
Theoretical Study of CO Adsorption and Activation on Orthorhombic Fe7C3(001) Surfaces for Fischer–Tropsch Synthesis Using Density Functional Theory Calculations


Abstract
:1. Introduction
2. Computational Method
3. Results and Discussion
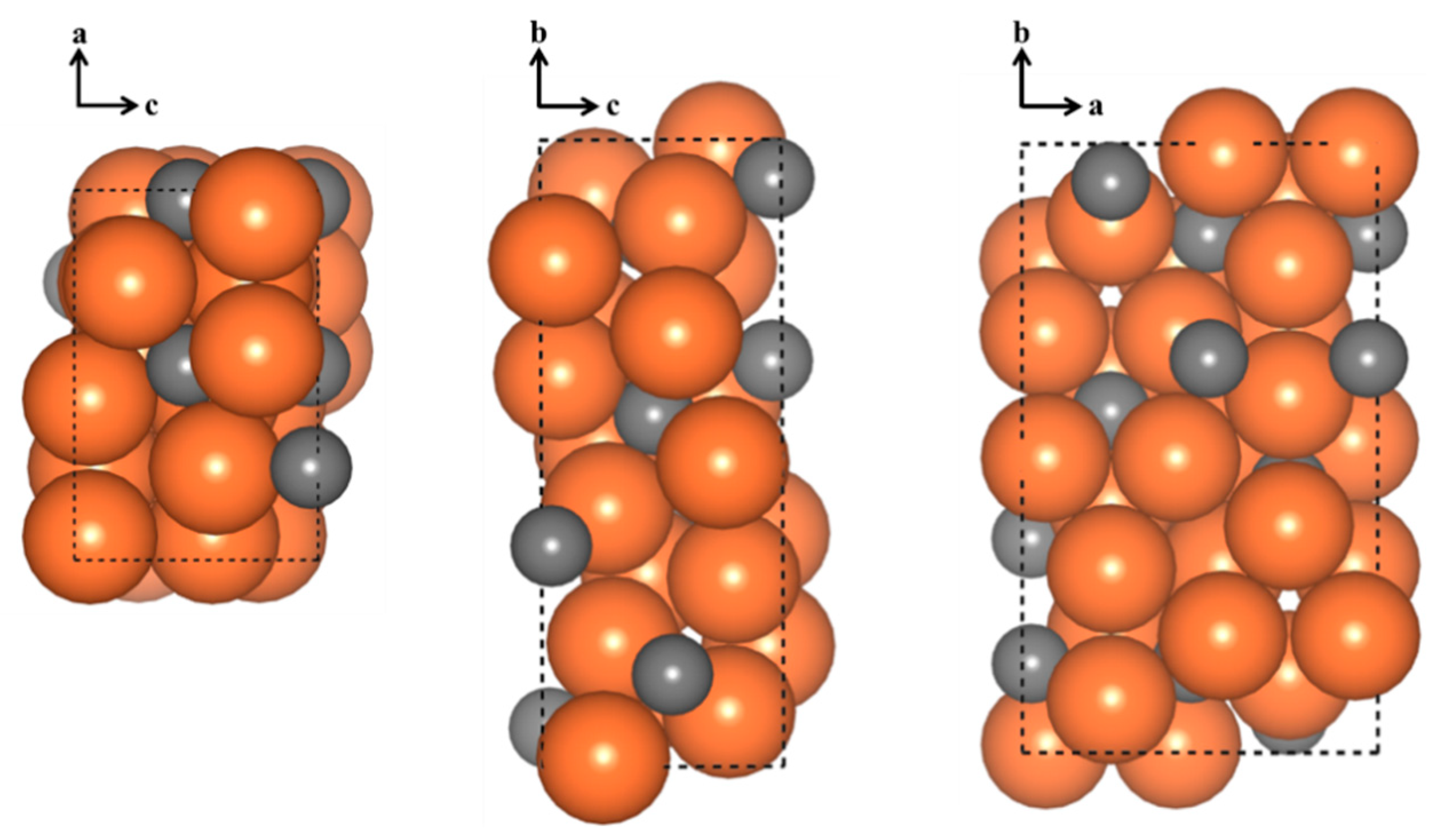
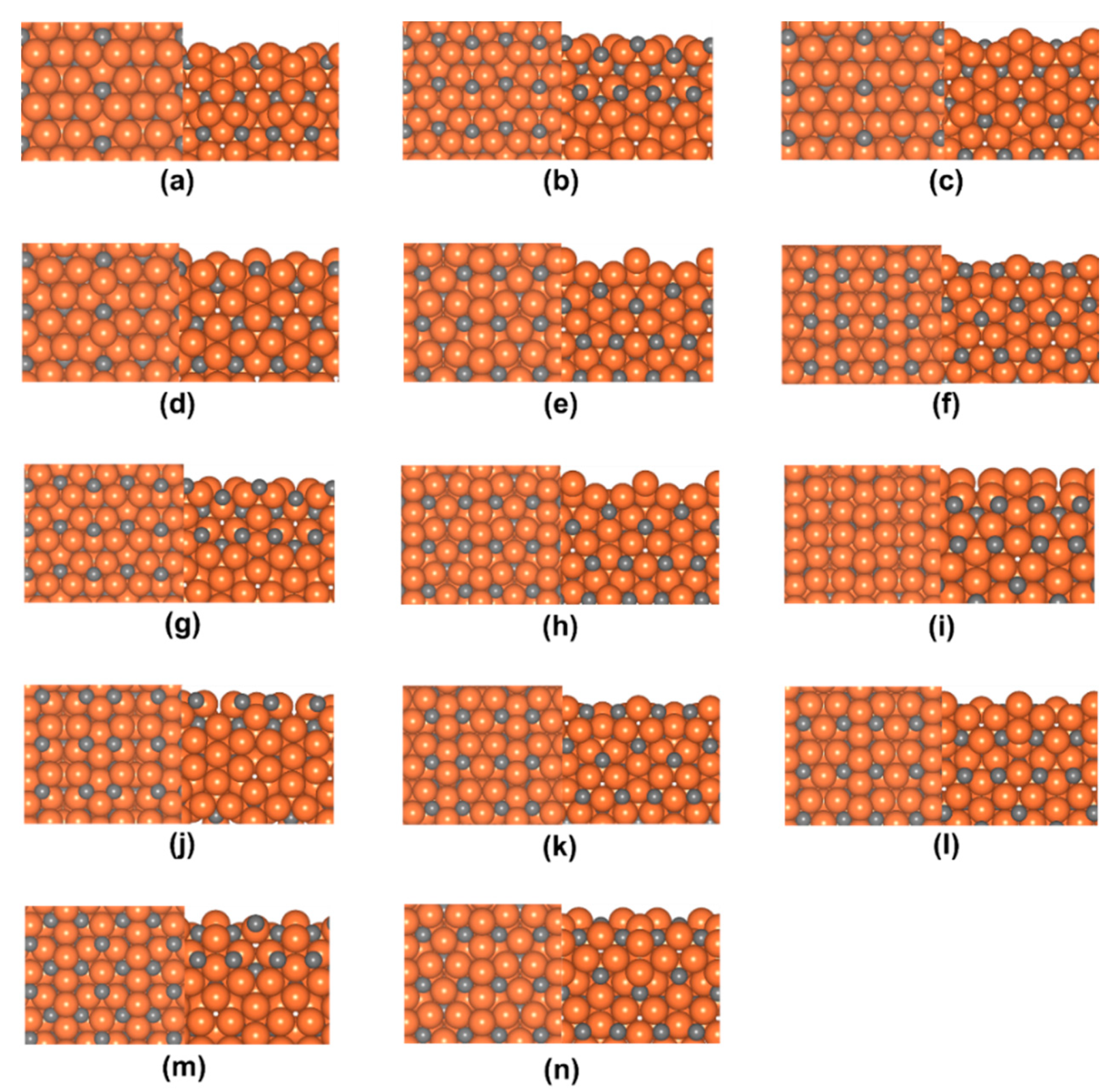
3.1. Surface Energies
3.2. CO* Activation
3.2.1. CO*, C*, and O* Adsorption
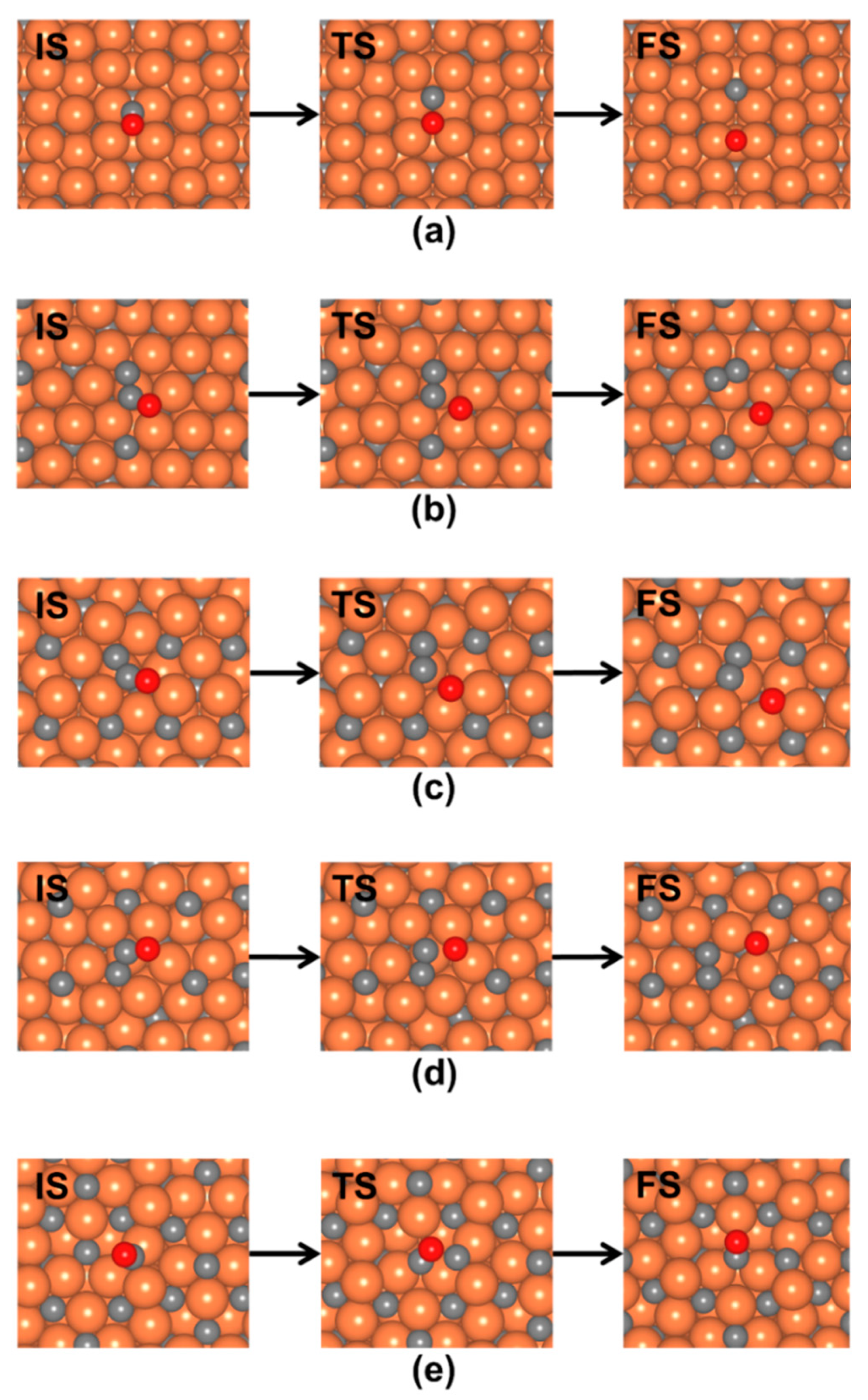
3.2.2. Direct CO* Dissociation
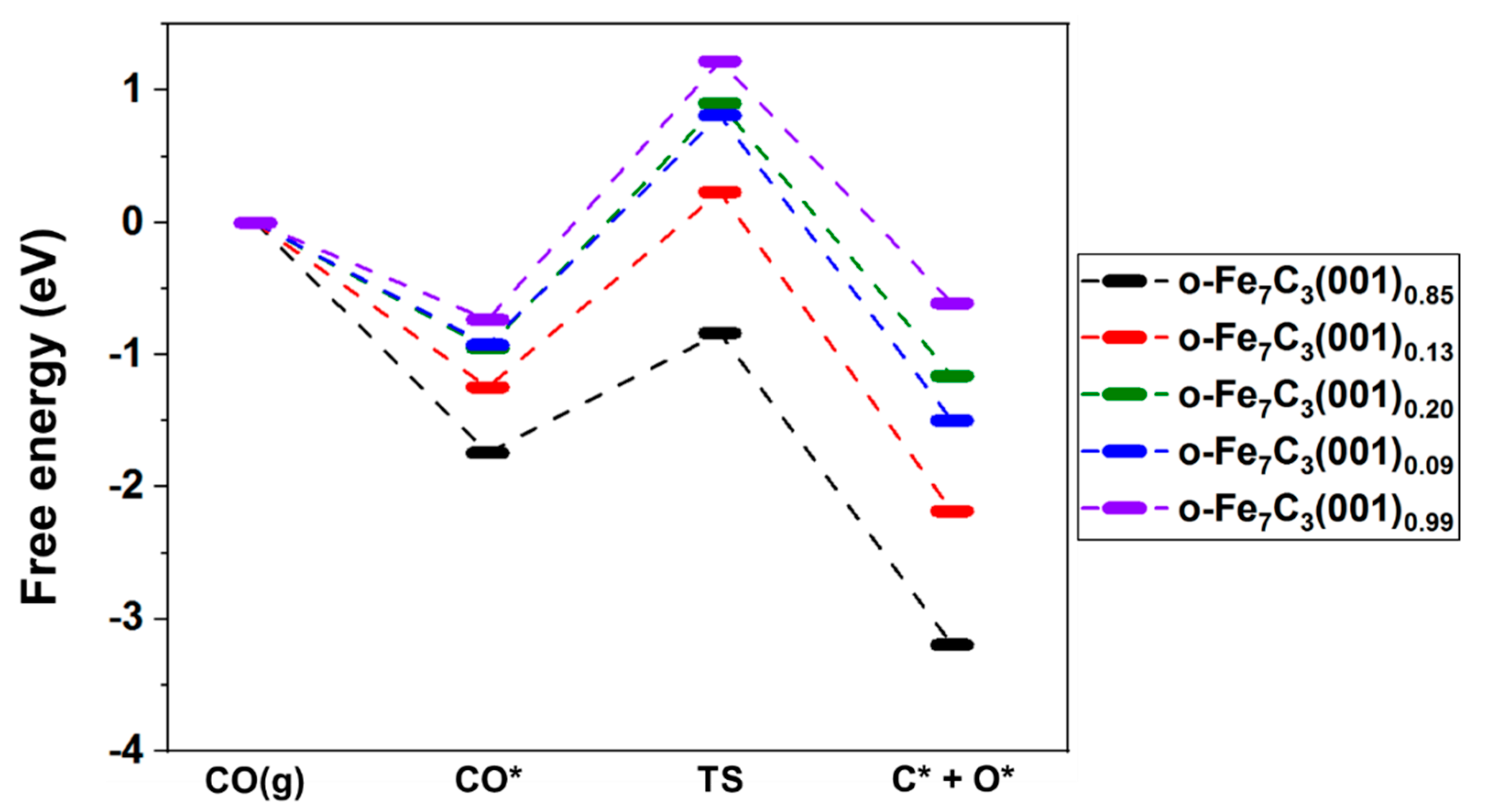
3.2.3. Free Energy Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dry, M.E. The Fischer-Tropsch process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Steynberg, A.P. Introduction to Fischer-Tropsch Technology. Stud. Surf. Sci. Catal. 2004, 152, 1–63. [Google Scholar]
- Huang, C.-S.; Xu, L.; Davis, B.H. Fischer-Tropsch synthesis: Impact of pretreatment of ultrafine iron oxide upon catalyst structure and selectivity. Fuel Sci. Technol. Int. 1993, 11, 639–664. [Google Scholar] [CrossRef]
- Jin, Y.; Datye, A.K. Phase transformations in iron Fischer-Tropsch catalysts during temperature-programmed reduction. J. Catal. 2000, 196, 8–17. [Google Scholar] [CrossRef]
- Zhang, Q.; Kang, J.; Wang, Y. Development of novel catalysts for Fischer-Tropsch synthesis: Tuning the product selectivity. ChemCatChem 2010, 2, 1030–1058. [Google Scholar] [CrossRef]
