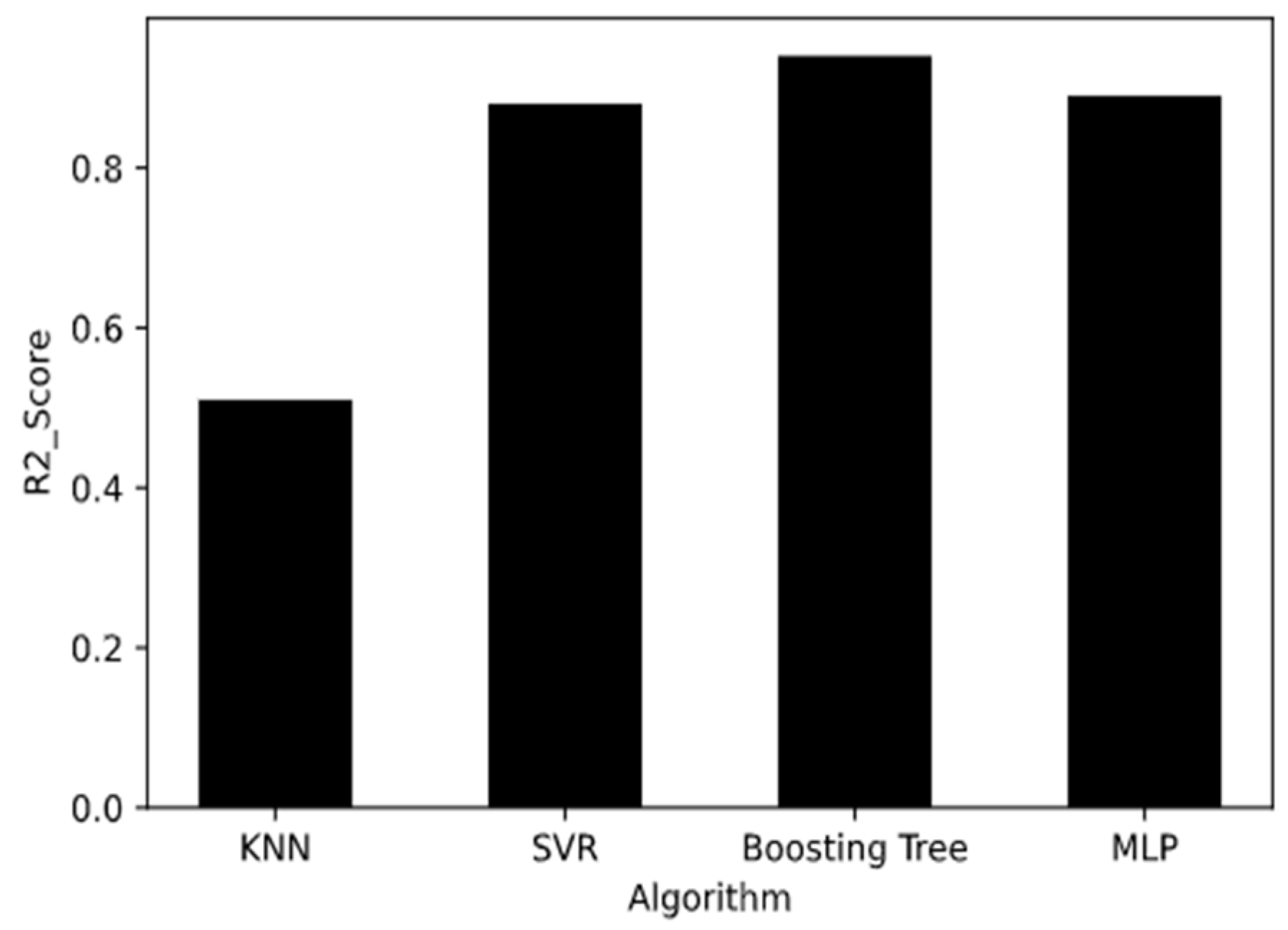
2.1. K-Nearest Neighbor Learning Algorithm
The K-Nearest Neighbor (KNN) algorithm is a simple and commonly used supervised learning method, and it was recognized as one of the top 10 algorithms [
31]. KNN is mainly used for classification.
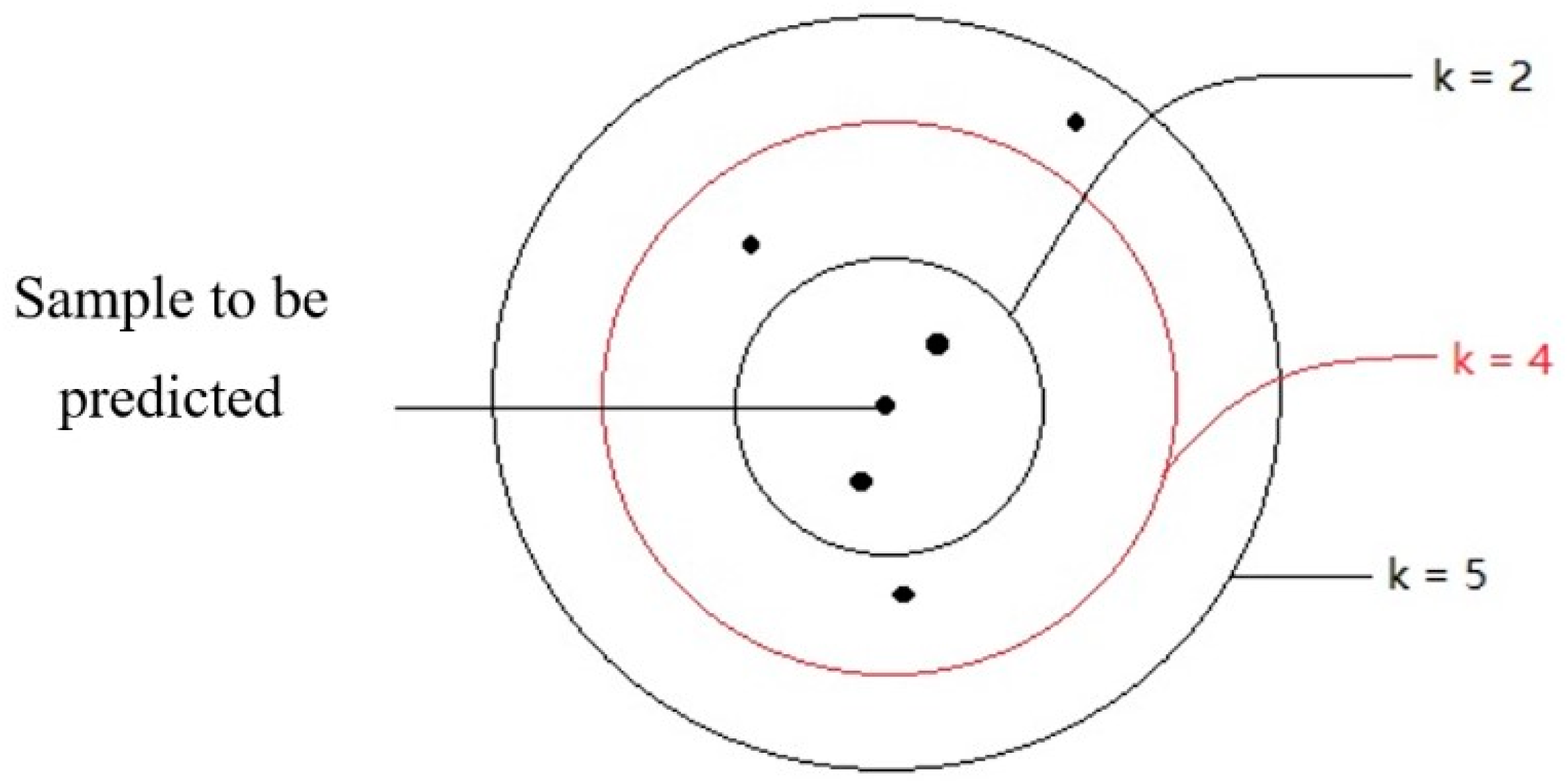
Figure 1 shows a schematic diagram of KNN. The working principle of KNN is to find out the K training samples closest to a new test data point in the training set by using some distance measurement, and then to use the label of the K similar points to predict the test samples. In the progress of a regression, the average of the label values of the K sample is used as the prediction result. The weighted average can also be based on the distance, so the closer the sample weight is, the more accurate the prediction can be when the sample distribution is uneven. The advantage of the KNN algorithm the avoidance of the training process before classification. Instead of a training process, it simply saves the samples and calculates the distance after receiving the samples to be predicted [
32]. On the other side of the coin, the time complexity of the prediction is large. In addition, the other main challenges in KNN include the computation of K, nearest neighbor selection, nearest neighbor search, and classification rule [
33]. Despite these shortcomings, KNN is still an efficient artificial intelligence (AI) algorithm according to the comparison of 16 different algorithms by Li et al. [
34].
