N.; Lee, K.; Choi, S. Promotion Effect of Modified Ni/C by La-Ce Oxide for Durable Hydrogen Evolution Reaction. ACS Sustain. Chem. Eng. 2021, 9, 12508–12513. [Google Scholar] [CrossRef]
- Zhao, X.; Yin, F.; He, X.; Chen, B.; Li, G. Enhancing hydrogen evolution reaction activity on cobalt oxide in alkaline electrolyte by doping inactive rare-earth metal. Electrochim. Acta 2020, 363, 137230. [Google Scholar] [CrossRef]
- Park, Y.; Lee, J.; Jang, M.; Jeong, J.; Park, S.; Choi, W.S.; Kim, Y.; Yang, J.; Choi, S. Co3S4 nanosheets on Ni foam via electrodeposition with sulfurization as highly active electrocatalysts for anion exchange membrane electrolyzer. Int. J. Hydrogen Energy 2020, 45, 36–45. [Google Scholar] [CrossRef]
- Paraknowitsch, J.; Thomas, A. Doping carbons beyond nitrogen: An overview of advanced heteroatom doped carbons with boron, sulfur and phosphorus for energy applications. Energy Environ. Sci. 2013, 6, 2839–2855. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Miao, R.; Huan, T.D.; Mosa, I.; Poyraz, A.; Zhong, W.; Cloud, J.; Kriz, D.; Thanneeru, S.; He, J.; et al. Mesoporous MoO3-x material as an efficient electrocatalyst for hydrogen evolution reactions. Adv. Energy Mater. 2016, 6, 1600528. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Ci, S.; Ma, Y.; Liu, T.; Gao, L.; Qian, P.; Ji, C.; Su, Y. Accelerating 2D MXenes Catalyst Discovery for Hydrogen Evolution Reaction by Computer-Driven Workflow and Ensemble Learning Strategy. J. Mater. Chem. A 2020, 8, 23488–23497. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Zhou, W.; Zhu, Z.; Su, C.; Liu, M.; Shao, Z. A Perovskite Electrocatalyst for Efficient Hydrogen Evolution Reaction. Adv. Mater. 2016, 28, 6442–6448. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, W.; Zhong, Y.; Bu, Y.; Chen, X.; Zhong, Q.; Liu, M.; Shao, Z. A Perovskite Nanorod as Bifunctional Electrocatalyst for Overall Water Splitting. Adv. Energy Mater. 2017, 7, 1602122. [Google Scholar] [CrossRef]
- Laursen, A.; Wexler, R.; Whitaker, M.; Izett, E.; Calvinho, K.; Hwang, S.; Rucker, R.; Wang, H.; Li, J.; Garfunkel, E.; et al. Climbing the Volcano of Electrocatalytic Activity while Avoiding Catalyst Corrosion: Ni3P, a Hydrogen Evolution Electrocatalyst Stable in Both Acid and Alkali. ACS Catal. 2018, 8, 4408–4419. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, H.C.; Hsu, C.S.; Lin, T.S.; Chang, C.J.; Chang, S.C.; Tsai, L.D.; Chen, H.M. Operando unraveling of the structural and chemical stability of P-substituted CoSe2 electrocatalysts toward hydrogen and oxygen evolution reactions in alkaline electrolyte. ACS Energy Lett. 2019, 4, 987–994. [Google Scholar] [CrossRef]
- Zhang, W.; Zou, Y.; Liu, H.; Chen, S.; Wang, X.; Zhang, H.; She, X.; Yang, D. Single-crystalline (FexNi1-x)2P nanosheets with dominant {011¯1¯} facets: Efficient electrocatalysts for hydrogen evolution reaction at all pH values. Nano Energy 2019, 56, 813–822. [Google Scholar] [CrossRef]
- Li, Z.; Niu, W.; Yang, Z.; Kara, A.; Wang, Q.; Wang, M.; Gu, M.; Feng, Z.; Du, Y.; Yang, Y. Boosting alkaline hydrogen evolution: The dominating role of interior modification in surface electrocatalysis. Energy Environ. Sci. 2020, 13, 3110–3118. [Google Scholar] [CrossRef]
