
Hydrogen Production with In Situ CO2 Capture at High and Medium Temperatures Using Solid Sorbents

 ,
,  ,
, 
 and
and 
Abstract
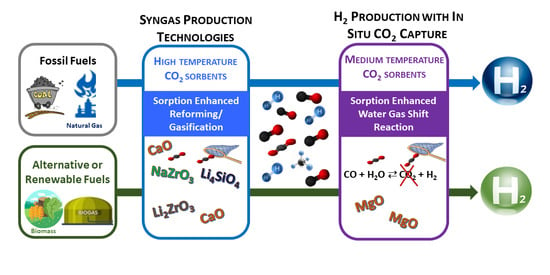
:
1. Introduction
2. High-Temperature CO2 Sorbents: Syngas Production
2.1. Sorption-Enhanced Steam Reforming
2.2. Sorption-Enhanced Gasification
2.3. Enhancement of the H2 Production with Ca-Based Sorbents
2.3.1. Effect of Reforming/Gasification Temperature on Ca-Based Sorbents
2.3.2. Effect of Reforming/Gasification Steam on Ca-Based Sorbents
2.3.3. Effect of Reforming/Gasification High-Pressure on Ca-Sorbents
2.4. Enhancement of the H2 Production with Alkali-Based Sorbents
2.4.1. Effect of Reforming/Gasification Temperature on Alkali-Based Sorbents
2.4.2. Effect of Reforming/Gasification Steam on Alkali-Based Sorbents
2.5. High-Temperature Catalyst–Sorbent: Hybrid/Mixed Materials and Sequential Arrangement
3. Medium-Temperature CO2 Sorbents: Syngas Upgrade for Better H2 Yields
3.1. Sorption-Enhanced Water–Gas Shift Reaction
3.2. Enhancement of the H2 Production with Mg-Based Sorbents
3.2.1. Effect of Temperature on Mg-Based Sorbents
3.2.2. Effect of Steam on Mg-Based Sorbents
3.2.3. Effect of Pressure on Mg-Based Sorbents
3.3. Medium-Temperature Catalyst–Sorbent: Hybrid/Mixed Materials and Sequential Arrangement
4. Conclusions and Recommendations
- Sorbents’ regeneration under realistic conditions, i.e., CO2 concentration higher than 70%.
- Sorbents obtained or synthetized using waste resources (e.g., CaO from paper and pulp industry sludges or CaO-rich biomass ashes; lithium from ores, brine, sea water or recycled batteries; MgO recovered from magnesite mine sludges or from desalination reject brine).
- Sorbents or sorbents–catalyst stability when a high number of carbonation–calcination cycles are performed (>50 carbonation–calcination cycles).
- Effect of granulation methodologies on sorbents or sorbents–catalyst reactivity and mechanical properties. The act of pressing and binders used during the pellet’s preparation can affect the porosity and reduce the catalysts–sorbents specific surface area. Then, the use of materials that can improve the materials porosity should be evaluated (e.g., biomass templates, ethylene glycol).
- Synergies between the hybrid, mixed or sequential arrangement of sorbents–catalyst. Since the carbonation of sorbents can block the access to the catalyst’s active sites, especially in the case of hybrid materials, the use of support materials or the increase in materials porosity that can reduce the occurrence of these problems should be evaluated.
- Tecno-economic viability of reforming, gasification and water–gas shift processes, considering, for example, the cost of the raw material, operating costs, energy requirements, and retrofit of existent industrial plants.
- Life cycle assessment of all the processes and the fulfilment of the circular economy concept. The use of different wastes as raw matter for the sorbents or catalyst synthesis should be compared.
- Modelling and numerical simulation of reforming, gasification and water–gas shift reactors for different catalyst/sorbent formulations foreseeing the processes’ upscale potential.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMS | Alkali molten salts |
| CaL | Calcium looping |
| CCS | Carbon capture and storage |
| CCU | Carbon capture and utilization |
| CHL | Chemical looping hydrogen |
| CP | Conventional process |
| HTlc | Hydrotalcite-like-compound |
| HTS | High-temperature shift |
| IGCC | Integrated gasification combined cycles |
| LTS | Low-temperature shift |
| S/C | Steam/carbon |
| SE | Sorption enhanced |
| SEG | Sorption-enhanced gasification |
| SEP | Sorption-enhanced process |
| SER | Sorption-enhanced reforming |
| SESG | Sorption-enhanced steam gasification |
| SESR | Sorption-enhanced steam reforming |
| SEWGS | Sorption-enhanced water–gas shift |
| SMR | Steam methane reforming |
| TRL | Technology readiness level |
| WGS | Water–gas shift |
References
- European Commission. COM(2019) 640 final. Eur. Green Deal 2019, 53, 1689–1699. [Google Scholar] [CrossRef]
- Parkinson, B.; Balcombe, P.; Speirs, J.F.; Hawkes, A.D.; Hellgardt, K. Levelized cost of CO2 mitigation from hydrogen production routes. Energy Environ. Sci. 2019, 12, 19–40. [Google Scholar] [CrossRef]
- Thomas, H.; Armstrong, F.; Brandon, N.; David, B.; Barron, A.; Durrant, J.; Guwy, A.; Kucernak, A.; Lewis, M.; Maddy, J.; et al. Options for Producing Low-Carbon Hydrogen at Scale; Royal Society: London, UK, 2018; ISBN 9781782523185. [Google Scholar]
- Wang, Y.; Memon, M.Z.; Seelro, M.A.; Fu, W.; Gao, Y.; Dong, Y.; Ji, G. A review of CO2 sorbents for promoting hydrogen production in the sorption-enhanced steam reforming process. Int. J. Hydrogen Energy 2021, 46, 23358–23379. [Google Scholar] [CrossRef]
- Dou, B.; Zhang, H.; Song, Y.; Zhao, L.; Jiang, B.; He, M.; Ruan, C.; Chen, H.; Xu, Y. Hydrogen production from the thermochemical conversion of biomass: Issues and challenges. Sustain. Energy Fuels 2019, 3, 314–342. [Google Scholar] [CrossRef]
- Hu, Y.; He, Q.; Xu, C. Catalytic conversion of glycerol into hydrogen and value-added chemicals: Recent research advances. Catalysts 2021, 11, 1455. [Google Scholar] [CrossRef]
- Dang, C.; Yu, H.; Wang, H.; Peng, F.; Yang, Y. A bi-functional Co-CaO-Ca12Al14O33 catalyst for sorption-enhanced steam reforming of glycerol to high-purity hydrogen. Chem. Eng. J. 2016, 286, 329–338. [Google Scholar] [CrossRef]
- Macedo, M.S.; Soria, M.A.; Madeira, L.M. Process intensification for hydrogen production through glycerol steam reforming. Renew. Sustain. Energy Rev. 2021, 146, 111151. [Google Scholar] [CrossRef]
- Alves, H.J.; Bley Junior, C.; Niklevicz, R.R.; Frigo, E.P.; Frigo, M.S.; Coimbra-Araújo, C.H. Overview of hydrogen production technologies from biogas and the applications in fuel cells. Int. J. Hydrogen Energy 2013, 38, 5215–5225. [Google Scholar] [CrossRef]
- Wu, X.; Wu, S. Production of high-purity hydrogen by sorption-enhanced steam reforming process of methanol. J. Energy Chem. 2015, 24, 315–321. [Google Scholar] [CrossRef]
- Aceves Olivas, D.Y.; Baray Guerrero, M.R.; Escobedo Bretado, M.A.; Marques Da Silva Paula, M.; Salinas Gutiérrez, J.; Guzmán Velderrain, V.; López Ortiz, A.; Collins-Martínez, V. Enhanced ethanol steam reforming by CO2 absorption using CaO, CaO∗MgO or Na2ZrO3. Int. J. Hydrogen Energy 2014, 39, 16595–16607. [Google Scholar] [CrossRef]
- Dewoolkar, K.D.; Vaidya, P.D. Tailored hydrotalcite-based hybrid materials for hydrogen production via sorption-enhanced steam reforming of ethanol. Int. J. Hydrogen Energy 2016, 41, 6094–6106. [Google Scholar] [CrossRef]