- Pham, T.H.; Duan, X.; Qian, G.; Zhou, X.; Chen, D. CO activation pathways of Fischer-Tropsch synthesis on χ-Fe5C2 (510): Direct versus hydrogen-assisted CO dissociation. J. Phys. Chem. C 2014, 118, 10170–10176. [Google Scholar] [CrossRef]
- Chen, B.; Wang, D.; Duan, X.; Liu, W.; Li, Y.; Qian, G.; Yuan, W.; Holmen, A.; Zhou, X.; Chen, D. Charge-tuned CO activation over a χ-Fe5C2 Fischer-Tropsch catalyst. ACS Catal. 2018, 8, 2709–2714. [Google Scholar] [CrossRef]
- Datye, A.K.; Jin, Y.; Mansker, L.; Motjope, R.T.; Dlamini, T.H.; Coville, N.J. The nature of the active phase in iron Fischer-Tropsch catalysts. Stud. Surf. Sci. Catal. 2000, 130, 1139–1144. [Google Scholar]
- Rivera de la Cruz, J.G.; Sabbe, M.K.; Reyniers, M.F. First principle study on the adsorption of hydrocarbon chains involved in Fischer-Tropsch synthesis over iron carbides. J. Phys. Chem. C 2017, 121, 25052–25063. [Google Scholar] [CrossRef]
- Chang, Q.; Zhang, C.; Liu, C.; Wei, Y.; Cheruvathur, A.V.; Dugulan, A.I.; Niemantsverdriet, J.W.; Liu, X.; He, Y.; Qing, M.; et al. Relationship between iron carbide phases (ε-Fe2C, Fe7C3, and χ-Fe5C2) and catalytic performances of Fe/SiO2 Fischer-Tropsch catalysts. ACS Catal. 2018, 8, 3304–3316. [Google Scholar] [CrossRef]