As mentioned above, K is an important super-parameter in KNN, and it determines the precision of the prediction. The prediction results will be different when K takes different values. The computation of K relies on the sample’s distribution [
35]. The selection of K can be based on either different sample subspaces [
36] or different test samples [
37]. Small ranges of training samples are used for prediction if a small K value is selected. In this way, the prediction error of KNN will be reduced if the sample size is large enough. This is because only the training samples close to the samples to be predicted play a role in the prediction results. Meanwhile, it is easier to overfit because the prediction results are very sensitive to adjacent samples. The prediction makes mistakes quite easily if an adjacent sample contains incorrect data. If a larger K value is selected, it is equivalent to using a wide range of samples for prediction. The advantage is that it can reduce the possibility of overfitting by the learner, but in this way, the training samples that are far away from the samples to be predicted will also play a role in prediction, resulting in a decline in the prediction accuracy. Generally, in practice, a K value with a smaller value is still selected. On the other hand, if different distance calculation methods are used, the “neighbors” found will be different, and the prediction results will, of course, be significantly different. The most commonly used distance measure is the
distance or Minkowski distance.
2.2. Support Vector Regression Algorithm
Support Vector Regression (SVR) is also commonly accepted as one of the standard machine learning algorithms, and it falls under the category of supervised learning methods [
38]. Its algorithmic principle can be summarized in the following two main points: (1) Firstly, it is a linear fitting algorithm that is only suitable for linearly distributed data. For data with a nonlinear distribution, a special nonlinear mapping is used to make their high-dimensional distribution linear, and the linear algorithm is used in the high-dimensional feature space to try to learn to fit the training data. (2) Secondly, the algorithm constructs an optimal hyperplane in the whole feature space based on the theory of structural risk minimization (including regularization). SVR is not a traditional regression fitting algorithm based on its basic principle. The main advantage of the SVR algorithm is the capability of achieving a global optimum and avoiding overfitting. The main feature of SVR comes from the different kernel functions that are used to fit different types of data. The computational complexity depends only on the number of support vectors. Therefore, a small number of support vectors determine the final result, not the entire dataset.
In the given training sample set
, the objective is to have a regression model
that makes
as close as possible to
. Both
and
are the model parameters that need to be determined. For a certain sample
, traditional regression models usually calculate the loss based on the difference between the model output
and the real value
. The loss is zero only when
and
are exactly the same. In contrast, SVR can tolerate the maximum deviation between
and
. The loss is calculated only when the absolute value of the difference between
and
is greater than
. The loss is not calculated for a sample with a prediction error falling to
, while the samples on and outside the spacer are called support vectors. Thus, the SVR problem can be written as:
where C is the regularization constant, which is a super-parameter;
is the
− insensitive loss function, which can be determined with the following equation:
Introducing slack variables
and
and rewriting the above formula gives:
After introducing the Lagrange multiplier
, the Lagrange function of Equation (3) can be obtained with the Lagrange multiplier method:
Let the partial derivative of
to
,
,
, and
be zero:
Substituting Equations (5)–(8) into Equation (4), the dual form of SVR can be obtained:
Applying the Karush–Kuhn–Tucker (KKT) conditions when searching for the optimized value yields [
39]:
Andreani et al. applied a sequential minimum optimization algorithm (SMO) to solve the above optimization problem [
40]. Substituting Equation (5) into Equation (10), the solution of SVR is:
According to the KKT conditions in Equation (10), the samples falling in the −spacer satisfy and . Therefore, the samples of in Equation (11) can be the support vector of SVR, which falls outside the −spacing band. Obviously, the support vector of SVR is only a part of the training samples, and its solution is still sparse.
In addition, it can also be seen from the KKT conditions (Equation (10)) that
and
for each sample
. Therefore, after getting
, it must be that
for
, which yields:
In practical problems, multiple samples satisfying the condition are often selected to solve b, and then the average is taken.
The sample is assumed to have a linear distribution in the above derivation; however, data are often nonlinear in real applications [
41]. For such problems, the samples can be mapped from the original space to a higher-dimensional feature space so that they are linearly distributed in this new space.
Let
denote the vector after mapping
to a high-dimensional space, so the corresponding model of the linear regression equation in the new high-dimensional space is:
So, the solution of SVR is:
However, there are still two problems: (1) Different data need different mapping functions
for different scenarios, which makes it hard to predict how high the dimension at which the original sample is to be mapped should be to create a linear distribution. Therefore, the first problem comes from the selection of the mapping function
. (2) The solution involves the calculation of
, which is the inner product of the sample
and
mapped to a high-dimensional space. Since the dimension may be very high or even infinite, a direct calculation is particularly difficult. A kernel function can help to solve these problems. Kernel functions show good performance in solving the optimization problems of increasing complexity [
42,
43]. In Equation (14),
and
always appear in pairs. Then, the following relationship can be derived:
The inner product of
and
in a high--dimensional space is equal to the result calculated with the function
in the original sample space. There is no need to pay attention to the selection of mapping functions with such a function. Therefore, the inner product in a high-dimensional or even infinite-dimensional feature space can be directly calculated, which involves mapping the input data into a higher-dimensional space [
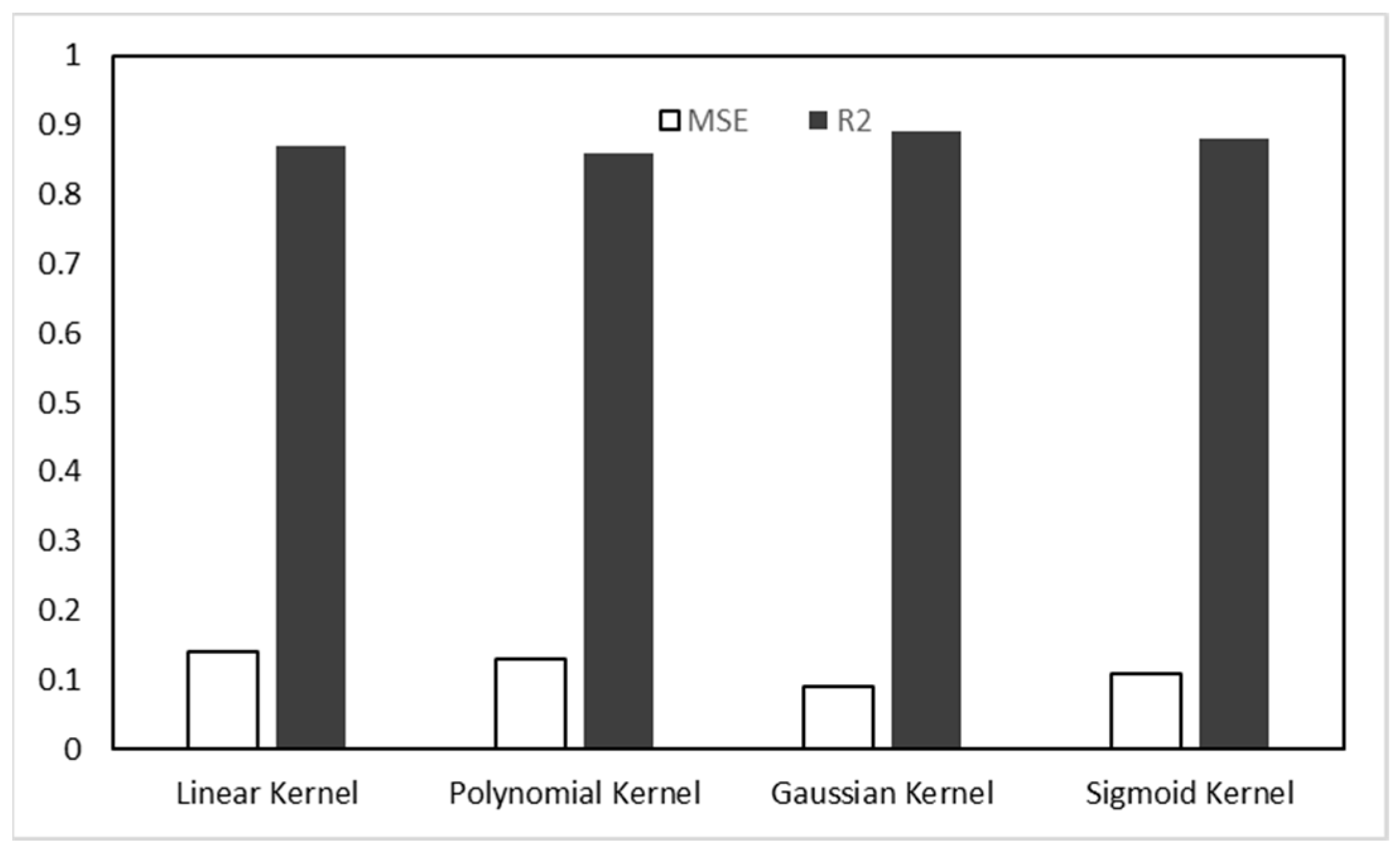
44]. The most commonly used kernel functions are shown in
Table 1.