- Chimpae, S.; Wongsakulphasatch, S.; Vivanpatarakij, S. Syngas production from combined steam gasification of biochar and a sorption-enhanced. Processes 2019, 7, 349. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zheng, H.; Zheng, Y.; Tian, J.; Jing, T.; Chang, J.S.; Ho, S.H. Recent advances in hydrogen production by thermo-catalytic conversion of biomass. Int. J. Hydrogen Energy 2019, 44, 14266–14278. [Google Scholar] [CrossRef]
- Yan, J. Negative-emissions hydrogen energy. Nat. Clim. Chang. 2018, 8, 560–561. [Google Scholar] [CrossRef]
- Nazir, H.; Louis, C.; Jose, S.; Prakash, J.; Muthuswamy, N.; Buan, M.E.M.; Flox, C.; Chavan, S.; Shi, X.; Kauranen, P.; et al. Is the H2 economy realizable in the foreseeable future? Part I: H2 production methods. Int. J. Hydrogen Energy 2020, 45, 13777–13788. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Efstathiou, A.M. Hydrogen production technologies: Current state and future developments. Conf. Pap. Energy 2013, 2013, 690627. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, G.; Sun, Z.; Zhang, Y.; Xu, D. Review of renewable energy-based hydrogen production processes for sustainable energy innovation. Glob. Energy Interconnect. 2019, 2, 436–443. [Google Scholar] [CrossRef]
- Maggio, G.; Nicita, A.; Squadrito, G. How the hydrogen production from RES could change energy and fuel markets: A review of recent literature. Int. J. Hydrogen Energy 2019, 44, 11371–11384. [Google Scholar] [CrossRef]
- Baykara, S.Z. Hydrogen: A brief overview on its sources, production and environmental impact. Int. J. Hydrogen Energy 2018, 43, 10605–10614. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Voldsund, M.; Jordal, K.; Anantharaman, R. Hydrogen production with CO2 capture. Int. J. Hydrogen Energy 2016, 41, 4969–4992. [Google Scholar] [CrossRef]
- Cantuarias-Villessuzanne, C.; Weinberger, B.; Roses, L.; Vignes, A.; Brignon, J.M. Social cost-benefit analysis of hydrogen mobility in Europe. Int. J. Hydrogen Energy 2016, 41, 19304–19311. [Google Scholar] [CrossRef] [Green Version]
- Pal, D.B.; Chand, R.; Upadhyay, S.N.; Mishra, P.K. Performance of water gas shift reaction catalysts: A review. Renew. Sustain. Energy Rev. 2018, 93, 549–565. [Google Scholar] [CrossRef]
- Miller, J.E. Initial Case for Splitting Carbon Dioxide to Carbon Monoxide and Oxygen; Sandia National Laboratories: Albuquerque, NM, USA, 2007. [Google Scholar]
- Sun, H.; Wu, C.; Shen, B.; Zhang, X.; Huang, J.; Zhang, Y. Progress in the development and application of CaO-based adsorbents for CO2 capture—A review. Mater. Today Sustain. 2018, 1–2, 1–27. [Google Scholar] [CrossRef]
- Hu, Y.; Guo, Y.; Sun, J.; Li, H.; Liu, W. Progress in MgO sorbents for cyclic CO2 capture: A comprehensive review. J. Mater. Chem. A 2019, 7, 20103–20120. [Google Scholar] [CrossRef]
- Yan, X.; Li, Y.; Ma, X.; Zhao, J.; Wang, Z. Performance of Li4SiO4 material for CO2 capture: A review. Int. J. Mol. Sci. 2019, 20, 928. [Google Scholar] [CrossRef] [Green Version]
- Dunstan, M.T.; Donat, F.; Bork, A.H.; Grey, C.P.; Müller, C.R. CO2 capture at medium to high temperature using solid oxide-based sorbents: Fundamental aspects, mechanistic insights, and recent advances. Chem. Rev. 2021, 121, 12681–12745. [Google Scholar] [CrossRef]
- Dubinin, A.M.; Tuponogov, V.G.; Ikonnikov, I.S. Modeling the process of producing hydrogen from methane. Theor. Found. Chem. Eng. 2013, 47, 697–701. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Wang, T. Investigation of low rank coal gasification in a two-stage downdraft entrained-flow gasifier. Int. J. Clean Coal Energy 2014, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Raibhole, V.N.; Sapali, S.N. Simulation and parametric analysis of cryogenic oxygen plant for biomass gasification. Mech. Eng. Res. 2012, 2, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Plis, P.; Wilk, R.K. Theoretical and experimental investigation of biomass gasification process in a fixed bed gasifier. Energy 2011, 36, 3838–3845. [Google Scholar] [CrossRef]
- Chen, L.; Qi, Z.; Zhang, S.; Su, J.; Somorjai, G.A. Catalytic hydrogen production from methane: A review on recent progress and prospect. Catalysts 2020, 10, 858. [Google Scholar] [CrossRef]
- Capa, A.; García, R.; Chen, D.; Rubiera, F.; Pevida, C.; Gil, M.V. On the effect of biogas composition on the H2 production by sorption enhanced steam reforming (SESR). Renew. Energy 2020, 160, 575–583. [Google Scholar] [CrossRef]
- Tomishige, K.; Asadullah, M.; Kunimori, K. Syngas production by biomass gasification using Rh/CeO2/SiO2 catalysts and fluidized bed reactor. Catal. Today 2004, 89, 389–403. [Google Scholar] [CrossRef]
- Antzaras, A.N.; Lemonidou, A.A. Recent advances on materials and processes for intensified production of blue hydrogen. Renew. Sustain. Energy Rev. 2021, 155, 111917. [Google Scholar] [CrossRef]
- Ma, X.; Li, Y.; Huang, X.; Feng, T.; Mu, M. Sorption-enhanced reaction process using advanced Ca-based sorbents for low-carbon hydrogen production. Process Saf. Environ. Prot. 2021, 155, 325–342. [Google Scholar] [CrossRef]
- Chisalita, D.A.; Cormos, C.C. Techno-economic assessment of hydrogen production processes based on various natural gas chemical looping systems with carbon capture. Energy 2019, 181, 331–344. [Google Scholar] [CrossRef]
- Kumar, G.; Eswari, A.P.; Kavitha, S.; Kumar, M.D.; Kannah, R.Y.; How, L.C.; Muthukaruppan, G.; Banu, J.R. Thermochemical conversion routes of hydrogen production from organic biomass: Processes, challenges and limitations. Biomass Convers. Biorefinery 2020. [Google Scholar] [CrossRef]
- Doranehgard, M.H.; Samadyar, H.; Mesbah, M.; Haratipour, P.; Samiezade, S. High-purity hydrogen production with in situ CO2 capture based on biomass gasification. Fuel 2017, 202, 29–35. [Google Scholar] [CrossRef]
- Parvez, A.M.; Hafner, S.; Hornberger, M.; Schmid, M.; Scheffknecht, G. Sorption enhanced gasification (SEG) of biomass for tailored syngas production with in-situ CO2 capture: Current status, process scale-up experiences and outlook. Renew. Sustain. Energy Rev. 2021, 141, 110756. [Google Scholar] [CrossRef]
- Mongkolsiri, P.; Jitkeaw, S.; Patcharavorachot, Y.; Arpornwichanop, A.; Assabumrungrat, S.; Authayanun, S. Comparative analysis of biomass and coal based co-gasification processes with and without CO2 capture for HT-PEMFCs. Int. J. Hydrogen Energy 2019, 44, 2216–2229. [Google Scholar] [CrossRef]
- Fermoso, J.; Rubiera, F.; Chen, D. Sorption enhanced catalytic steam gasification process: A direct route from lignocellulosic biomass to high purity hydrogen. Energy Environ. Sci. 2012, 5, 6358–6367. [Google Scholar] [CrossRef]