- Ojeda, M.; Nabar, R.; Nilekar, A.U.; Ishikawa, A.; Mavrikakis, M.; Iglesia, E. CO activation pathways and the mechanism of Fischer-Tropsch synthesis. J. Catal. 2010, 272, 287–297. [Google Scholar] [CrossRef]
- Petersen, M.A.; van Rensburg, W.J. CO dissociation at vacancy sites on Hägg iron carbide: Direct versus hydrogen-assisted routes investigated with DFT. Top. Catal. 2015, 58, 665–674. [Google Scholar] [CrossRef]
- Ozbek, M.O.; Niemantsverdriet, J.H. Elementary reactions of CO and H2 on C-terminated χ-Fe5C2(001) surfaces. J. Catal. 2014, 317, 158–166. [Google Scholar] [CrossRef]
- Cao, D.B.; Li, Y.W.; Wang, J.; Jiao, H. Adsorption and reaction of surface carbon species on Fe5C2(001). J. Phys. Chem. C 2008, 112, 14883–14890. [Google Scholar] [CrossRef]
- Yang, S.; Chun, H.-J.; Lee, S.; Han, S.J.; Lee, K.Y.; Kim, Y.T. Comparative study of olefin production from CO and CO2 using Na- and K-promoted zinc ferrite. ACS Catal. 2020, 10, 10742–10759. [Google Scholar] [CrossRef]
- Fang, C.M.; van Huis, M.A.; Zandbergen, H.W. Structural, electronic, and magnetic properties of iron carbide Fe7C3 phases from first-principles theory. Phys. Rev. B 2009, 80, 224108. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkelman, G. A climbing image nudged elastic band method for finding saddle points and mini-mum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.A.; Chen, D.; Zhou, X.G.; Yuan, W.K. DFT studies of dry reforming of methane on Ni catalyst. Catal. Today 2009, 148, 260–267. [Google Scholar] [CrossRef]