2.3. Boosting Tree Algorithm
Boosting is an integration method; within this category, the Boosting Tree is one of the most commonly used algorithms. The principle of ensemble learning is to learn repeatedly with a series of weak learners, which are later integrated into a strong learner to get better generalization performance [
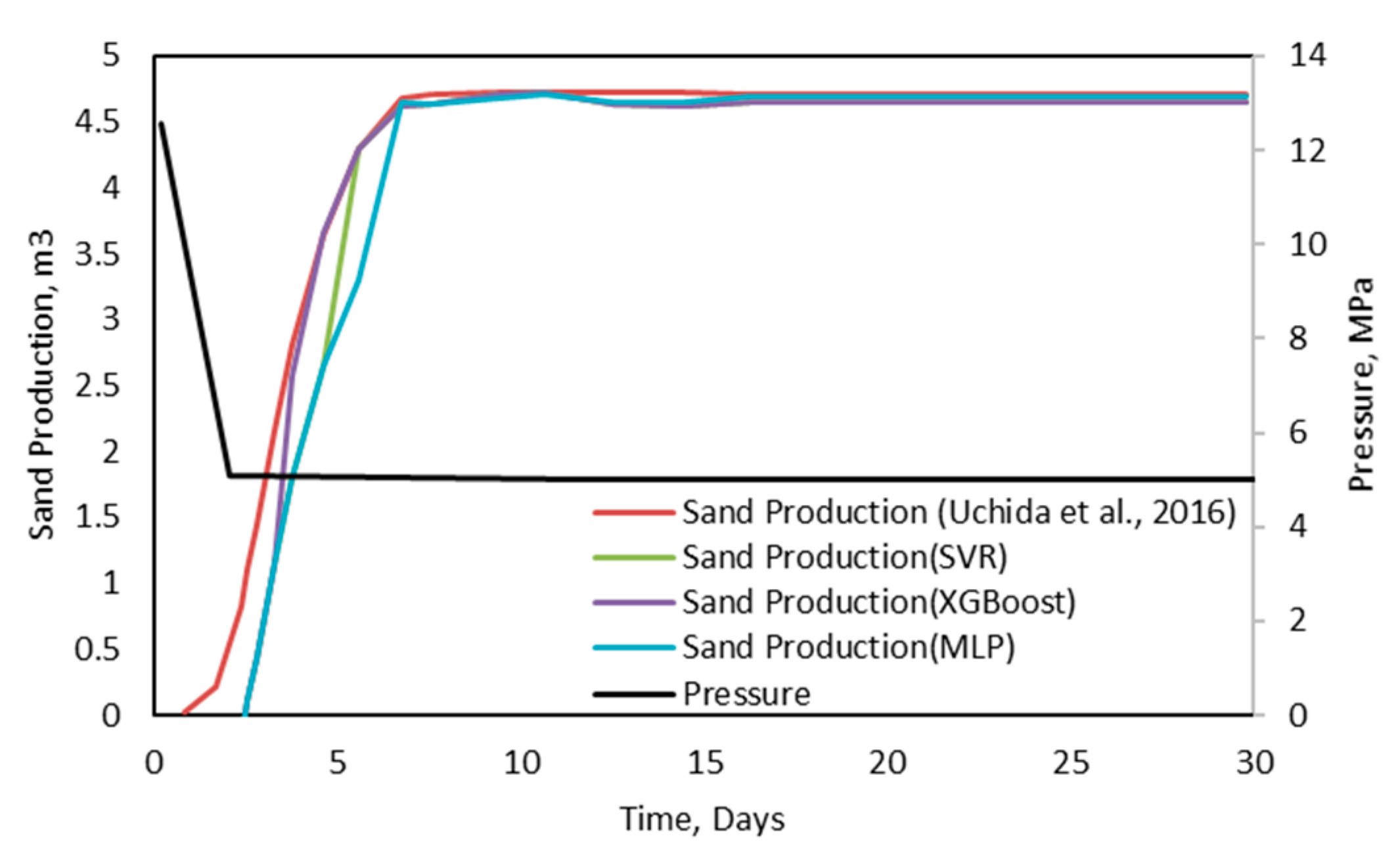
46]. As the name implies, the Lifting Tree is an algorithm that selects a weak learner as a decision tree and then integrates it. A weak learner is a machine learning model with performance that is a little better than that of chance. Some researchers used field datasets to prove that the Boosting Tree algorithm was better than a neural network algorithm [
47]. The Boosting Tree mainly comprises two powerful algorithms, which are the Gradient Boosting Tree [
48] and Extreme Gradient Boosting [
49]. According to a case study provided by Tixier et al., the Gradient Boosting Tree (GBT) was proven to have better performance than that of other machine learning methods, since it can determine nonlinear and local relationships [
50]. Extreme Gradient Boosting (XGB) is treated as an implementation of a Gradient Boosting Machine (GBM), but it is more accurate and efficient [
51]. Some researchers also found that XGB algorithms could significantly reduce the risk of overfitting [
52].
Given the training sample set
, a regression tree corresponds to a partition of the input space and the output value on all partition units. Now, assuming that the space has been divided into
units
, and there is a fixed output value
on each unit. The regression tree model can be expressed as:
In machine learning, the square error is applied to represent the prediction error of the regression tree for the training data, and the minimum square error criterion is used to solve the optimal output value on each unit, which is transformed into an optimization problem [
53]. The optimal value
of
on unit
is the mean of output
corresponding to all output instances
on
:
The
jth feature of the data (
x(j)) and a certain value (or values) are assumed to be selected as the segmentation variable and segmentation point. Two regions are defined as follows:
Then, the optimal cut feature
and the optimal cut point
are found:
Traversing all features, the optimal segmentation features and optimal segmentation points are found. According to this rule, the input space is divided into two regions. Then, the above process is repeated for each region until the stop condition is satisfied, where a regression decision tree can be generated. Combining Equation (18) with (17), the final regression tree model is:
where the parameter
represents the regional division of the tree and the optimal value of each region;
is the number of leaf nodes of the regression tree.
In the lifting tree, we use an additive model and a forward distribution algorithm. First, the initial lifting tree
is set, and the model in step t is:
which is the current model
. The required solution is:
The parameter of the t-tree is determined through empirical risk minimization.
When the square error loss function is used,
Here,
is the residual of the predicted value of the
t−1 tree:
So, the optimization goal of the
t tree becomes:
When the lifting tree solves the regression problem, it only needs to fit the residual of the predicted value of the previous tree. This means that the algorithm becomes quite simple, and the final model of the lifting tree can be expressed as an additive model.
where
represents the decision tree,
is the parameter of the decision tree, and
is the number of decision trees.