- Stanmore, B.R.; Gilot, P. Review-calcination and carbonation of limestone during thermal cycling for CO2 sequestration. Fuel Process. Technol. 2005, 86, 1707–1743. [Google Scholar] [CrossRef]
- Halliday, C.; Hatton, T.A. Sorbents for the capture of CO2 and other acid gases: A review. Ind. Eng. Chem. Res. 2021, 60, 9313–9346. [Google Scholar] [CrossRef]
- Radfarnia, H.R.; Iliuta, M.C. Metal oxide-stabilized calcium oxide CO2 sorbent for multicycle operation. Chem. Eng. J. 2013, 232, 280–289. [Google Scholar] [CrossRef]
- Chi, C.; Li, Y.; Zhang, W.; Wang, Z. Synthesis of a hollow microtubular Ca/Al sorbent with high CO2 uptake by hard templating. Appl. Energy 2019, 251, 113382. [Google Scholar] [CrossRef]
- Skoufa, Z.; Antzara, A.; Heracleous, E.; Lemonidou, A.A. Evaluating the activity and stability of CaO-based sorbents for post-combustion CO2 capture in fixed-bed reactor experiments. Energy Procedia 2016, 86, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Drese, J.H.; Jones, C.W. Adsorbent materials for carbon dioxide capture from large anthropogenic point sources. ChemSusChem 2009, 2, 796–854. [Google Scholar] [CrossRef]
- Teixeira, P.; Mohamed, I.; Fernandes, A.; Silva, J.; Ribeiro, F.; Pinheiro, C.I.C. Enhancement of sintering resistance of CaO-based sorbents using industrial waste resources for Ca-looping in the cement industry. Sep. Purif. Technol. 2020, 235, 116190. [Google Scholar] [CrossRef]
- Peng, W.; Xu, Z.; Zhao, H. Batch fluidized bed test of SATS-derived CaO/TiO2–Al2O3 sorbent for calcium looping. Fuel 2016, 170, 226–234. [Google Scholar] [CrossRef]
- Valverde, J.M.; Medina, S. Crystallographic transformation of limestone during calcination under CO2. Phys. Chem. Chem. Phys. 2015, 17, 21912–21926. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Feng, Y.; Liu, L.; Guo, X. Effects of preparation methods on the structure and property of Al-stabilized CaO-based sorbents for CO2 capture. Fuel Process. Technol. 2018, 173, 276–284. [Google Scholar] [CrossRef]
- Valverde, J.M.; Sanchez-Jimenez, P.E.; Perez-Maqueda, L.A. Ca-looping for postcombustion CO2 capture: A comparative analysis on the performances of dolomite and limestone. Appl. Energy 2015, 138, 202–215. [Google Scholar] [CrossRef] [Green Version]
- Arstad, B.B.; Lind, A.; Andreassen, K.A.; Pierchala, J.; Thorshaug, K.; Blom, R. In-situ XRD studies of dolomite based CO2 sorbents. Energy Procedia 2014, 63, 2082–2091. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.C.; Phalak, N.; Sun, Z.; Fan, L.S. Activation strategies for calcium-based sorbents for CO2 capture: A perspective. Ind. Eng. Chem. Res. 2012, 51, 2133–2142. [Google Scholar] [CrossRef]
- Santos, E.T.; Alfonsín, C.; Chambel, A.J.S.; Fernandes, A.; Soares Dias, A.P.; Pinheiro, C.I.C.; Ribeiro, M.F. Investigation of a stable synthetic sol-gel CaO sorbent for CO2 capture. Fuel 2012, 94, 624–628. [Google Scholar] [CrossRef]
- Teixeira, P.; Hipólito, J.; Fernandes, A.; Ribeiro, F.; Pinheiro, C.I.C. Tailoring synthetic gol-gel CaO sorbents with high reactivity or high stability for Ca-Looping CO2 Capture. Ind. Eng. Chem. Res. 2019, 58, 8484–8494. [Google Scholar] [CrossRef]
- Pinheiro, C.I.C.; Fernandes, A.; Freitas, C.; Santos, E.T.; Ribeiro, M.F. Waste Marble Powders as Promising Inexpensive Natural CaO-Based Sorbents for Post-Combustion CO2 Capture. Ind. Eng. Chem. Res. 2016, 55, 7860–7872. [Google Scholar] [CrossRef]
- Nawar, A.; Ghaedi, H.; Ali, M.; Zhao, M.; Iqbal, N.; Khan, R. Recycling waste-derived marble powder for CO2 capture. Process Saf. Environ. Prot. 2019, 132, 214–225. [Google Scholar] [CrossRef]
- He, S.; Hu, Y.; Hu, T.; Ma, A.; Jia, Q.; Su, H.; Shan, S. Investigation of CaO-based sorbents derived from eggshells and red mud for CO2 capture. J. Alloys Compd. 2017, 701, 828–833. [Google Scholar] [CrossRef]
- Li, Y.; Sun, R. Studies on adsorption of carbon dioxide on alkaline paper mill waste using cyclic process. Energy Convers. Manag. 2014, 82, 46–53. [Google Scholar] [CrossRef]
- Nawar, A.; Ali, M.; Mahmood, M.; Anwar, M.; Khan, Z.A. Effect of structural promoters on calcium based sorbents from waste derived sources. Mater. Today Commun. 2020, 24, 101075. [Google Scholar] [CrossRef]
- Li, Y.; Liu, H.; Sun, R.; Wu, S.; Lu, C. Thermal analysis of cyclic carbonation behavior of CaO derived from carbide slag at high temperature. J. Therm. Anal. Calorim. 2012, 110, 685–694. [Google Scholar] [CrossRef]
- Chi, C.; Li, Y.; Ma, X.; Duan, L. CO2 capture performance of CaO modified with by-product of biodiesel at calcium looping conditions. Chem. Eng. J. 2017, 326, 378–388. [Google Scholar] [CrossRef]
- Yasipourtehrani, S.; Tian, S.; Strezov, V.; Kan, T.; Evans, T. Development of robust CaO-based sorbents from blast furnace slag for calcium looping CO2 capture. Chem. Eng. J. 2020, 387, 124140. [Google Scholar] [CrossRef]
- Shan, S.Y.; Ma, A.H.; Hu, Y.C.; Jia, Q.M.; Wang, Y.M.; Peng, J.H. Development of sintering-resistant CaO-based sorbent derived from eggshells and bauxite tailings for cyclic CO2 capture. Environ. Pollut. 2015, 208, 546–552. [Google Scholar] [CrossRef]
- Witoon, T. Characterization of calcium oxide derived from waste eggshell and its application as CO2 sorbent. Ceram. Int. 2011, 37, 3291–3298. [Google Scholar] [CrossRef]
- Castilho, S.; Kiennemann, A.; Costa Pereira, M.F.; Soares Dias, A.P. Sorbents for CO2 capture from biogenesis calcium wastes. Chem. Eng. J. 2013, 226, 146–153. [Google Scholar] [CrossRef]
- Salaudeen, S.A.; Tasnim, S.H.; Heidari, M.; Acharya, B.; Dutta, A. Eggshell as a potential CO2 sorbent in the calcium looping gasification of biomass. Waste Manag. 2018, 80, 274–284. [Google Scholar] [CrossRef]
- Yan, F.; Jiang, J.; Li, K.; Liu, N.; Chen, X.; Gao, Y.; Tian, S. Green synthesis of nanosilica from coal fly ash and its stabilizing effect on CaO sorbents for CO2 capture. Environ. Sci. Technol. 2017, 51, 7606–7615. [Google Scholar] [CrossRef]
- Nawar, A.; Ali, M.; Waqas, A.; Javed, A.; Iqbal, N.; Khan, R.; Ash, F. Effect of different activation processes on CaO/fly ash mixture for CO2 capture. Energy Fuels 2020, 34, 2035–2044. [Google Scholar] [CrossRef]
- Mohamed, M.; Yusup, S.; Bustam, M.A.; Azmi, N. Effect of coal bottom ash and binder addition into CaO-based sorbent on CO2 capture performance. Chem. Eng. Trans. 2017, 56, 325–330. [Google Scholar] [CrossRef]