- Kim, J.C.; Kim, J.; Park, J.; Ahn, S.; Kim, D.W. Ru2P Nanofibers for High-Performance Anion Exchange Membrane Water Electrolyzer. Chem. Eng. J. 2021, 420, 130491. [Google Scholar] [CrossRef]
- Xin, H.; Dai, Z.; Zhao, Y.; Guo, S.; Sun, J.; Luo, Q.; Zhang, P.; Sun, L.; Ogiwara, N.; Kitagawa, H.; et al. Recording the Pt-beyond Hydrogen Production Electrocatalysis by Dirhodium Phosphide with an Overpotential of Only 4.3 mV in Alkaline Electrolyte. Appl. Catal. B Environ. 2021, 297, 120457. [Google Scholar] [CrossRef]
- Ding, M.; Xu, H.; Liu, M.; Wang, Y.; Wang, A.; Lin, T.; Zhang, L.; Zhang, K. Interface construction and porosity engineering to trigger efficient hydrogen evolution of two-dimensional porous molybdenum phosphide/dioxide heterojunction nanosheets in acidic and alkaline electrolytes. Chem. Eng. J. 2022, 430, 132674. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, C.; Xu, S.; Liu, Y.; Jiang, D.; Zhu, J. Facile synthesis of hierarchical NiCoP nanosheets/NiCoP nanocubes homojunction electrocatalyst for highly efficient and stable hydrogen evolution reaction. Appl. Surf. Sci. 2021, 565, 150537. [Google Scholar] [CrossRef]
- Men, Y.; Li, P.; Zhou, J.; Chen, S.; Luo, W. Trends in Alkaline Hydrogen Evolution Activity on Cobalt Phosphide Electrocatalysts Doped with Transition Metals. Cell Rep. Phys. Sci. 2020, 1, 100136. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, L.; Hensen, E.; Hofmann, J. Evaluating the stability of Co2P electrocatalysts in the hydrogen evolution reaction for both acidic and alkaline electrolytes. ACS Energy Lett. 2018, 3, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lv, Q.; Li, G.; Ge, J.; Liu, C. and Xing, W. Core-shell structured Ni12P5/Ni3(PO4)2 hollow spheres as difunctional and efficient electrocatalysts for overall water electrolysis. Appl. Catal. B Environ. 2017, 204, 486–496. [Google Scholar] [CrossRef]
- Sinha, A.; Ojha, U. Recent Trends in Development of Metal Nitride Nanocatalysts for Water Electrolysis Application. In Electrocatalysis; IntechOpen: London, UK, 2021. [Google Scholar]
- Xing, Z.; Li, Q.; Wang, D.; Yang, X.; Sun, X. Self-supported nickel nitride as an efficient high-performance three-dimensional cathode for the alkaline hydrogen evolution reaction. Electrochim. Acta 2016, 191, 841–845. [Google Scholar] [CrossRef]
- Liao, J.W.; Lu, X.; Huang, B.Y.; Yu, G.Q.; Li, X.B. Hydrogen evolution on different facets of δ1-MoN and δ3-MoN: Considering the adsorbed oxygen and hydroxyl by Surface Pourbaix diagrams. Int. J. Hydrogen Energy 2021, 46, 9077–9086. [Google Scholar] [CrossRef]
- Lv, Z.; Tahir, M.; Lang, X.; Yuan, G.; Pan, L.; Zhang, X.; Zou, J.J. Well-dispersed molybdenum nitrides on a nitrogen-doped carbon matrix for highly efficient hydrogen evolution in alkaline media. J. Mater. Chem. A 2017, 5, 20932–20937. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Li, Y.; He, B.; Wang, R.; Gong, Y. MOFs-derived hybrid nanosheet arrays of nitrogen-rich CoS2 and nitrogen-doped carbon for efficient hydrogen evolution in both alkaline and acidic media. Int. J. Hydrogen Energy 2018, 43, 23319–23326. [Google Scholar] [CrossRef]