2.4. Multi-Layer Perceptron
The neural network is a representative algorithm in machine learning, and it is the most popular and widely used machine learning model [
54]. A Multi-Layer Perceptron (MLP) is a supplement to feed-forward neural networks [
55]. Similarly to a biological neural network, the most basic constituent unit of an artificial neural network is the ‘neuron’. Each neuron is connected to other neurons. It multiplies the weight on each edge and adds the bias of the neuron itself when it receives an input. The output of a neuron is finally generated through an activation function, which is a nonlinear function, such as a sigmoid function, arc-tangent function, or hyperbolic-tangent function [
56]. A sigmoid function with many excellent properties is often selected as the most common activation function [
57]. The perceptron model consists of two layers, which are the input layer and output layer. However, the perceptron has only two layers and only one output layer. One layer of functional neurons restricts the model to fitting data. To solve complex problems, multi-layer functional neurons are needed. The neuron layer between the input layer and the output layer is called the hidden layer. In other words, the Multi-Layer Perceptron (MLP) is a neural network model with multiple hidden layers. Each layer of neurons is completely connected to the next layer of neurons, and there are no connections within the same layer or cross-layer connections. Some researchers proved that a neural network with more than three layers could simulate any continuous function with arbitrary accuracy [
58,
59]. The learning of MLP takes place by adjusting the weights and biases between neurons according to the training data. An error