- Yan, F.; Jiang, J.; Li, K.; Tian, S.; Zhao, M.; Chen, X. Performance of coal fly ash stabilized, CaO-based sorbents under different carbonation-calcination conditions. ACS Sustain. Chem. Eng. 2015, 3, 2092–2099. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, C.; Ren, Q.; Duan, L.; Chen, H.; Chen, X. Effect of rice husk ash addition on CO2 capture behavior of calcium-based sorbent during calcium looping cycle. Fuel Process. Technol. 2009, 90, 825–834. [Google Scholar] [CrossRef]
- Hu, Y.C.; Liu, W.Q.; Yang, Y.D.; Sun, J.; Zhou, Z.J.; Xu, M.H. Enhanced CO2 capture performance of limestone by industrial waste sludge. Chem. Eng. Technol. 2017, 40, 2322–2328. [Google Scholar] [CrossRef]
- Su, C.; Duan, L.; Donat, F.; Anthony, E.J. From waste to high value utilization of spent bleaching clay in synthesizing high-performance calcium-based sorbent for CO2 capture. Appl. Energy 2018, 210, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Salaudeen, S.A.; Acharya, B.; Dutta, A. CaO-based CO2 sorbents: A review on screening, enhancement, cyclic stability, regeneration and kinetics modelling. J. CO2 Util. 2018, 23, 179–199. [Google Scholar] [CrossRef]
- Esence, T.; Guillot, E.; Tessonneaud, M.; Sans, J.L.; Flamant, G. Solar calcination at pilot scale in a continuous flow multistage horizontal fluidized bed. Sol. Energy 2020, 207, 367–378. [Google Scholar] [CrossRef]
- Tregambi, C.; Troiano, M.; Montagnaro, F.; Solimene, R.; Salatino, P. Fluidized beds for concentrated solar thermal technologies—A review. Front. Energy Res. 2021, 9, 618421. [Google Scholar] [CrossRef]
- Rivero, M.A.; Rodrigues, D.; Pinheiro, C.I.C.; Cardoso, J.P.; Mendes, L.F. Solid – gas reactors driven by concentrated solar energy with potential application to calcium looping: A comparative review. Renew. Sustain. Energy Rev. 2022, 158, 112048. [Google Scholar] [CrossRef]
- Dang, C.; Wu, S.; Cao, Y.; Wang, H.; Peng, F.; Yu, H. Co-production of high quality hydrogen and synthesis gas via sorption-enhanced steam reforming of glycerol coupled with methane reforming of carbonates. Chem. Eng. J. 2019, 360, 47–53. [Google Scholar] [CrossRef]
- Dang, C.; Long, J.; Li, H.; Cai, W.; Yu, H. Pd-promoted Ni-Ca-Al bi-functional catalyst for integrated sorption-enhanced steam reforming of glycerol and methane reforming of carbonate. Chem. Eng. Sci. 2021, 230, 116226. [Google Scholar] [CrossRef]
- Mahishi, M.R.; Goswami, D.Y. An experimental study of hydrogen production by gasification of biomass in the presence of a CO2 sorbent. Int. J. Hydrogen Energy 2007, 32, 2803–2808. [Google Scholar] [CrossRef]
- Irfan, M.; Li, A.; Zhang, L.; Wang, M.; Chen, C.; Khushk, S. Production of hydrogen enriched syngas from municipal solid waste gasification with waste marble powder as a catalyst. Int. J. Hydrogen Energy 2019, 44, 8051–8061. [Google Scholar] [CrossRef]
- Irfan, M.; Li, A.; Zhang, L.; Javid, M.; Wang, M.; Khushk, S. Enhanced H2 production from municipal solid waste gasification using Ni-CaO-TiO2 bifunctional catalyst prepared by dc arc plasma melting. Ind. Eng. Chem. Res. 2019, 58, 13408–13419. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, C.; Ying, Z.; Wang, B. Experimental study on gasification performance of bamboo and PE from municipal solid waste in a bench-scale fixed bed reactor. Energy Convers. Manag. 2016, 117, 393–399. [Google Scholar] [CrossRef]
- Wang, J.; Kang, D.; Shen, B.; Sun, H.; Wu, C. Enhanced hydrogen production from catalytic biomass gasification with in-situ CO2 capture. Environ. Pollut. 2020, 267, 115487. [Google Scholar] [CrossRef]
- Shahbaz, M.; Yusup, S.; Inayat, A.; Patrick, D.O.; Ammar, M.; Pratama, A. Cleaner production of hydrogen and syngas from catalytic steam palm kernel shell gasification using CaO sorbent and coal bottom ash as a catalyst. Energy Fuels 2017, 31, 13824–13833. [Google Scholar] [CrossRef]
- Arnold, R.A.; Hill, J.M. Catalysts for gasification: A review. Sustain. Energy Fuels 2019, 3, 656–672. [Google Scholar] [CrossRef]
- Detchusananard, T.; Im-orb, K.; Ponpesh, P.; Arpornwichanop, A. Biomass gasification integrated with CO2 capture processes for high-purity hydrogen production: Process performance and energy analysis. Energy Convers. Manag. 2018, 171, 1560–1572. [Google Scholar] [CrossRef]
- Hanak, D.P.; Michalski, S.; Manovic, V. From post-combustion carbon capture to sorption-enhanced hydrogen production: A state-of-the-art review of carbonate looping process feasibility. Energy Convers. Manag. 2018, 177, 428–452. [Google Scholar] [CrossRef]
- Rosa, L.G. Solar heat for materials processing: A review on recent achievements and a prospect on future trends. ChemEngineering 2019, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Fuchs, J.; Schmid, J.C.; Benedikt, F.; Hofbauer, H. Experimental development of sorption enhanced reforming by the use of an advanced gasification test plant. Int. J. Hydrogen Energy 2017, 42, 29694–29707. [Google Scholar] [CrossRef]
- Guoxin, H.; Hao, H. Hydrogen rich fuel gas production by gasification of wet biomass using a CO2 sorbent. Biomass Bioenergy 2009, 33, 899–906. [Google Scholar] [CrossRef]
- Criado, Y.A.; Arias, B.; Abanades, J.C. Effect of the carbonation temperature on the CO2 carrying capacity of CaO. Ind. Eng. Chem. Res. 2018, 57, 12595–12599. [Google Scholar] [CrossRef] [Green Version]
- Donat, F.; Florin, N.H.; Anthony, E.J.; Fennell, P.S. Influence of high-temperature steam on the reactivity of CaO sorbent for CO2 capture. Environ. Sci. Technol. 2012, 46, 1262–1269. [Google Scholar] [CrossRef]
- Dong, J.; Tang, Y.; Nzihou, A.; Weiss-Hortala, E. Effect of steam addition during carbonation, calcination or hydration on re-activation of CaO sorbent for CO2 capture. J. CO2 Util. 2020, 39, 101167. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Guo, X.; Van Duin, A.C.T. Insights into the Role of H2O in the Carbonation of CaO Nanoparticle with CO2. J. Phys. Chem. C 2018, 122, 21401–21410. [Google Scholar] [CrossRef]
- Dou, B.; Song, Y.; Liu, Y.; Feng, C. High temperature CO2 capture using calcium oxide sorbent in a fixed-bed reactor. J. Hazard. Mater. 2010, 183, 759–765. [Google Scholar] [CrossRef]
- Donat, F. The Influence of High-Temperature Steam on the Rate and Extent of Reaction of CaO-Sorbent for CO2 Capture-and-Release. Master’s Thesis, TU Bergakademie Freiberg, Freiberg, Germany, 2011. [Google Scholar]
- Champagne, S.; Lu, D.Y.; MacChi, A.; Symonds, R.T.; Anthony, E.J. Influence of steam injection during calcination on the reactivity of CaO-based sorbent for carbon capture. Ind. Eng. Chem. Res. 2013, 52, 2241–2246. [Google Scholar] [CrossRef]
- Champagne, S.; Lu, D.Y.; Symonds, R.T.; Macchi, A.; Anthony, E.J.J. The effect of steam addition to the calciner in a calcium looping pilot plant. Powder Technol. 2015, 290, 114–123. [Google Scholar] [CrossRef]