- Hanif, S.; Iqbal, N.; Shi, X.; Noor, T.; Ali, G.; Kannan, A. NiCo-N-doped carbon nanotubes based cathode catalyst for alkaline membrane fuel cell. Renew. Energy 2020, 154, 508–516. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, T.; Chen, M.; Tong, Y.; Zhang, N.; Peng, X.; Chu, W.; Wu, X.; Wu, C.; Xie, Y. Enhanced catalytic activity in nitrogen-anion modified metallic cobalt disulfide porous nanowire arrays for hydrogen evolution. ACS Catal. 2017, 7, 7405–7411. [Google Scholar] [CrossRef]
- Tang, C.; Hu, Q.; Li, F.; He, C.; Chai, X.; Zhu, C.; Liu, J.; Zhang, Q.; Zhu, B.; Fan, L. Coupled molybdenum carbide and nitride on carbon nanosheets: An efficient and durable hydrogen evolution electrocatalyst in both acid and alkaline media. Electrochim. Acta 2018, 280, 323–331. [Google Scholar] [CrossRef]
- Liu, W.; Xia, T.; Ye, Y.; Wang, H.; Fang, Z.; Du, Z.; Hou, X. Self-supported Ni3N nanoarray as an efficient nonnoble-metal catalyst for alkaline hydrogen evolution reaction. Int. J. Hydrogen Energy 2021, 46, 27037–27043. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, L.; Han, C.; Zong, H.; Yang, G.; Lin, S.; Kumar, A.; Jadhav, A.; Tran, N.; Hwang, Y.; et al. Identifying the Activity Origin of a Cobalt Single-Atom Catalyst for Hydrogen Evolution Using Supervised Learning. Adv. Funct. Mater. 2021, 31, 2100547. [Google Scholar] [CrossRef]
- Cao, L.; Luo, Q.; Liu, W.; Lin, Y.; Liu, X.; Cao, Y.; Zhang, W.; Wu, Y.; Yang, J.; Yao, T.; et al. Identification of single-atom active sites in carbon-based cobalt catalysts during electrocatalytic hydrogen evolution. Nat. Catal. 2019, 2, 134–141. [Google Scholar] [CrossRef]
- Shen, H.; Liang, S.; Adimi, S.; Guo, X.; Zhu, Y.; Guo, H.; Thomas, T.; Attfield, T.; Yang, M. Supporting Nickel on Vanadium Nitride for Platinum Comparable Hydrogen Evolution in Alkaline Solution. J. Mater. Chem. 2021, 9, 19669–19674. [Google Scholar] [CrossRef]
- Yang, Y.; Lai, L.; Wei, L.; Chen, Y. Degradation: A Critical Challenge for M–N–C Electrocatalysts. Available online: https://www.sciencedirect.com/science/article/abs/pii/S2095495621005714 (accessed on 15 November 2021).
- Xie, X.; He, C.; Li, B.; He, Y.; Cullen, D.; Wegener, E.; Kropf, A.; Martinez, U.; Cheng, Y.; Engelhard, M.; et al. Performance enhancement and degradation mechanism identification of a single-atom Co-N-C catalyst for proton exchange membrane fuel cells. Nat. Catal. 2020, 3, 1044–1054. [Google Scholar] [CrossRef]
- Kim, J.; Yoo, J.; Lee, H.; Sung, Y.E.; Hyeon, T. Single-atom M–N–C catalysts for oxygen reduction electrocatalysis. Trends Chem. 2021, 3, 779–794. [Google Scholar] [CrossRef]
- Jin, H.; Liu, X.; Chen, S.; Vasileff, A.; Li, L.; Jiao, Y.; Song, L.; Zheng, Y.; Qiao, S.Z. Heteroatom-Doped Transition Metal Electrocatalysts for Hydrogen Evolution Reaction. ACS Energy Lett. 2019, 4, 805–810. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, B.; Zhu, J.; Liu, Z.; Zhang, H.; Li, Y. Callistemon-like Zn and S co-doped CoP nanorod clusters as highly efficient electrocatalysts for neutral-pH overall water splitting. J. Mater. Chem. A 2019, 7, 22453–22462. [Google Scholar] [CrossRef]