- Cao, X.M.; Burch, R.; Hardacre, C.; Hu, P. An understanding of chemoselective hydrogenation on crotonaldehyde over Pt(111) in the free energy landscape: The microkinetics study based on first-principles calculations. Catal. Today 2011, 165, 71–79. [Google Scholar] [CrossRef]
- Chase, M.W. NIST-JANAF Thermochemical Tables, 4th ed.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 1998; p. 643.
- Scholl, D.S.; Steckel, J.A. Density Functional Theory: A Practical Introduction, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 131–161. [Google Scholar]
- Steynberg, P.J.; Van den Berg, J.A.; van Rensburg, W.J. Bulk and surface analysis of Hägg Fe carbide (Fe5C2): A density functional theory study. J. Phys. Condens. Matter 2008, 20, 064238. [Google Scholar] [CrossRef]
- Zhang, J.M.; Pang, Q.; Xu, K.W.; Ji, V. First-principles study of the (001) surface of cubic PbTiO3. Surf. Interface Anal. 2008, 40, 1382–1387. [Google Scholar] [CrossRef]
- Tian, X.; Wang, T.; Fan, L.; Wang, Y.; Lu, H.; Mu, Y. A DFT based method for calculating the surface energies of asymmetric MoP facets. Appl. Surf. Sci. 2018, 427, 357–362. [Google Scholar] [CrossRef]