- Tan, R.S.; Tuan Abdullah, T.A.; Ripin, A.; Ahmad, A.; Md Isa, K. Hydrogen-rich gas production by steam reforming of gasified biomass tar over Ni/dolomite/La2O3 catalyst. J. Environ. Chem. Eng. 2019, 7, 103490. [Google Scholar] [CrossRef]
- Iulianelli, A.; Manzolini, G.; De Falco, M.; Campanari, S.; Longo, T.; Liguori, S.; Basile, A. H2 production by low pressure methane steam reforming in a Pd-Ag membrane reactor over a Ni-based catalyst: Experimental and modeling. Int. J. Hydrogen Energy 2010, 35, 11514–11524. [Google Scholar] [CrossRef]
- Butler, J.W.; Grace, J.R. High-Pressure Systems and Processes for Calcium Looping; Elsevier Ltd.: Amsterdam, The Netherlands, 2015; ISBN 9780857097606. [Google Scholar]
- Yu, F.C.; Fan, L.S. Kinetic study of high-pressure carbonation reaction of calcium-based sorbents in the calcium looping process (CLP). Ind. Eng. Chem. Res. 2011, 50, 11528–11536. [Google Scholar] [CrossRef]
- Shahid, M.M.; Abbas, S.Z.; Maqbool, F.; Ramirez-Solis, S.; Dupont, V.; Mahmud, T. Modeling of sorption enhanced steam methane reforming in an adiabatic packed bed reactor using various CO2 sorbents. J. Environ. Chem. Eng. 2021, 9, 105863. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Y.; Cheng, Z.; Luo, X.; Jia, L.; Song, M.; Jiang, B.; Dou, B. Sorption-enhanced steam reforming of glycerol for hydrogen production over a NiO/NiAl2O4 Catalyst and Li2ZrO3-Based Sorbent. Energy Fuels 2015, 29, 7408–7418. [Google Scholar] [CrossRef]
- Ochoa-Fernández, E.; Lacalle-Vilà, C.; Zhao, T.; Rønning, M.; Chen, D. Experimental demonstration of H2 production by CO2 sorption enhanced steam methane reforming using ceramic acceptors. Stud. Surf. Sci. Catal. 2007, 167, 159–164. [Google Scholar] [CrossRef]
- Ni, Y.; Wang, C.; Chen, Y.; Cai, X.; Dou, B.; Chen, H.; Xu, Y.; Jiang, B.; Wang, K. High purity hydrogen production from sorption enhanced chemical looping glycerol reforming: Application of NiO-based oxygen transfer materials and potassium promoted Li2ZrO3 as CO2 sorbent. Appl. Therm. Eng. 2017, 124, 454–465. [Google Scholar] [CrossRef]
- Yang, X.; Liu, W.; Sun, J.; Hu, Y.; Wang, W.; Chen, H.; Zhang, Y.; Li, X.; Xu, M. Preparation of Novel Li4SiO4 sorbents with superior performance at low CO2 Concentration. ChemSusChem 2016, 9, 1607–1613. [Google Scholar] [CrossRef]
- Chen, S.; Dai, J.; Qin, C.; Yuan, W.; Manovic, V. Adsorption and desorption equilibrium of Li4SiO4-based sorbents for high-temperature CO2 capture. Chem. Eng. J. 2022, 429, 132236. [Google Scholar] [CrossRef]
- Abanades, J.C.; Rubin, E.S.; Anthony, E.J. Sorbent cost and performance in CO2 capture systems. Ind. Eng. Chem. Res. 2004, 43, 3462–3466. [Google Scholar] [CrossRef]
- Xiang, M.; Zhang, Y.; Hong, M.; Liu, S.; Zhang, Y.; Liu, H.; Gu, C. CO2 absorption properties of Ti- and Na-doped porous Li4SiO4 prepared by a sol–gel process. J. Mater. Sci. 2015, 50, 4698–4706. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, S.; Hu, Y.; Sun, J.; Cao, J.; Li, Q.; Liu, W. Mechanochemically activated Li4SiO4-based adsorbent with enhanced CO2 capture performance and its modification mechanisms. Fuel 2020, 273, 117749. [Google Scholar] [CrossRef]
- Subha, P.V.; Nair, B.N.; Visakh, V.; Achu, R.; Shikhila, S.; Peer Mohamed, A.; Yamaguchi, T.; Hareesh, U.S. Wet chemically derived Li4SiO4 nanowires as efficient CO2 sorbents at intermediate temperatures. Chem. Eng. J. 2021, 406, 126731. [Google Scholar] [CrossRef]
- Zhang, T.; Li, M.; Ning, P.; Jia, Q.; Wang, Q.; Wang, J. K2CO3 promoted novel Li4SiO4-based sorbents from sepiolite with high CO2 capture capacity under different CO2 partial pressures. Chem. Eng. J. 2020, 380, 122515. [Google Scholar] [CrossRef]
- Lee, S.C.; Kim, M.J.; Kwon, Y.M.; Chae, H.J.; Cho, M.S.; Park, Y.K.; Seo, H.M.; Kim, J.C. Novel regenerable solid sorbents based on lithium orthosilicate for carbon dioxide capture at high temperatures. Sep. Purif. Technol. 2019, 214, 120–127. [Google Scholar] [CrossRef]
- Li, H.; Qu, M.; Hu, Y. High-temperature CO2 capture by Li4SiO4 adsorbents: Effects of pyroligneous acid (PA) modification and existence of CO2 at desorption stage. Fuel Process. Technol. 2020, 197, 106186. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Y.; Pfeiffer, H.; Louis, B.; Sun, L.; O’Hare, D.; Wang, Q. Recent advances in lithium containing ceramic based sorbents for high-temperature CO2 capture. J. Mater. Chem. A 2019, 7, 7962–8005. [Google Scholar] [CrossRef]
- Radfarnia, H.R.; Iliuta, M.C. Surfactant-template/ultrasound-assisted method for the preparation of porous nanoparticle lithium zirconate. Ind. Eng. Chem. Res. 2011, 50, 9295–9305. [Google Scholar] [CrossRef]
- Martinez-Cruz, L.; Pfeiffer, H. Microstructural Thermal Evolution of the Na2CO3 Phase Produced during a Na2ZrO3−CO2 Chemisorption Process. J. Phys. Chem. 2012, 49, 9038–9042. [Google Scholar]
- Shokrollahi Yancheshmeh, M.; Radfarnia, H.R.; Iliuta, M.C. High temperature CO2 sorbents and their application for hydrogen production by sorption enhanced steam reforming process. Chem. Eng. J. 2016, 283, 420–444. [Google Scholar] [CrossRef]
- Ochoa-Fernández, E.; Haugen, G.; Zhao, T.; Rønning, M.; Aartun, I.; Børresen, B.; Rytter, E.; Rønnekleiv, M.; Chen, D. Process design simulation of H2 production by sorption enhanced steam methane reforming: Evaluation of potential CO2 acceptors. Green Chem. 2007, 9, 654–666. [Google Scholar] [CrossRef]
- Xiao, Q.; Tang, X.; Liu, Y.; Zhong, Y.; Zhu, W. Citrate route to prepare K-doped Li2ZrO3 sorbents with excellent CO2 capture properties. Chem. Eng. J. 2011, 174, 231–235. [Google Scholar] [CrossRef]
- Kwon, Y.M.; Lee, S.C.; Chae, H.J.; Cho, M.S.; Park, Y.K.; Seo, H.M.; Chang Kim, J. Regenerable sodium-based lithium silicate sorbents with a new mechanism for CO2 capture at high temperature. Renew. Energy 2019, 144, 180–187. [Google Scholar] [CrossRef]
- Essaki, K.; Muramatsu, T.; Kato, M. Effect of equilibrium shift by using lithium silicate pellets in methane steam reforming. Int. J. Hydrogen Energy 2008, 33, 4555–4559. [Google Scholar] [CrossRef]
- Sanna, A.; Thompson, S.; Whitty, K.J.; Maroto-Valer, M.M. Fly ash derived lithium silicate for in-situ pre-combustion CO2 capture. Energy Procedia 2017, 114, 2401–2404. [Google Scholar] [CrossRef]
- Xiong, R.; Ida, J.; Lin, Y.S. Kinetics of carbon dioxide sorption on potassium-doped lithium zirconate. Chem. Eng. Sci. 2003, 58, 4377–4385. [Google Scholar] [CrossRef]