- Xu, K.; Sun, Y.; Sun, Y.; Zhang, Y.; Jia, G.; Zhang, Q.; Gu, L.; Li, S.; Li, Y.; Fan, H. Yin-Yang Harmony: Metal and Non-metal Dual-Doping Boosts Electrocatalytic Activity for Alkaline Hydrogen Evolution. ACS Energy Lett. 2018, 3, 2750–2756. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Y.; Liu, Q.; Guo, J.; Zhang, X. Vanadium and nitrogen co-doped CoP nanoleaf array as pH-universal electrocatalyst for efficient hydrogen evolution. J. Alloys Compd. 2019, 791, 1070–1078. [Google Scholar] [CrossRef]
- Liu, W.; Hu, E.; Jiang, H.; Xiang, Y.; Weng, Z.; Li, M.; Fan, Q.; Yu, X.; Altman, E.; Wang, H. A highly active and stable hydrogen evolution catalyst based on pyrite-structured cobalt phosphosulfide. Nat. Commun. 2016, 7, 10771. [Google Scholar] [CrossRef]
- Verma, C.; Verma, D.; Ebenso, E.; Quraishi, M. Sulfur and phosphorus heteroatom-containing compounds as corrosion inhibitors: An overview. Heteroat. Chem. 2018, 29, e21437. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tan, X.; Hocking, R.; Bo, X.; Ren, H.; Johannessen, B.; Smith, S.; Zhao, C. Implanting Ni-O-VOx sites into Cu-doped Ni for low-overpotential alkaline hydrogen evolution. Nat. Commun. 2020, 11, 2720. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, Y.; Liu, H.; Shao, M.; Chen, Z.; Ma, T.; Wu, Z.; Wang, L. N, P-doped carbon supported ruthenium doped Rhenium phosphide with porous nanostructure for hydrogen evolution reaction using sustainable energies. J. Colloid Interface Sci. 2022, 606, 1874–1881. [Google Scholar] [CrossRef]
- Zhang, H.; Li, W.; Feng, X.; Zhu, L.; Fang, Q.; Li, S.; Wang, L.; Li, Z.; Kou, Z. A chainmail effect of ultrathin N-doped carbon shell on Ni2P nanorod arrays for efficient hydrogen evolution reaction catalysis. J. Colloid Interface Sci. 2022, 607, 281–289. [Google Scholar] [CrossRef]
- Kumar, A.; Bui, V.; Lee, J.; Jadhav, A.; Hwang, Y.; Kim, M.; Kawazoe, Y.; Lee, H. Modulating Interfacial Charge Density of NiP2-FeP2 via Coupling with Metallic Cu for Accelerating Alkaline Hydrogen Evolution. ACS Energy Lett. 2021, 6, 354–363. [Google Scholar] [CrossRef]
- Ling, T.; Zhang, T.; Ge, B.; Han, L.; Zheng, L.; Lin, F.; Xu, Z.; Hu, W.B.; Du, X.W.; Davey, K.; et al. Well-dispersed nickel-and zinc-tailored electronic structure of a transition metal oxide for highly active alkaline hydrogen evolution reaction. Adv. Mater. 2019, 31, 1807771. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.; Bonde, J.; Chorkendorff, I.; Nørskov, J. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef]
- Mamun, O.; Winther, K.; Boes, J.; Bligaard, T. A bayesian framework for adsorption energy prediction on bimetallic alloy catalysts. Npj Comput. Mater. 2020, 6, 177. [Google Scholar] [CrossRef]
- Jung, Y.; Zhou, Y.; Cha, J. Intercalation in two-dimensional transition metal chalcogenides. Inorg. Chem. Front. 2016, 3, 452–463. [Google Scholar] [CrossRef]
- Wang, C.; Tian, B.; Wu, M.; Wang, J. Revelation of the excellent intrinsic activity of MoS2|NiS|MoO3 nanowires for hydrogen evolution reaction in alkaline medium. ACS Appl. Mater. Interfaces 2017, 9, 7084–7090. [Google Scholar] [CrossRef]