back-propagation (BP) algorithm is commonly used to train multi-layer neural networks. These are based on a gradient descent strategy and are iterative optimization algorithms, which adjust the parameters in the negative gradient direction of the target parameters [
60]. According to the gradient descent strategy, let the loss function be
and a pair of parameters be
. The initial values
are randomly selected; in the (
n + 1)th iteration,
where the learning rate
determines the update step size of each iteration of the algorithm. The oscillation causes the model to be unable to converge normally if the value of
is too large. The convergence speed of the algorithm is slow if too small of a value of
is used. The learning rate is usually set to 0.8 [
60]. Newton’s method is applied for rapid convergence [
61]. Then, Equations (29) and (30) yield:
Loss functions (or objective functions) may have multiple local extremums, but there is only one global minimum [
62]. The global minimum value is the final objective of the calculations. However, for the gradient descent algorithm, in each iteration, the gradient of the loss function is calculated at a certain point, and then the optimal solution is determined along the negative gradient direction [
63]. The gradient at the current point is zero if the loss function has reached the local minimum, where the updating of the parameters is terminated. This leads to a local extremum in the parameter optimization. The local minimum can be avoided through the following strategies in order to further approach the global minimum [
64,
65,
66]: (1) The neural network is initialized with multiple sets of different parameters. The parameters with the loss function are the final solution after training. This process