- Park, Y.C.; Jo, S.-H.; Ryu, C.K.; Yi, C.-K. Demonstration of pilot scale carbon dioxide capture system using dry regenerable sorbents to the real coal-fired power plant in Korea. Energy Procedia 2011, 4, 1508–1512. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Shen, C.; Zhang, S.; Wu, Y. Steam methane reforming reaction enhanced by a novel K2CO3-Doped Li4SiO4 sorbent: Investigations on the sorbent and catalyst coupling behaviors and sorbent regeneration strategy. Int. J. Hydrogen Energy 2016, 41, 4831–4842. [Google Scholar] [CrossRef]
- Ochoa-Fernández, E.; Zhao, T.; Rønning, M.; Chen, D. Effects of steam addition on the properties of high temperature ceramic CO2 acceptors. J. Environ. Eng. 2009, 135, 397–403. [Google Scholar] [CrossRef]
- Xie, M.; Zhou, Z.; Qi, Y.; Cheng, Z.; Yuan, W. Sorption-enhanced steam methane reforming by in situ CO2 capture on a CaO-Ca9Al6O18 sorbent. Chem. Eng. J. 2012, 207–208, 142–150. [Google Scholar] [CrossRef]
- Pecharaumporn, P.; Wongsakulphasatch, S.; Glinrun, T.; Maneedaeng, A.; Hassan, Z.; Assabumrungrat, S. Synthetic CaO-based sorbent for high-temperature CO2 capture in sorption-enhanced hydrogen production. Int. J. Hydrogen Energy 2019, 44, 20663–20677. [Google Scholar] [CrossRef]
- Kim, S.M.; Abdala, P.M.; Hosseini, D.; Armutlulu, A.; Margossian, T.; Copéret, C.; Müller, C. Bi-functional Ru/Ca3Al2O6-CaO catalyst-CO2 sorbent for the production of high purity hydrogen: Via sorption-enhanced steam methane reforming. Catal. Sci. Technol. 2019, 9, 5745–5756. [Google Scholar] [CrossRef] [Green Version]
- Di Giuliano, A.; Girr, J.; Massacesi, R.; Gallucci, K.; Courson, C. Sorption enhanced steam methane reforming by Ni–CaO materials supported on mayenite. Int. J. Hydrogen Energy 2017, 42, 13661–13680. [Google Scholar] [CrossRef]
- Di Giuliano, A.; Gallucci, K.; Kazi, S.S.; Giancaterino, F.; Di Carlo, A.; Courson, C.; Meyer, J.; Di Felice, L. Development of Ni- and CaO-based mono- and bi-functional catalyst and sorbent materials for sorption enhanced steam methane reforming: Performance over 200 cycles and attrition tests. Fuel Process. Technol. 2019, 195, 106160. [Google Scholar] [CrossRef]
- Masoudi Soltani, S.; Lahiri, A.; Bahzad, H.; Clough, P.; Gorbounov, M.; Yan, Y. Sorption-enhanced steam methane reforming for combined CO2 capture and hydrogen production: A state-of-the-art review. Carbon Capture Sci. Technol. 2021, 1, 100003. [Google Scholar] [CrossRef]
- Di Giuliano, A.; Giancaterino, F.; Courson, C.; Foscolo, P.U.; Gallucci, K. Development of a Ni-CaO-mayenite combined sorbent-catalyst material for multicycle sorption enhanced steam methane reforming. Fuel 2018, 234, 687–699. [Google Scholar] [CrossRef]
- Phromprasit, J.; Powell, J.; Wongsakulphasatch, S.; Kiatkittipong, W.; Bumroongsakulsawat, P.; Assabumrungrat, S. Activity and stability performance of multifunctional catalyst (Ni/CaO and Ni/Ca12Al14O33-CaO) for bio-hydrogen production from sorption enhanced biogas steam reforming. Int. J. Hydrogen Energy 2016, 41, 7318–7331. [Google Scholar] [CrossRef]
- Santos, D.B.L.; Oliveira, A.C.P.; Hori, C.E. Performance of Na2CO3-CaO sorbent in sorption-enhanced steam methane reforming. J. CO2 Util. 2021, 51, 101634. [Google Scholar] [CrossRef]
- Wu, Y.J.; Li, P.; Yu, J.G.; Cunha, A.F.; Rodrigues, A.E. Progress on sorption-enhanced reaction process for hydrogen production. Rev. Chem. Eng. 2016, 32, 271–303. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Chen, Y.; Guo, X. Lithium-based sorbent from rice husk materials for hydrogen production via sorption-enhanced steam reforming of ethanol. Fuel 2019, 245, 263–273. [Google Scholar] [CrossRef]
- Swain, B. Recovery and recycling of lithium: A review. Sep. Purif. Technol. 2017, 172, 388–403. [Google Scholar] [CrossRef]
- Coman, V.; Robotin, B.; Ilea, P. Nickel recovery/removal from industrial wastes: A review. Resour. Conserv. Recycl. 2013, 73, 229–238. [Google Scholar] [CrossRef]
- Chen, C.H.; Yu, C.T.; Chen, W.H.; Kuo, H.T. Effect of in-situ carbon dioxide sorption on methane reforming by nickel-calcium composite catalyst for hydrogen production. IOP Conf. Ser. Earth Environ. Sci. 2020, 463, 012102. [Google Scholar] [CrossRef]
- Dewoolkar, K.D.; Vaidya, P.D. Improved hydrogen production by sorption-enhanced steam methane reforming over hydrotalcite- and calcium-based hybrid materials. Energy Fuels 2015, 29, 3870–3878. [Google Scholar] [CrossRef]
- Johnsen, K.; Ryu, H.J.; Grace, J.R.; Lim, C.J. Sorption-enhanced steam reforming of methane in a fluidized bed reactor with dolomite as CO2-acceptor. Chem. Eng. Sci. 2006, 61, 1195–1202. [Google Scholar] [CrossRef]
- Rahmanzadeh, L.; Taghizadeh, M. Sorption-enhanced ethanol steam reforming on Ce-Ni/MCM-41 with simultaneous CO2 adsorption over Na- and Zr- promoted CaO based sorbent. Int. J. Hydrogen Energy 2019, 44, 21238–21250. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Guo, X.; Ni, S. Continuous high-purity hydrogen production by sorption enhanced steam reforming of ethanol over modified lithium silicate. Int. J. Hydrogen Energy 2021, 46, 10119–10130. [Google Scholar] [CrossRef]
- Phromprasit, J.; Powell, J.; Wongsakulphasatch, S.; Kiatkittipong, W.; Bumroongsakulsawat, P.; Assabumrungrat, S. H2 production from sorption enhanced steam reforming of biogas using multifunctional catalysts of Ni over Zr-, Ce- and La-modified CaO sorbents. Chem. Eng. J. 2017, 313, 1415–1425. [Google Scholar] [CrossRef]
- Xie, H.; Yu, Q.; Zuo, Z.; Han, Z.; Yao, X.; Qin, Q. Hydrogen production via sorption-enhanced catalytic steam reforming of bio-oil. Int. J. Hydrogen Energy 2016, 41, 2345–2353. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yang, H.; Wei, L.; Shao, J.; Wang, X.; Chen, H. Absorption-enhanced steam gasification of biomass for hydrogen production: Effects of calcium-based absorbents and NiO-based catalysts on corn stalk pyrolysis-gasification. Int. J. Hydrogen Energy 2017, 42, 5840–5848. [Google Scholar] [CrossRef]
- Mostafavi, E.; Mahinpey, N.; Rahman, M.; Sedghkerdar, M.H.; Gupta, R. High-purity hydrogen production from ash-free coal by catalytic steam gasification integrated with dry-sorption CO2 capture. Fuel 2016, 178, 272–282. [Google Scholar] [CrossRef]
- Zamboni, I.; Debal, M.; Matt, M.; Girods, P.; Kiennemann, A.; Rogaume, Y.; Courson, C. Catalytic gasification of biomass (Miscanthus) enhanced by CO2 sorption. Environ. Sci. Pollut. Res. 2016, 23, 22253–22266. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Chen, C.Y. Water gas shift reaction for hydrogen production and carbon dioxide capture: A review. Appl. Energy 2020, 258, 114078. [Google Scholar] [CrossRef]
- Baraj, E.; Ciahotný, K.; Hlinčik, T.; Šnajdrová, V. Hydrogen production via water gas shift reaction on a nickel based catalyst. PALIVA 2016, 8, 138–142. [Google Scholar] [CrossRef]