- Qu, Y.; Yang, M.; Chai, J.; Tang, Z.; Shao, M.; Kwok, C.; Yang, M.; Wang, Z.; Chua, D.; Wang, S.; et al. Facile Synthesis of Vanadium-Doped Ni3S2 Nanowire Arrays as Active Electrocatalyst for Hydrogen Evolution Reaction. ACS Appl. Mater. Interfaces 2017, 9, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Jiang, Y.; Liu, S.; Zhang, M.; Guo, Q.; Shen, W.; He, R.; Su, W.; Li, M. Regulating the electron density of dual transition metal sulfide heterostructures for highly efficient hydrogen evolution in alkaline electrolytes. Nanoscale 2019, 11, 14016–14023. [Google Scholar] [CrossRef]
- Liu, T.; Asiri, A.; Sun, X. Electrodeposited Co-doped NiSe2 nanoparticles film: A good electrocatalyst for efficient water splitting. Nanoscale 2016, 8, 3911–3915. [Google Scholar] [CrossRef]
- Anantharaj, S.; Karthick, K.; Kundu, S. NiTe2 Nanowire Outperforms Pt/C in High-Rate Hydrogen Evolution at Extreme pH Conditions. Inorg. Chem. 2018, 57, 3082–3096. [Google Scholar] [CrossRef]
- Li, Y.; Tan, X.; Tan, H.; Ren, H.; Chen, S.; Yang, W.; Smith, S.; Zhao, C. Phosphine vapor-assisted construction of heterostructured Ni2P/NiTe2 catalysts for efficient hydrogen evolution. Energy Environ. Sci. 2020, 13, 1799–1807. [Google Scholar] [CrossRef]
- Mingli, F.; Xue, L.; Dandan, W.; Yinling, W.; Maoguo, L. Fabrication of Te@NiTe2/NiS heterostructures for electrocatalytic hydrogen evolution reaction. Electrochim. Acta 2019, 328, 135075. [Google Scholar] [CrossRef]
- Bhat, K.S.; Nagaraja, H. Performance evaluation of molybdenum dichalcogenide (MoX2; X = S, Se, Te) nanostructures for hydrogen evolution reaction. Int. J. Hydrogen Energy 2019, 44, 17878–17886. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, S.; Pan, H.; Xiao, S.; Guo, Z.; Tang, L.; Khan, U.; Ding, B.F.; Li, M.; Cai, Z.; et al. Unsaturated Single Atoms on Monolayer Transition Metal Dichalcogenides for Ultrafast Hydrogen Evolution. ACS Nano 2020, 14, 767–776. [Google Scholar]
- Ran, N.; Sun, B.; Qiu, W.; Song, E.; Chen, T.; Liu, J. Identifying Metallic Transition-Metal Dichalcogenides for Hydrogen Evolution through Multilevel High-Throughput Calculations and Machine Learning. J. Phys. Chem. Lett. 2021, 12, 2102–2111. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, D.; Gong, L.; Zhang, L.; Xia, Z. Catalytic Activity Origin and Design Principles of Graphitic Carbon Nitride Electrocatalysts for Hydrogen Evolution. Front. Mater. 2019, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Ling, T.; Jaroniec, M.; Qiao, S.Z. Recent Progress in Engineering the Atomic and Electronic Structure of Electrocatalysts via Cation Exchange Reactions. Adv. Mater. 2020, 32, 2001866. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhou, M.; Long, A.; Xue, Y.; Liao, H.; Wei, C.; Xu, Z. Heterostructured electrocatalysts for hydrogen evolution reaction under alkaline conditions. Nano-Micro Lett. 2018, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.F.; Lu, B.A.; Qu, X.M.; Ye, J.Y.; Zhang, J.M.; Yin, S.H.; Wu, Q.H.; Wang, R.X.; Shen, S.Y.; Sheng, T.; et al. Does the oxophilic effect serve the same role for hydrogen evolution/oxidation reaction in alkaline media? Nano Energy 2019, 62, 601–609. [Google Scholar] [CrossRef]