- García-García, F.R.; Guerrero-Ruiz, A.; Rodriguez-Ramos, I. Role of B5-type sites in Ru catalysts used for the NH3 decomposition reaction. Top. Catal. 2009, 52, 758–764. [Google Scholar] [CrossRef]
- Arevalo, R.L.; Aspera, S.M.; Escaño, M.C.S.; Nakanishi, H.; Kasai, H. First principles study of methane decomposition on B5 step-edge type site of Ru surface. J. Phys. Condens. Matter 2017, 29, 184001. [Google Scholar] [CrossRef]
- Ge, Q.; Neurock, M. Structure dependence of NO adsorption and dissociation on platinum surfaces. J. Am. Chem. Soc. 2004, 126, 1551–1559. [Google Scholar] [CrossRef]
- Rempel, J.; Greeley, J.; Hansen, L.B.; Nielsen, O.H.; Nørskov, J.K.; Mavrikakis, M. Step effects on the dissociation of NO on close-packed rhodium surfaces. J. Phys. Chem. C 2009, 113, 20623–20631. [Google Scholar] [CrossRef]
- Broos, R.J.; Zijlstra, B.; Filot, I.A.; Hensen, E.J. Quantum-chemical DFT study of direct and H- and C-assisted CO dissociation on the χ-Fe5C2 Hägg carbide. J. Phys. Chem. C 2018, 122, 9929–9938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozbek, M.O.; Niemantsverdriet, J.H. Methane, formaldehyde and methanol formation pathways from carbon monoxide and hydrogen on the (001) surface of the iron carbide χ-Fe5C2. J. Catal. 2015, 325, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Lin, T.; Dai, Y.; An, Y.; Yu, F.; Zhong, L.; Li, S.; Sun, Y. Recent advances in the investigation of nanoeffects of Fischer-Tropsch catalysts. Catal. Today 2018, 311, 8–22. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Popov, A.I. Systematic trends in (0 0 1) surface ab initio calculations of ABO3 perovskites. J. Saudi Chem. Soc. 2018, 22, 459–468. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Purans, J.; Gabrusenoks, J.; Popov, A.; Jia, R. Comparative ab initio calculations of ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces. Crystals 2020, 10, 745. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Surface Termination | Surface Energy (eV·Å−2) | Surface C Atom Coverage |
|---|---|---|---|
| 1 | (001)0.20 | 0.140 (2.247) a | 0.33 |
| 2 b | (001)0.09 | 0.153 (2.456) | 0.33 |
| (001)0.59 | 0.153 (2.456) | 0.33 | |
| 4 | (001)0.87 | 0.153 (2.458) | 0.33 |
| 5 | (001)0.81 | 0.156 (2.495) | 0.40 |
| 6 | (001)0.99 | 0.158 (2.529) | 0.60 |
| 7 c | (001)0.41 | 0.160 (2.557) | 0.40 |
| (001)0.91 | 0.160 (2.557) | 0.40 | |
| 9 | (001)0.94 | 0.161 (2.582) | 0.40 |
| 10 | (001)1.00 | 0.164 (2.633) | 0.50 |
| 11 | (001)0.13 | 0.166 (2.657) | 0.17 |
| 12 | (001)0.06 | 0.179 (2.875) | 0.17 |
| 13 | (001)0.85 | 0.189 (3.032) | 0.00 |
| 14 | (001)0.15 | 0.192 (3.069) | 0.17 |
| Surface | Pre-Exponential Factor (ν) (s−1) | Kinetic Barrier Energy (eV) | Reaction Rate Constant (k) (s−1) |
|---|---|---|---|