- Newsome, D.S. The water-gas shift reaction. Catal. Rev. Technol. 1980, 21, 275–318. [Google Scholar] [CrossRef]
- Smith J, B.R.; Loganathan, M.; Shekhar Shantha, M. A Review of the water gas shift reaction kinetics. Int. J. Chem. React. Eng. 2010, 8, 49. [Google Scholar] [CrossRef]
- Jin, S.; Ko, K.J.; Lee, C.H. Direct formation of hierarchically porous MgO-based sorbent bead for enhanced CO2 capture at intermediate temperatures. Chem. Eng. J. 2019, 371, 64–77. [Google Scholar] [CrossRef]
- Oliveira, N.M.B.; Valença, G.P.; Vieira, R. Water gas shift reaction on copper catalysts supported on alumina and carbon nanofibers. Chem. Eng. Trans. 2015, 43, 931–936. [Google Scholar] [CrossRef]
- Hwang, B.W.; Lim, J.H.; Chae, H.J.; Ryu, H.J.; Lee, D.; Lee, J.B.; Kim, H.; Lee, S.C.; Kim, J.C. CO2 capture and regeneration properties of MgO-based sorbents promoted with alkali metal nitrates at high pressure for the sorption enhanced water gas shift process. Process Saf. Environ. Prot. 2018, 116, 219–227. [Google Scholar] [CrossRef]
- Hu, Y.; Cui, H.; Cheng, Z.; Zhou, Z. Sorption-enhanced water gas shift reaction by in situ CO2 capture on an alkali metal salt-promoted MgO-CaCO3 sorbent. Chem. Eng. J. 2019, 377, 119823. [Google Scholar] [CrossRef]
- Lee, C.H.; Lee, K.B. Sorption-enhanced water gas shift reaction for high-purity hydrogen production: Application of a Na-Mg double salt-based sorbent and the divided section packing concept. Appl. Energy 2017, 205, 316–322. [Google Scholar] [CrossRef]
- Gao, W.; Zhou, T.; Gao, Y.; Wang, Q. Enhanced water gas shift processes for carbon dioxide capture and hydrogen production. Appl. Energy 2019, 254, 113700. [Google Scholar] [CrossRef]
- Soria, M.A.; Rocha, C.; Tosti, S.; Mendes, A.; Madeira, L.M. COx free hydrogen production through water-gas shift reaction in different hybrid multifunctional reactors. Chem. Eng. J. 2019, 356, 727–736. [Google Scholar] [CrossRef]
- Halabi, M.H.; De Croon, M.H.J.M.; Van Der Schaaf, J.; Cobden, P.D.; Schouten, J.C. High capacity potassium-promoted hydrotalcite for CO2 capture in H2 production. Int. J. Hydrogen Energy 2012, 37, 4516–4525. [Google Scholar] [CrossRef]
- Lee, C.H.; Kim, S.; Yoon, H.J.; Yoon, C.W.; Lee, K.B. Water gas shift and sorption-enhanced water gas shift reactions using hydrothermally synthesized novel Cu–Mg–Al hydrotalcite-based catalysts for hydrogen production. Renew. Sustain. Energy Rev. 2021, 145, 111064. [Google Scholar] [CrossRef]
- García-Moncada, N.; González-Castaño, M.; Ivanova, S.; Centeno, M.Á.; Romero-Sarria, F.; Odriozola, J.A. New concept for old reaction: Novel WGS catalyst design. Appl. Catal. B Environ. 2018, 238, 1–5. [Google Scholar] [CrossRef]
- Vovchok, D.; Guild, C.J.; Llorca, J.; Xu, W.; Jafari, T.; Toloueinia, P.; Kriz, D.; Waluyo, I.; Palomino, R.M.; Rodriguez, J.A.; et al. Cu supported on mesoporous ceria: Water gas shift activity at low Cu loadings through metal-support interactions. Phys. Chem. Chem. Phys. 2017, 19, 17708–17717. [Google Scholar] [CrossRef] [Green Version]
- Jeong, C.H.; Byeon, H.J.; Jang, W.J.; Jeon, K.W.; Jeong, D.W. The optimization of Nb loading amount over Cu–Nb–CeO2 catalysts for hydrogen production via the low-temperature water gas shift reaction. Int. J. Hydrogen Energy 2020, 45, 9648–9657. [Google Scholar] [CrossRef]
- Petallidou, K.C.; Polychronopoulou, K.; Efstathiou, A.M. Novel catalytic systems for hydrogen production via the water-gas shift reaction. Conf. Pap. Energy 2013, 2013, 426980. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Gao, W. MgO-Based Intermediate-Temperature CO2 Adsorbents. In Pre-Combustion Carbon Dioxide Capture Materials; Wang, Q., Ed.; Royal Society of Chemistry: Beijing, China, 2018. [Google Scholar]
- Wang, K.; Zhao, Y.; Clough, P.T.; Zhao, P.; Anthony, E.J. Structural and kinetic analysis of CO2 sorption on NaNO2-promoted MgO at moderate temperatures. Chem. Eng. J. 2019, 372, 886–895. [Google Scholar] [CrossRef]
- Fagerlund, J.; Highfield, J.; Zevenhoven, R. Kinetics studies on wet and dry gas-solid carbonation of MgO and Mg(OH)2 for CO2 sequestration. RSC Adv. 2012, 2, 10380–10393. [Google Scholar] [CrossRef]
- Zhang, K. Development of Molten Salt Promoted Metal Oxide Based Absorbents for CO2 Separation. Ph.D. Thesis, University of Connecticut, Storrs, CT, USA, 2014. [Google Scholar]
- Dal Pozzo, A.; Armutlulu, A.; Rekhtina, M.; Abdala, P.M.; Müller, C.R. CO2 uptake and cyclic stability of MgO-based CO2 sorbents promoted with alkali metal nitrates and their eutectic mixtures. ACS Appl. Energy Mater. 2019, 2, 1295–1307. [Google Scholar] [CrossRef]
- Zhang, K.; Li, X.S.; Li, W.Z.; Rohatgi, A.; Duan, Y.; Singh, P.; Li, L.; King, D.L. Phase transfer-catalyzed fast CO2 absorption by MgO-based absorbents with high cycling capacity. Adv. Mater. Interfaces 2014, 1, 1400030. [Google Scholar] [CrossRef]
- Zhao, X.; Ji, G.; Liu, W.; He, X.; Anthony, E.J.; Zhao, M. Mesoporous MgO promoted with NaNO3/NaNO2 for rapid and high-capacity CO2 capture at moderate temperatures. Chem. Eng. J. 2018, 332, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Abbasi, E.; Hassanzadeh, A.; Zarghami, S.; Arastoopour, H.; Abbasian, J. Regenerable MgO-based sorbent for high temperature CO2 removal from syngas: 3. CO2 capture and sorbent enhanced water gas shift reaction. Fuel 2014, 137, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Cheng, Z.; Zhou, Z. High-purity H2 production by sorption-enhanced water gas shift on a K2CO3-promoted Cu/MgO-Al2O3 difunctional material. Sustain. Energy Fuels 2021, 5, 3340–3350. [Google Scholar] [CrossRef]
- Hiremath, V.; Trivino, M.L.T.; Seo, J.G. Eutectic mixture promoted CO2 sorption on MgO-TiO2 composite at elevated temperature. J. Environ. Sci. (China) 2019, 76, 80–88. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, L.; Hu, Y.; Cheng, Z. Nanosheet MgO-based CO2 sorbent promoted by mixed-alkali-metal nitrate and carbonate: Performance and mechanism. Ind. Eng. Chem. Res. 2017, 56, 5802–5812. [Google Scholar] [CrossRef]
- Zarghami, S.; Hassanzadeh, A.; Arastoopour, H.; Abbasian, J. Effect of steam on the reactivity of MgO-based sorbents in precombustion CO2 capture processes. Ind. Eng. Chem. Res. 2015, 54, 8860–8866. [Google Scholar] [CrossRef]
- Ryu, D.Y.; Jo, S.; Kim, T.Y.; In, S.Y.; Kim, J.K.; Hwang, J.E.; Kim, J.C.; Lee, S.C. Influence of the sorption pressure and K2CO3 loading of a MgO-based sorbent for application to the SEWGS process. Korean J. Chem. Eng. 2022, 39, 1028–1035. [Google Scholar] [CrossRef]