- Strmcnik, D.; Uchimura, M.; Wang, C.; Subbaraman, R.; Danilovic, N.; Van Der Vliet, D.; Paulikas, A.; Stamenkovic, V.; Markovic, N. Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption. Nat. Chem. 2013, 5, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, S.; Jia, Q.; Bates, M.; Li, J.; Xu, C.; Gath, K.; Yang, J.; Waldecker, J.; Che, H.; Liang, W.; et al. Tuning Nb-Pt Interactions to Facilitate Fuel Cell Electrocatalysis. ACS Catal. 2017, 7, 4936–4946. [Google Scholar] [CrossRef]
- Xu, W.; Wang, B.; Ni, X.; Liu, H.; Wang, W.; Zhang, L.; Zhang, H.; Peng, Z.; Liu, Z. Heterogeneous Synergetic Effect of Metal–Oxide Interfaces for Efficient Hydrogen Evolution in Alkaline Solutions. ACS Appl. Mater. Interfaces 2021, 13, 13838–13847. [Google Scholar] [CrossRef]
- Chen, W.; Gu, J.; Du, Y.; Song, F.; Bu, F.; Li, J.; Yuan, Y.; Luo, R.; Liu, Q.; Zhang, D. Achieving Rich and Active Alkaline Hydrogen Evolution Heterostructures via Interface Engineering on 2D 1T-MoS2 Quantum Sheets. Adv. Funct. Mater. 2020, 30, 2000551. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Wang, J.; Ruan, Y.; Ji, X.; Xu, K.; Chen, C.; Wan, H.; Miao, L.; Jiang, J. Interface engineering: The Ni(OH)2/MoS2 heterostructure for highly efficient alkaline hydrogen evolution. Nano Energy 2017, 37, 74–80. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, X.; Zhang, J.; Wan, S.; Guo, S.; Lu, G.; Yao, J.; Huang, X. Precise tuning in platinum-nickel/nickel sulfide interface nanowires for synergistic hydrogen evolution catalysis. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Chen, J.; Jin, Q.; Li, Y.; Li, Y.; Cui, H.; Wang, C. Design Superior Alkaline Hydrogen Evolution Electrocatalyst by Engineering Dual Active Sites for Water Dissociation and Hydrogen Desorption. ACS Appl. Mater. Interfaces 2019, 11, 38771–38778. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T.; Liu, P.; Liao, Z.; Liu, S.; Zhuang, X.; Chen, M.; Zschech, E.; Feng, X. Efficient hydrogen production on MoNi4 electrocatalysts with fast water dissociation kinetics. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Wang, X.; Chen, X.; Zhang, Q.; Li, N.; Luo, H. Interfacial engineering of Ni(OH)2 on W2C for remarkable alkaline hydrogen production. Appl. Catal. Environ. 2021, 540, 120818. [Google Scholar] [CrossRef]
- Hua, W.; Sun, H.; Liu, H.; Li, Y.; Wang, J.G. Interface engineered NiMoN/Ni3N heterostructures for enhanced alkaline hydrogen evolution reaction. Appl. Surf. Sci. 2021, 540, 148407. [Google Scholar] [CrossRef]
- Feng, Y.; Li, Z.; Cheng, C.Q.; Kang, W.J.; Mao, J.; Shen, G.R.; Yang, J.; Dong, C.K.; Liu, H.; Du, X.W. Strawberry-like Co3O4-Ag bifunctional catalyst for overall water splitting. Appl. Catal. B Environ. 2021, 299, 120658. [Google Scholar] [CrossRef]
- Hu, Y.; Xiong, T.; Balogun Tang, M.-S.J.; Huang, Y.; Adekoya, D.; Zhang, S.; Tong, Y. Enhanced metallicity boosts hydrogen evolution capability of dual-bimetallic Ni–Fe nitride nanoparticles. Mater. Today Phys. 2020, 15, 100267. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, Y.; Zhao, Z.; Zhang, Q.H.; Huang, L.B.; Gu, L.; Lu, G.; Hu, J.S.; Wan, L.J. Steering elementary steps towards efficient alkaline hydrogen evolution via size-dependent Ni/NiO nanoscale heterosurfaces. Natl. Sci. Rev. 2020, 7, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.X.; Wu, J.Q.; Tong, Y.X.; Li, G.R. Efficient Hydrogen Evolution on Cu Nanodots-Decorated Ni3S2 Nanotubes by Optimizing Atomic Hydrogen Adsorption and Desorption. J. Am. Chem. Soc. 2018, 140, 610–617. [Google Scholar] [CrossRef]