| o-Fe7C3(001)0.85 | 1.24 × 1012 | 0.91 | 4.26 × 104 |
| o-Fe7C3(001)0.13 | 1.21 × 1012 | 1.48 | 8.66 × 10−1 |
| o-Fe7C3(001)0.20 | 1.30 × 1012 | 1.85 | 7.95 × 10−4 |
| o-Fe7C3(001)0.09 | 1.48 × 1012 | 1.74 | 7.72 × 10−3 |
| o-Fe7C3(001)0.99 | 1.22 × 1012 | 1.95 | 1.06 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chun, H.-J.; Kim, Y.T. Theoretical Study of CO Adsorption and Activation on Orthorhombic Fe7C3(001) Surfaces for Fischer–Tropsch Synthesis Using Density Functional Theory Calculations. Energies 2021, 14, 563. https://doi.org/10.3390/en14030563
Chun H-J, Kim YT. Theoretical Study of CO Adsorption and Activation on Orthorhombic Fe7C3(001) Surfaces for Fischer–Tropsch Synthesis Using Density Functional Theory Calculations. Energies. 2021; 14(3):563. https://doi.org/10.3390/en14030563
Chicago/Turabian StyleChun, Hee-Joon, and Yong Tae Kim. 2021. "Theoretical Study of CO Adsorption and Activation on Orthorhombic Fe7C3(001) Surfaces for Fischer–Tropsch Synthesis Using Density Functional Theory Calculations" Energies 14, no. 3: 563. https://doi.org/10.3390/en14030563
APA StyleChun, H. -J., & Kim, Y. T. (2021). Theoretical Study of CO Adsorption and Activation on Orthorhombic Fe7C3(001) Surfaces for Fischer–Tropsch Synthesis Using Density Functional Theory Calculations. Energies, 14(3), 563. https://doi.org/10.3390/en14030563