- Dong, H.; Yang, E.H.; Unluer, C.; Jin, F.; Al-Tabbaa, A. Investigation of the properties of MgO recovered from reject brine obtained from desalination plants. J. Clean. Prod. 2018, 196, 100–108. [Google Scholar] [CrossRef] [Green Version]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technology | Feedstock | Production Efficiency (%) | Production Costs (€/kg H2) | CO2 Emissions (kg CO2/kg H2) | Maturity | Ref |
|---|---|---|---|---|---|---|
| SMR without CCS | Hydrocarbons | 70–85 | 0.9–2.9 | 9.2–17.2 | TRL 9 (commercial) | [2,16,17,21,23] |
| SMR with CCS | Hydrocarbons | --- | 1.7–4.1 | 2.54–9.2 | TRL 7-8 | [2,16,23] |
| Reforming with biogas | Biogas | --- | 4–6.0 | 2.93 | --- | [23] |
| Partial Oxidation | Hydrocarbons | 60–75 | --- | --- | Commercial | [17] |
| Autothermal reforming | Hydrocarbons | 60–75 | --- | --- | Near Term | [17] |
| Plasma reforming | Hydrocarbons | 8–85 | --- | --- | Long Term | [17] |
| Coal Gasification without CCS | Coal | 0.9–1.7 | 15–31 | TRL 9 (commercial) | [2,21] | |
| Coal Gasification with CCS | Coal | -- | 1.4–2.4 | 1–10 | TRL 6-7 | [2,16,21] |
| Biomass gasification | Biomass | 35–52 | 1.3–2.7 | 0.3–9 | TRL 5-6 | [2,17,21] |
| Biomass gasification with CCS | Biomass | -- | 2.8–3.2 | −11.7 to −17.5 | TRL 3-5 | [2] |
| Electrolysis | H2O + electricity | 50–70 | 4.4–8 | -- | TRL 9 Commercial | [17,23] |
| Wind Electrolysis | H2O + wind | -- | 4.01–8.8 | 0.5–1.1 | TRL 9 | [2,16,21] |
| Solar Electrolysis | H2O + sunlight | -- | 4.5–12.4 | 1.3–2.5 | TRL 9 | [2,16,21] |
| Photo Electrolysis | H2O + sunlight | 0.2 | ~ 9 | ~2 | Long Term | [16,17] |
| Thermochemical water splitting | H2O + heat | NA | -- | -- | Long Term | [17] |
| mol % (Dry Basis) | SMR [30] | Coal Gasification [31] | Indian Coal Gasification [32] | Wood Pellets Gasification [32,33] | Rice Husk Gasification [32] |
|---|---|---|---|---|---|
| H2 | 71 | 13–18 | 9 | 7–34 | 25 |
| CO2 | 6 | 7–9 | 0.6 | 6–16 | 14 |
| CO | 16 | 55–62 | 42 | 16–31 | 20 |
| O2 | -- | -- | -- | 1–3 | -- |
| N2 | -- | ~7 | 32 | 48–58 | 40 |
| CH4 | 5 | 17 | 1–4 | 0.9 | |
| CxHy | -- | -- | 0.1–0.3 | -- |
| Process Name | Chemical Reaction | ||
|---|---|---|---|
| Steam methane reforming | (2) | ||
| Hydrocarbon steam reforming | (3) | ||
| Organic matters steam reforming | (4) |
| No Steam | Steam during Calcination | Steam during Carbonation | Steam during Carbonation and Calcination |
|---|---|---|---|
 |  |  |  |
|
|
|
|
| Carbonation Reactions | ΔH⁰ 25 °C (kJ/mol) | Theoretical CO2 Uptake Capacity (g CO2/g Sorbent) | Operating Carbonation Range (°C) | |
|---|---|---|---|---|
| Li2CO3 (s) + Li2SiO3 (s) | (16) | −143 | 0.367 | 450–600 |
| Li2CO3 (s) + ZrO2 (s) | (17) | −160 | 0.288 | 450–600 |
| Na2CO3 (s) + ZrO2 (s) | (18) | −149 | 0.234 | 400–800 |
| CaO | Li4SiO4 | Li2ZrO3 | KliZrO3 | NaZrO3 | |
|---|---|---|---|---|---|
| Capacity | G | F | F | F | F |
| Thermodynamic | G | F | F | F | F |
| Stability | P | G | G | F | G |
| Kinetics | G | F | F/P | F | G |
| Feedstock | Catalyst (CP) | Sorbent/Catalyst (SEP) | Pattern 1 | Technology 2 | Reactor 3 | Conditions | N Cycles | Maximum H2 (%) | χSE (%) | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T (°C) | S/C | CP | SEP | |||||||||
| CH4 | Ni/Al2O3 | Ni-CaO-Ca12Al14O33 | H | SR | FxB | 600 | 3 | 1 | ~70 | 93 | 33 | [136] |
| CH4 | Ni/Al2O3 | Ni/Al2O3-CaO | M | SR | FxB | 600 | 3 4 | ~72 | 95 98.4 | 32 37 | [148] | |
| CH4 | Ni/Al2O3 | Ni-CaO/Al2O3 Ni-CaO/Al2O3 | H H | SR | PB | 500 600 | 9 | ~60 | ~88 ~93 | 47 55 | [149] | |
| CH4 | Ni/Al2O3 Ni/Al2O3 Ni/Al2O3 | Ni/Al2O3-Li4SiO4 Ni/Al2O3-Li4SiO4 Ni/Al2O3-Li4SiO4 | M M M | SR | FB | 500 550 600 | 3.5 | 1 | 64.9 67.1 75.3 | 89.4 93.6 87.6 | 38 39 16 | [129] |
| CH4 | Ni/Al2O3 | Ni/Al2O3/K-Li4SiO4 | H | SR | FxB | 600 | 4 | 10 | ~85 | ~98 | 13 | [133] |
| CH4 | Ni commercial | Ni-Dolomite | M | SR | FB | 600 | 3 | 1 | ~73.4 | ~98 | 33 | [150] |
| Glycerol | NiO/NiAl2O4 NiO/ZrO2 | NiO/NiAl2O4 + (K-Li2ZrO3) NiO/ZrO2 + (K-Li2ZrO3) | n.a. n.a. | SR (Chemical looping) | PB | 550 | 3 | 10 | ~86 ~90 | ~90 ~93 | 5 3 | [112] |
| Ethanol | Ni/Al2O3 | Ni/Al2O3-CaO Ni/Al2O3-CaO-MgO Ni/Al2O3-Na2ZrO3 | M | SR | FxB | 600 | 6 | 1 | 64.7 | 97.0 96.2 96.5 | 50 49 49 | [11] |
| Ethanol | Ce-Ni/MCM-4 | Ce-Ni/MCM-4/Na-Zr-CaO Ce-Ni/MCM-4/Na-Zr-CaO | H M | SR | FxB | 600 | 3 | 1 | ~70 | ~94 ~80 | 34 14 | [151] |
| Ethanol | Ni/Al2O3 | Ni/Al2O3-Li4SiO 4Ni/Al2O3-K-Li4SiO4 | M M | SR | FxB | 575 | 9 | 10 | ~77 | ~98 >99 | 27 29 | [152] |
| Biogas | Ni/Al2O3 | Ni-Zr-Ca Ni-Ce-Ca Ni-La-Ca | H H H | SR | FB | 600 | 3 | 5 | 67 | ~85 ~85 ~80 | 27 27 19 | [153] |
| Bio-oil | Ce-Ni/Co-Al2O3 Ce-Ni/Co-Al2O3 Ce-Ni/Co-Al2O3 | Ce-Ni/Co-Al2O3-CaO Ce-Ni/Co-Al2O3-CaO Ce-Ni/Co-Al2O3-CaO | n.a. | SR | PB | 650 750 850 | 12 | 1 | ~65 ~70 ~75 | ~80 ~92 ~80 | 23 31 6 | [154] |
| Biogas: 50CH4/50CO2 100CH4/0CO2 | Pd/Ni-Co HT Pd/Ni-Co HT | Pd/Ni-Co HT/Dolomite Pd/Ni-Co HT/Dolomite | M | SR | FB | 600 650 | 3 | 1 | ~62 ~64 | ~98 ~98 | 58 53 | [35] |
| Biomass: corn stalk | NiO/γ-Al2O3 | NiO/γ-Al2O3-Calc. olivine NiO/γ-Al2O3-Calc. limestone NiO/γ-Al2O3-Calc. CaCO3NiO/γ-Al2O3-Calc. Dolomite | M | G | FxB | 650 | 2 | 1 | ~29 | ~47 ~50 ~67 ~72 | 62 72 131 148 | [155] |
| Coal | K2CO3 | K2CO3-Limestone | MM | G | FB | 675 | -- | 1 | ~65 | ~87 | 34 | [156] |
| Biomass: pine bark | No-catalyst CaO | No-sorbent CaO | n. a. | G | FB | 600 | -- | 1 | ~60 | ~83 | 38 | [85] |
| Biomass | Pd/Ni–Co-HT | Dolomite Pd/Ni–Co-Dolomite | M | G | FxB | 650 | -- | 1 | 69.7 | 91.1 99.0 | 31 42 | [44] |
| Biomass | Olivine | CaO-CaAl/olivine | H | G | FB | 700 | -- | 1 | 34.2 | 47.4 | 39 | [157] |
| Benefits | Challenges |
|---|---|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, P.; Bacariza, C.; Correia, P.; Pinheiro, C.I.C.; Cabrita, I. Hydrogen Production with In Situ CO2 Capture at High and Medium Temperatures Using Solid Sorbents. Energies 2022, 15, 4039. https://doi.org/10.3390/en15114039
Teixeira P, Bacariza C, Correia P, Pinheiro CIC, Cabrita I. Hydrogen Production with In Situ CO2 Capture at High and Medium Temperatures Using Solid Sorbents. Energies. 2022; 15(11):4039. https://doi.org/10.3390/en15114039
Chicago/Turabian StyleTeixeira, Paula, Carmen Bacariza, Patrícia Correia, Carla I. C. Pinheiro, and Isabel Cabrita. 2022. "Hydrogen Production with In Situ CO2 Capture at High and Medium Temperatures Using Solid Sorbents" Energies 15, no. 11: 4039. https://doi.org/10.3390/en15114039
APA StyleTeixeira, P., Bacariza, C., Correia, P., Pinheiro, C. I. C., & Cabrita, I. (2022). Hydrogen Production with In Situ CO2 Capture at High and Medium Temperatures Using Solid Sorbents. Energies, 15(11), 4039. https://doi.org/10.3390/en15114039