- Markovića, N.; Sarraf, S.; Gasteiger, H.; Ross, P. Hydrogen electrochemistry on platinum low-index single-crystal surfaces in alkaline solution. J. Chem. Soc. Faraday Trans. 1996, 92, 3719–3725. [Google Scholar] [CrossRef]
- Voiry, D.; Yamaguchi, H.; Li, J.; Silva, R.; Alves, D.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V.; Eda, G.; et al. Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution. Nat. Mater. 2013, 12, 850–855. [Google Scholar] [CrossRef] [PubMed]
- You, B.; Tang, M.; Tsai, C.; Abild-Pedersen, F.; Zheng, X.; Li, H. Enhancing electrocatalytic water splitting by strain engineering. Adv. Mater. 2019, 31, 1807001. [Google Scholar] [CrossRef]
- Khorshidi, A.; Violet, J.; Hashemi, J.; Peterson, A. How strain can break the scaling relations of catalysis. Nat. Catal. 2018, 1, 263–268. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Zhang, M.; Zhou, Y.; Rao, D.; Zhong, C.; Zhang, J.; Han, X.; Hu, W.; Zhang, Y.; et al. Lattice-Strain Engineering of Homogeneous NiS0.5Se0.5 Core-Shell Nanostructure as a Highly Efficient and Robust Electrocatalyst for Overall Water Splitting. Adv. Mater. 2020, 32, 2000231. [Google Scholar] [CrossRef] [PubMed]
- Schnur, S.; Groß, A. Strain and coordination effects in the adsorption properties of early transition metals: A density-functional theory study. Phys. Rev. B 2010, 81, 033402. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Wu, C.; Yang, G.; Yang, S. CO oxidation over strained Pt (100) surface: A DFT study. J. Phys. Chem. C 2015, 119, 15500–15505. [Google Scholar] [CrossRef]
- Rosen, A.; Notestein, J.; Snurr, R. Structure-Activity Relationships That Identify Metal-Organic Framework Catalysts for Methane Activation. ACS Catal. 2019, 9, 3576–3587. [Google Scholar] [CrossRef]
- Ling, T.; Yan, D.Y.; Wang, H.; Jiao, Y.; Hu, Z.; Zheng, Y.; Zheng, L.; Mao, J.; Liu, H.; Du, X.W.; et al. Activating cobalt (II) oxide nanorods for efficient electrocatalysis by strain engineering. Nat. Commun. 2017, 8, 1509. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Cheng, H.; Lv, H.; Wang, J.; Liu, L.; Liu, S.; Wu, X.; Chu, W.; Wu, C.; Xie, Y. Controllable Surface Reorganization Engineering on Cobalt Phosphide Nanowire Arrays for Efficient Alkaline Hydrogen Evolution Reaction. Adv. Mater. 2018, 30, 1703322. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Feng, Y.; Liang, Y.L.; Cheng, C.Q.; Dong, C.K.; Liu, H.; Du, X.W. Stable Rhodium (IV) Oxide for Alkaline Hydrogen Evolution Reaction. Adv. Mater. 2020, 32, 1908521. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhang, W.; Zeng, J.; He, L.; Li, D.; Gao, Q. Plasma-Engineered MoP with nitrogen doping: Electron localization toward efficient alkaline hydrogen evolution. Appl. Catal. B Environ. 2020, 268, 118441. [Google Scholar] [CrossRef]
- Anantharaj, S.; Sugime, H.; Noda, S. Surface amorphized nickel hydroxy sulfide for efficient hydrogen evolution reaction in alkaline medium. Chem. Eng. J. 2021, 408, 127275. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Z.; Cao, X.; Hu, J.; Wu, X.; Ng, A.; Lu, G.P.; Chen, Z. Elucidating the sources of activity and stability of FeP electrocatalyst for hydrogen evolution reactions in acidic and alkaline media. Appl. Catal. B Environ. 2020, 260, 118156. [Google Scholar] [CrossRef]
- Chen, W.Z.; Liu, P.Y.; Zhang, L.; Liu, Y.; Liu, Z.; He, J.; Wang, Y.Q. High-Efficient and Durable Overall Water Splitting Performance by Interfacial Engineering of Fe-Doped Urchin-like Ni2P/Ni3S2 Heterostructure. Chem. Eng. J. 2021, 424, 130434. [Google Scholar] [CrossRef]
- Luo, M.; Guo, S. Strain-controlled electrocatalysis on multimetallic nanomaterials. Nat. Rev. Mater. 2017, 2, 17059. [Google Scholar] [CrossRef]
- Ma, Q.; Hu, C.; Liu, K.; Hung, S.F.; Ou, D.; Chen, H.; Fu, G.; Zheng, N. Identifying the electrocatalytic sites of nickel disulfide in alkaline hydrogen evolution reaction. Nano Energy 2017, 41, 148–153. [Google Scholar] [CrossRef]
- Liu, L.; Chu, X.; Liao, J.; Huang, Y.; Li, Y.; Ge, Z.; Hickner, M.; Li, N. Tuning the properties of poly (2,6-dimethyl-1,4-phenylene oxide) anion exchange membranes and their performance in H2/O2 fuel cells. Energy Environ. Sci. 2018, 11, 435–446. [Google Scholar] [CrossRef]
- He, Y.; Liu, S.; Priest, C.; Shi, Q.; Wu, G. Atomically dispersed metal-nitrogen-carbon catalysts for fuel cells: Advances in catalyst design, electrode performance, and durability improvement. Chem. Soc. Rev. 2020, 49, 3484–3524. [Google Scholar] [CrossRef] [PubMed]
- Tkaczyk, A.; Bartl, A.; Amato, A.; Lapkovskis, V.; Petranikova, M. Sustainability evaluation of essential critical raw materials: Cobalt, niobium, tungsten and rare earth elements. J. Phys. D Appl. Phys. 2018, 51, 203001. [Google Scholar] [CrossRef] [Green Version]
- Entr, D. Report on Critical Raw Materials for the EU; European Commission: Brussels, Belgium, 2014. [Google Scholar]
- Faber, F.; Hutchison, L.; Huang, B.; Gilmer, J.; Schoenholz, S.; Dahl, G.; Vinyals, O.; Kearnes, S.; Riley, P.; Von Lilienfeld, O. Prediction errors of molecular machine learning models lower than hybrid DFT error. J. Chem. Theory Comput. 2017, 13, 5255–5264. [Google Scholar] [CrossRef] [PubMed]





















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferriday, T.B.; Middleton, P.H.; Kolhe, M.L. Review of the Hydrogen Evolution Reaction—A Basic Approach. Energies 2021, 14, 8535. https://doi.org/10.3390/en14248535
Ferriday TB, Middleton PH, Kolhe ML. Review of the Hydrogen Evolution Reaction—A Basic Approach. Energies. 2021; 14(24):8535. https://doi.org/10.3390/en14248535
Chicago/Turabian StyleFerriday, Thomas B., Peter Hugh Middleton, and Mohan Lal Kolhe. 2021. "Review of the Hydrogen Evolution Reaction—A Basic Approach" Energies 14, no. 24: 8535. https://doi.org/10.3390/en14248535
APA StyleFerriday, T. B., Middleton, P. H., & Kolhe, M. L. (2021). Review of the Hydrogen Evolution Reaction—A Basic Approach. Energies, 14(24), 8535. https://doi.org/10.3390/en14248535