is equivalent to starting from multiple different initial points for optimization; then, we may fall into different local extremums, from which we can select the results closer to the global minimum. (2) The random gradient descent method is used. This method adds a random factor, and only one sample is used in each update. There will be a certain possibility of making the direction deviate from the optimal direction so that it can jump out of the local extremum.
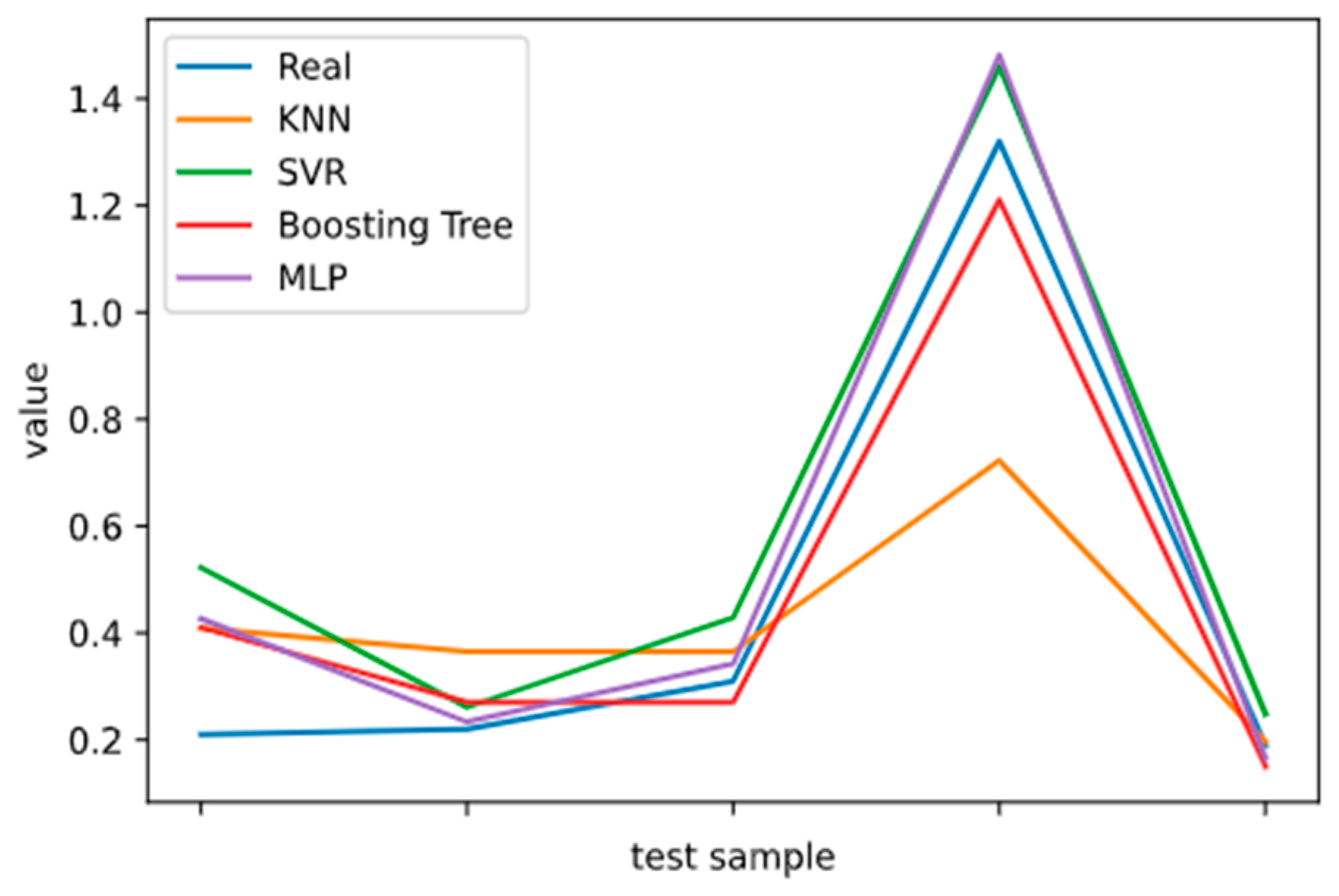
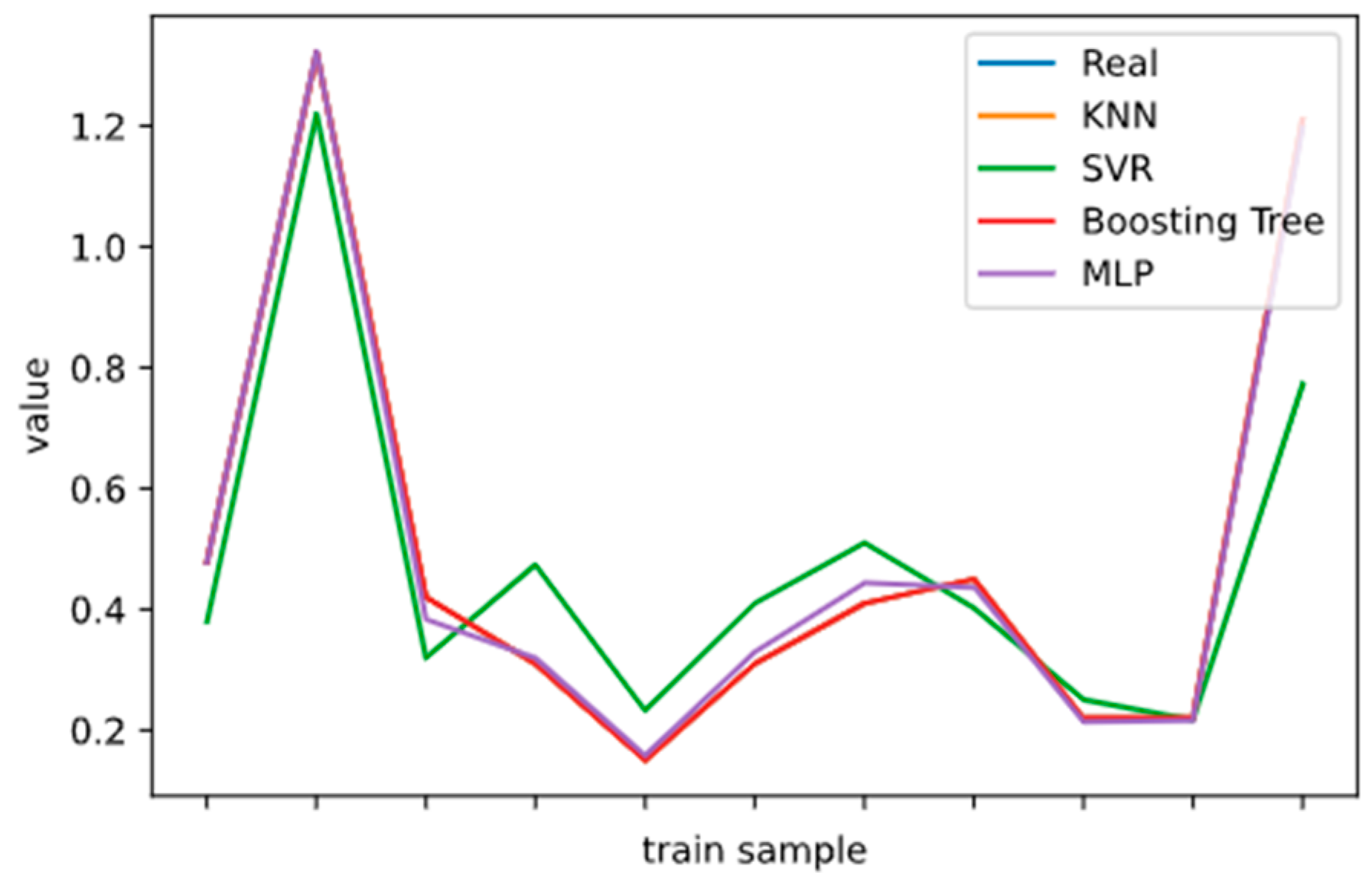
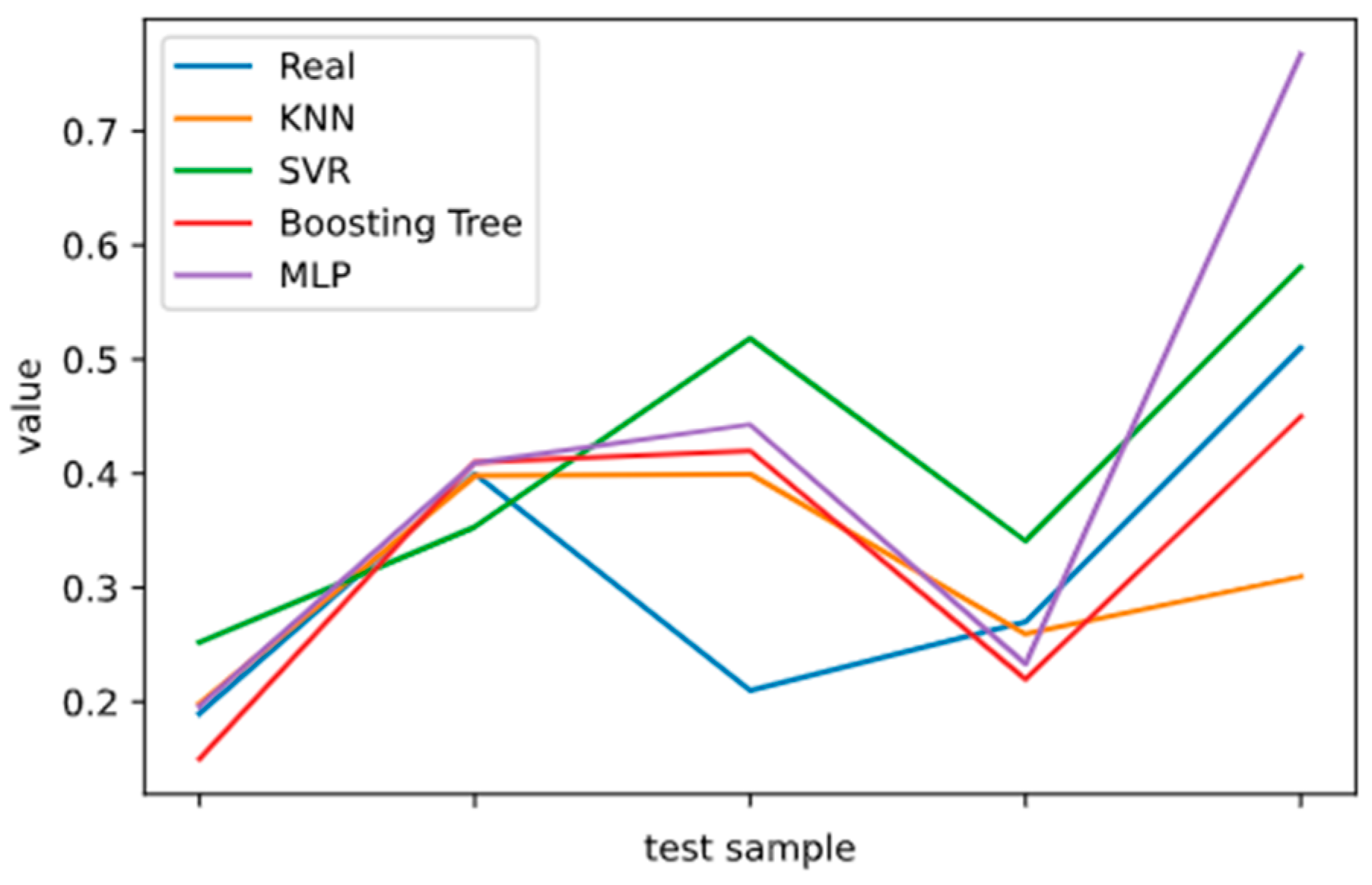
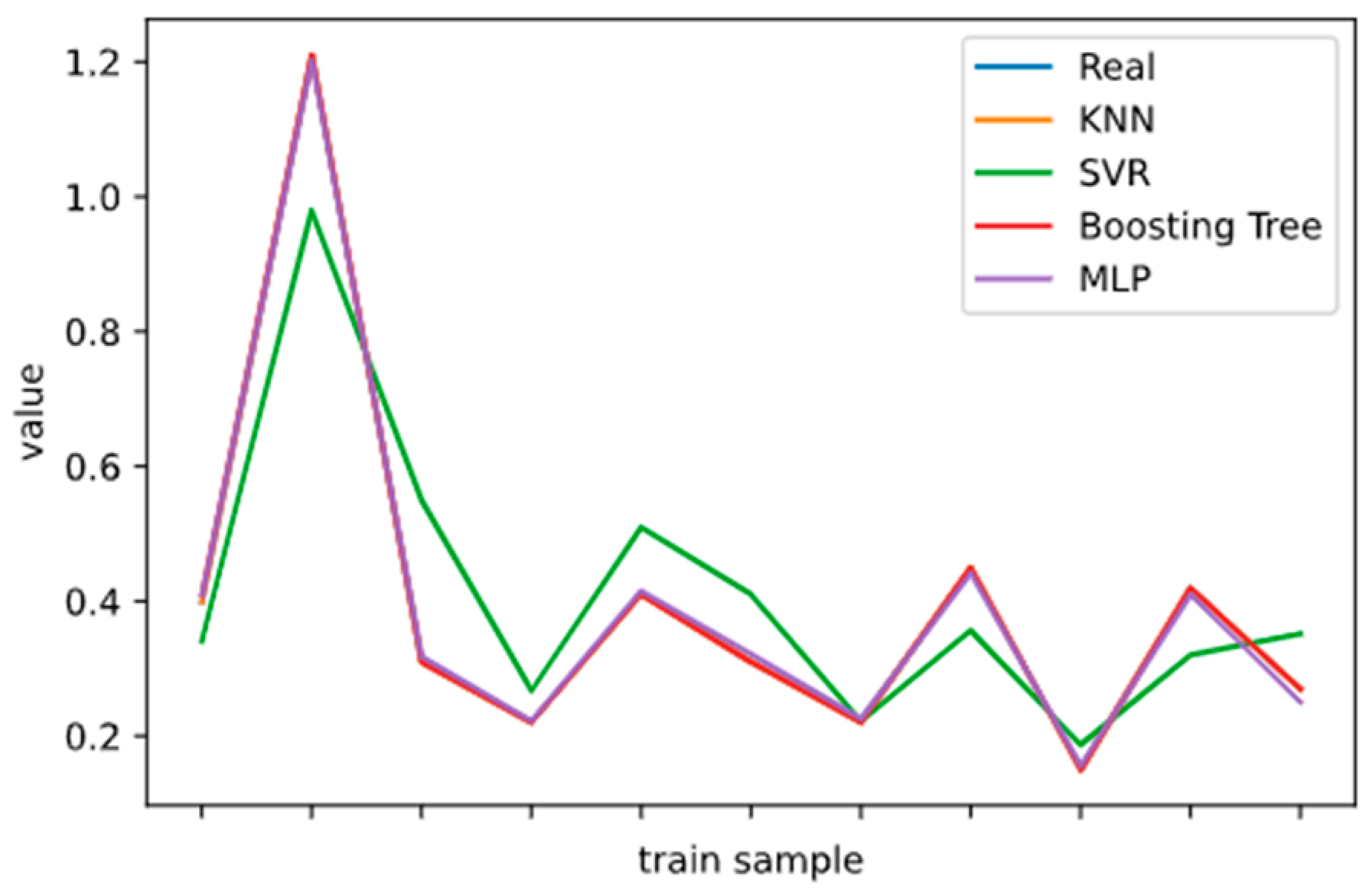
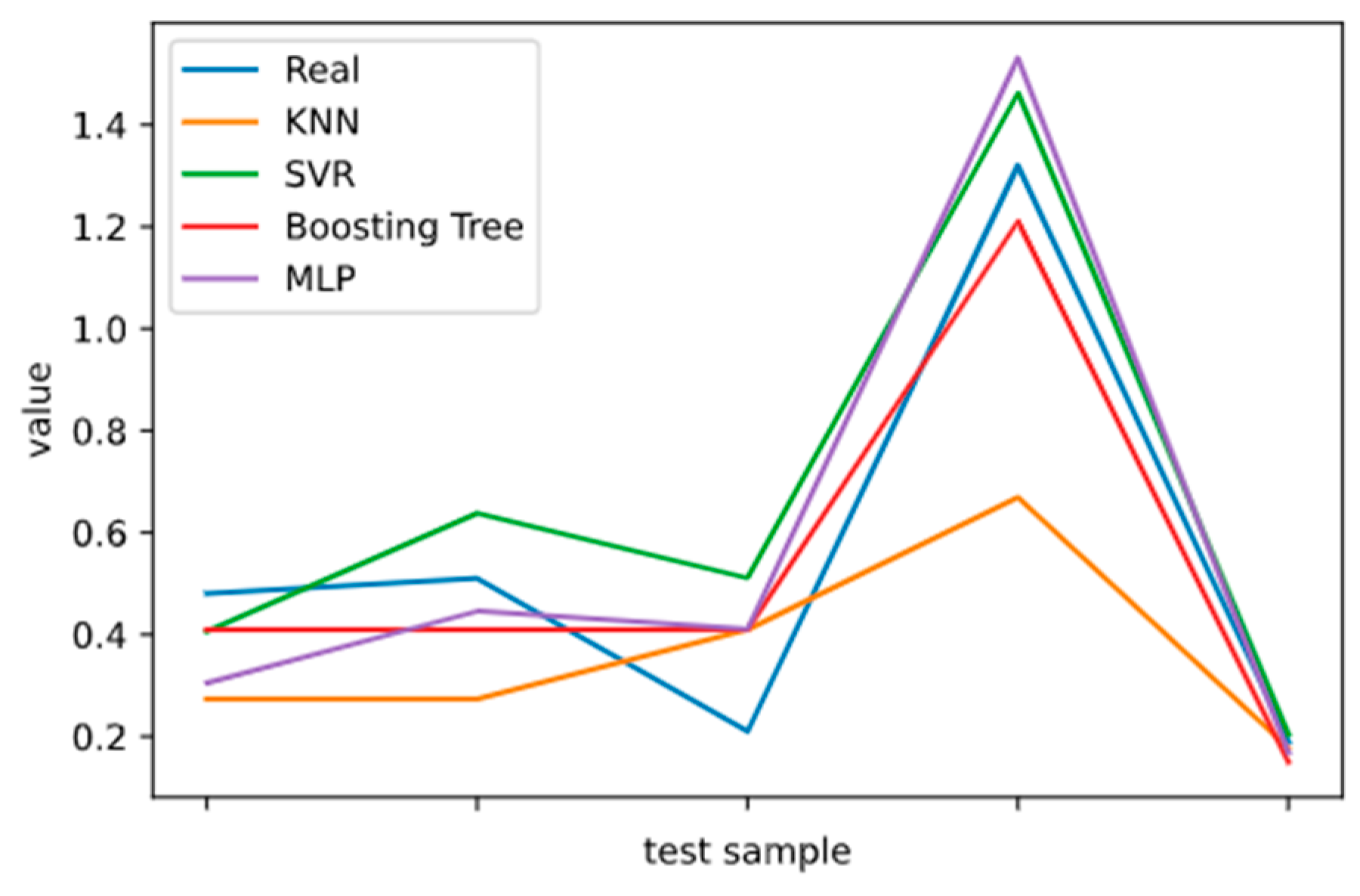
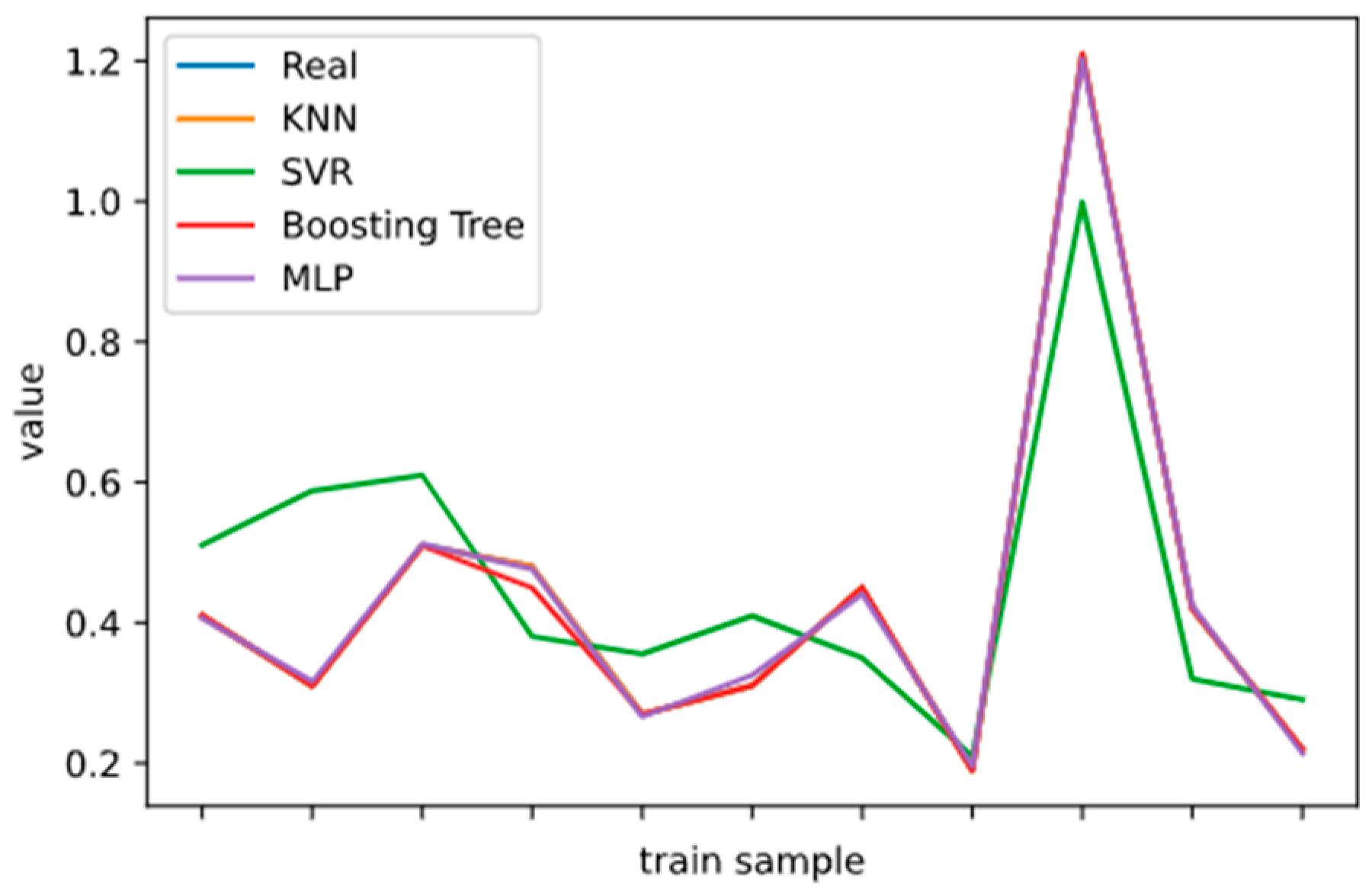
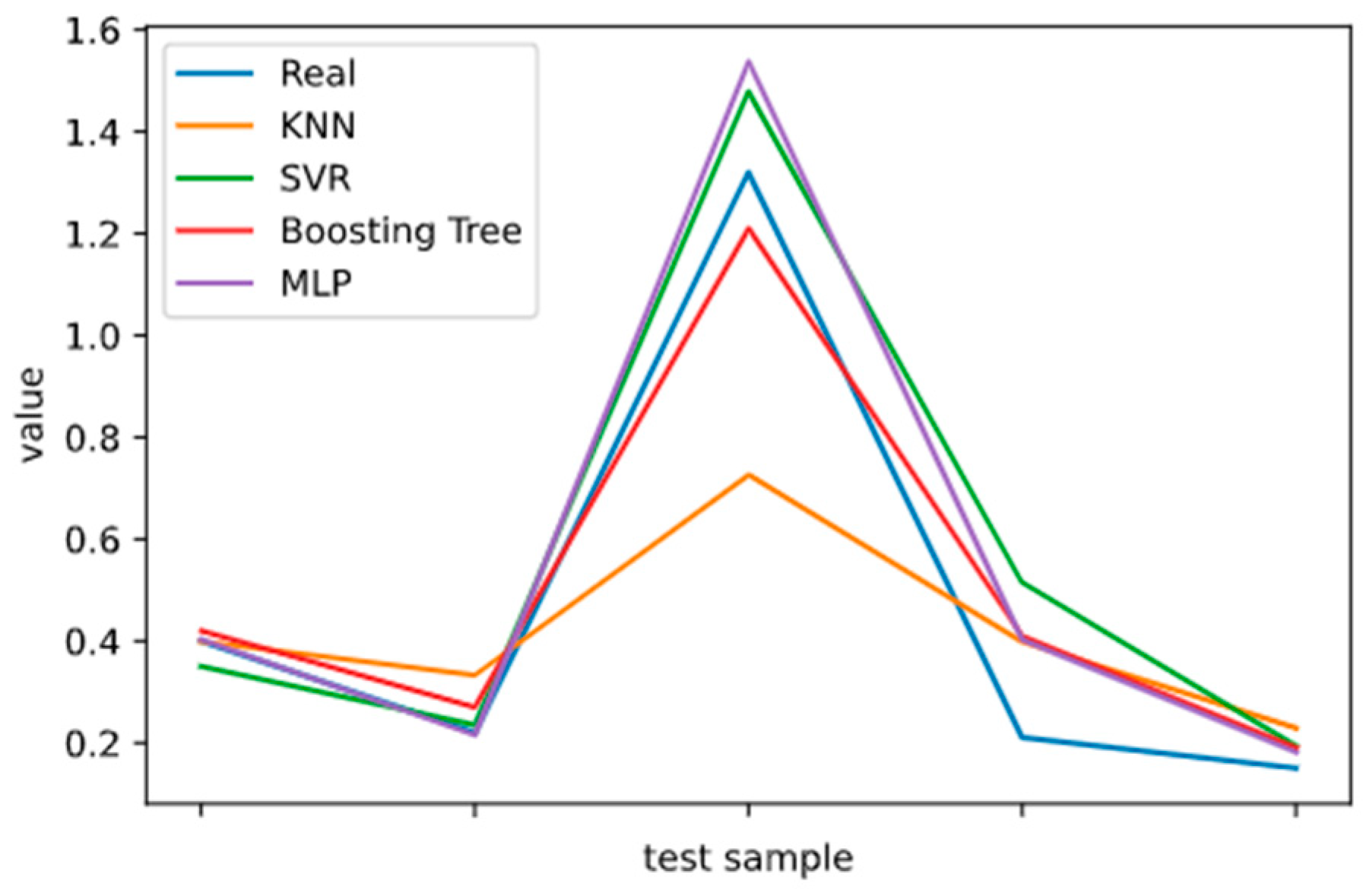
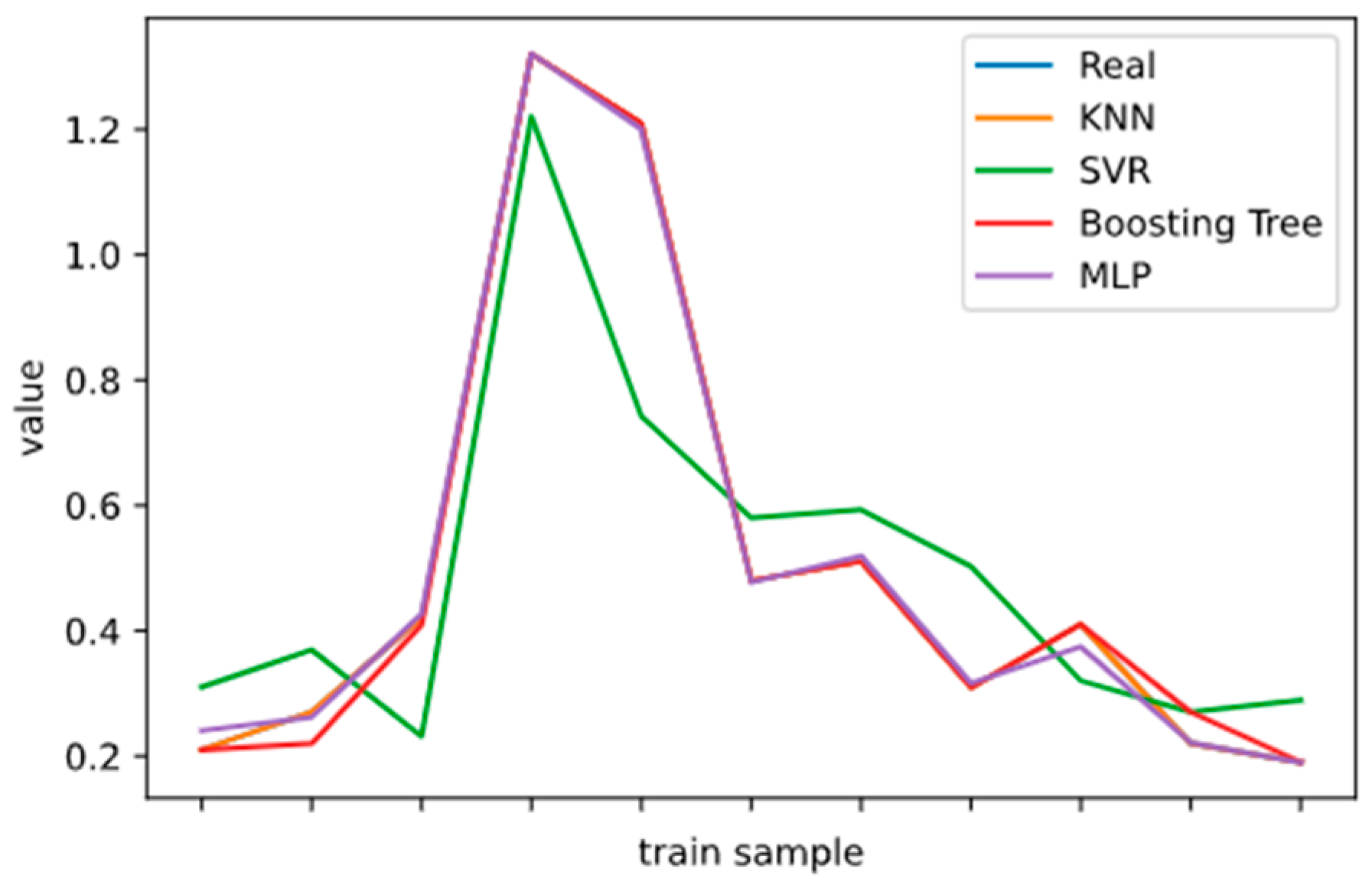
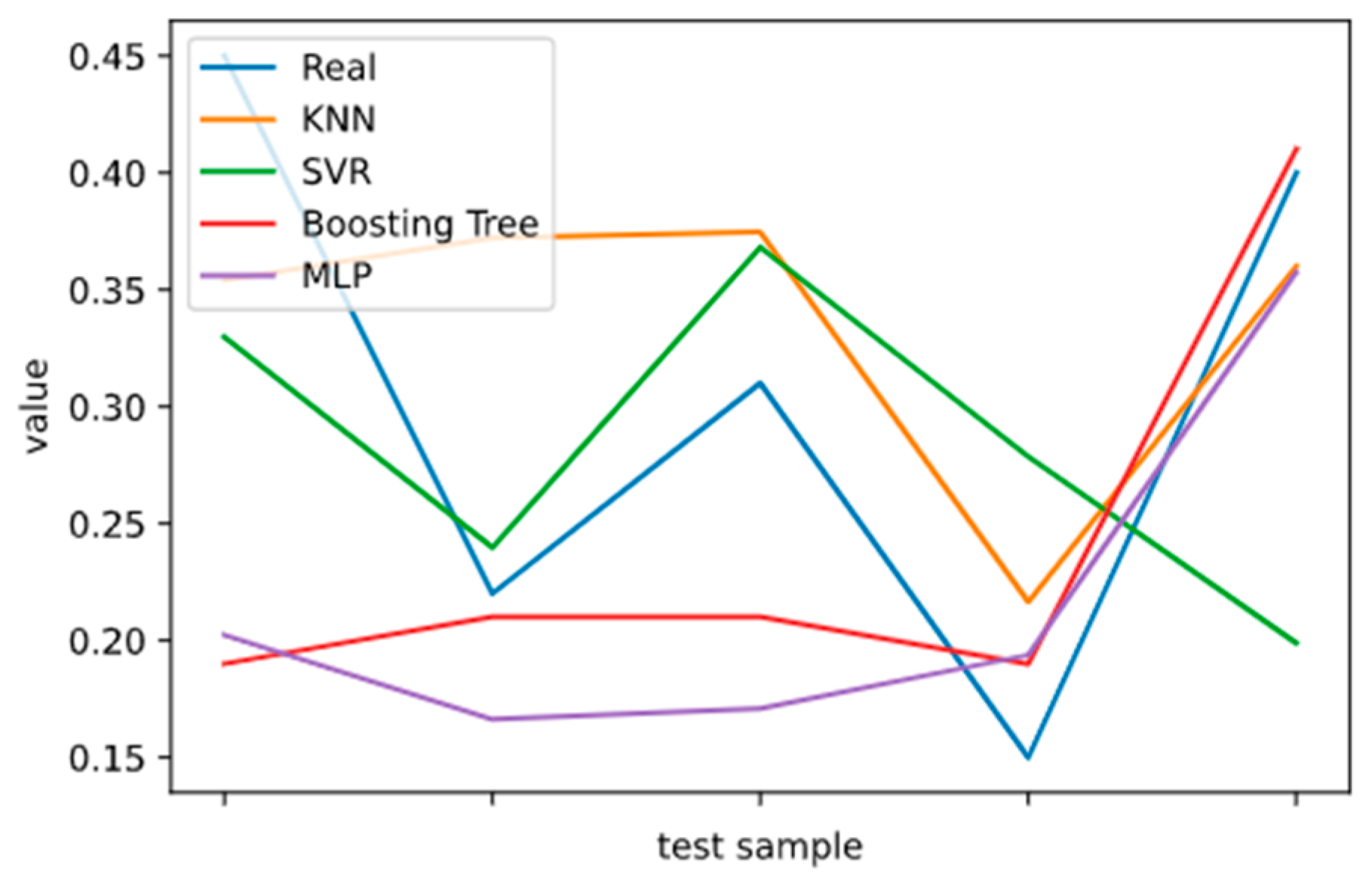
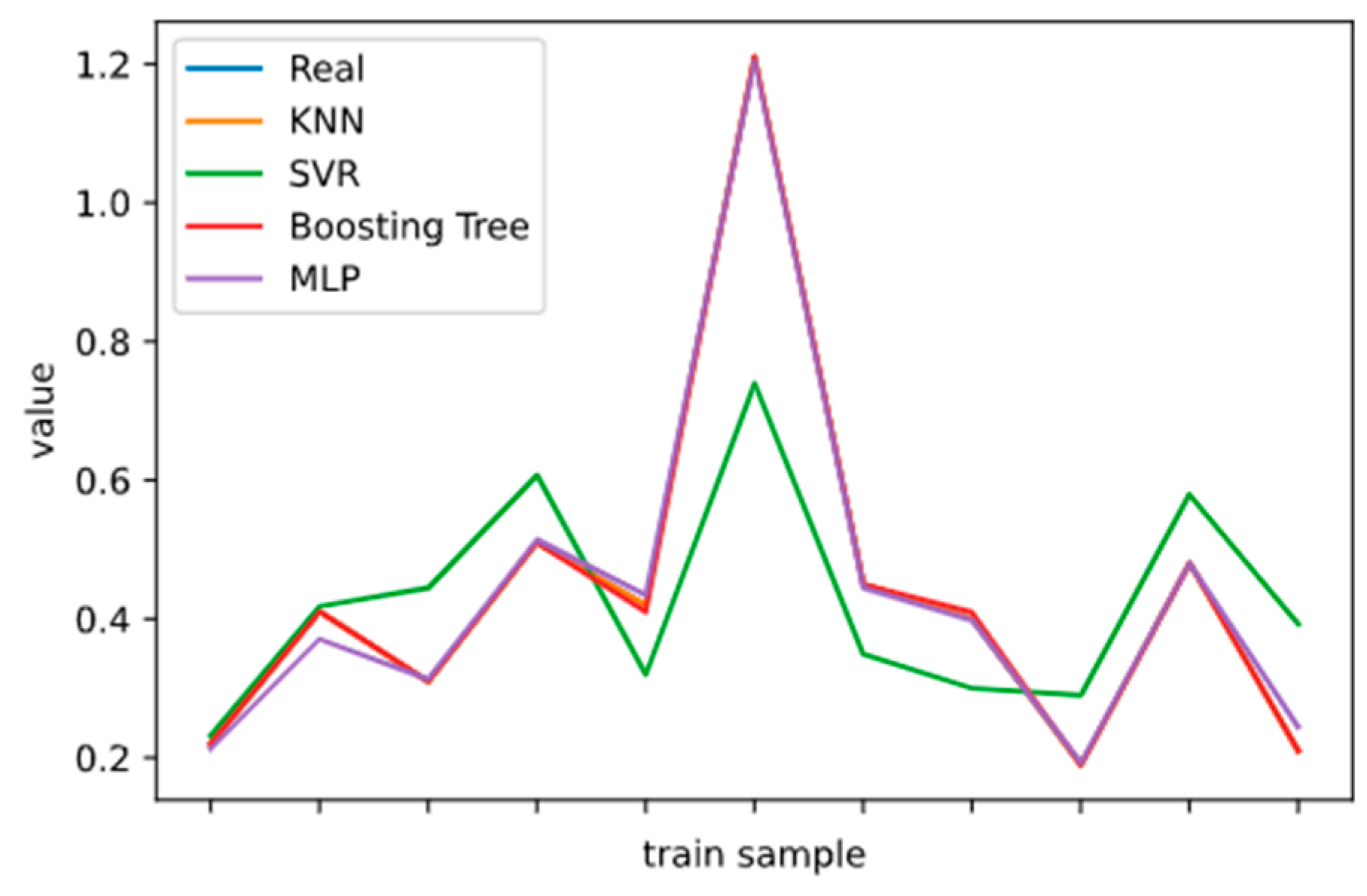
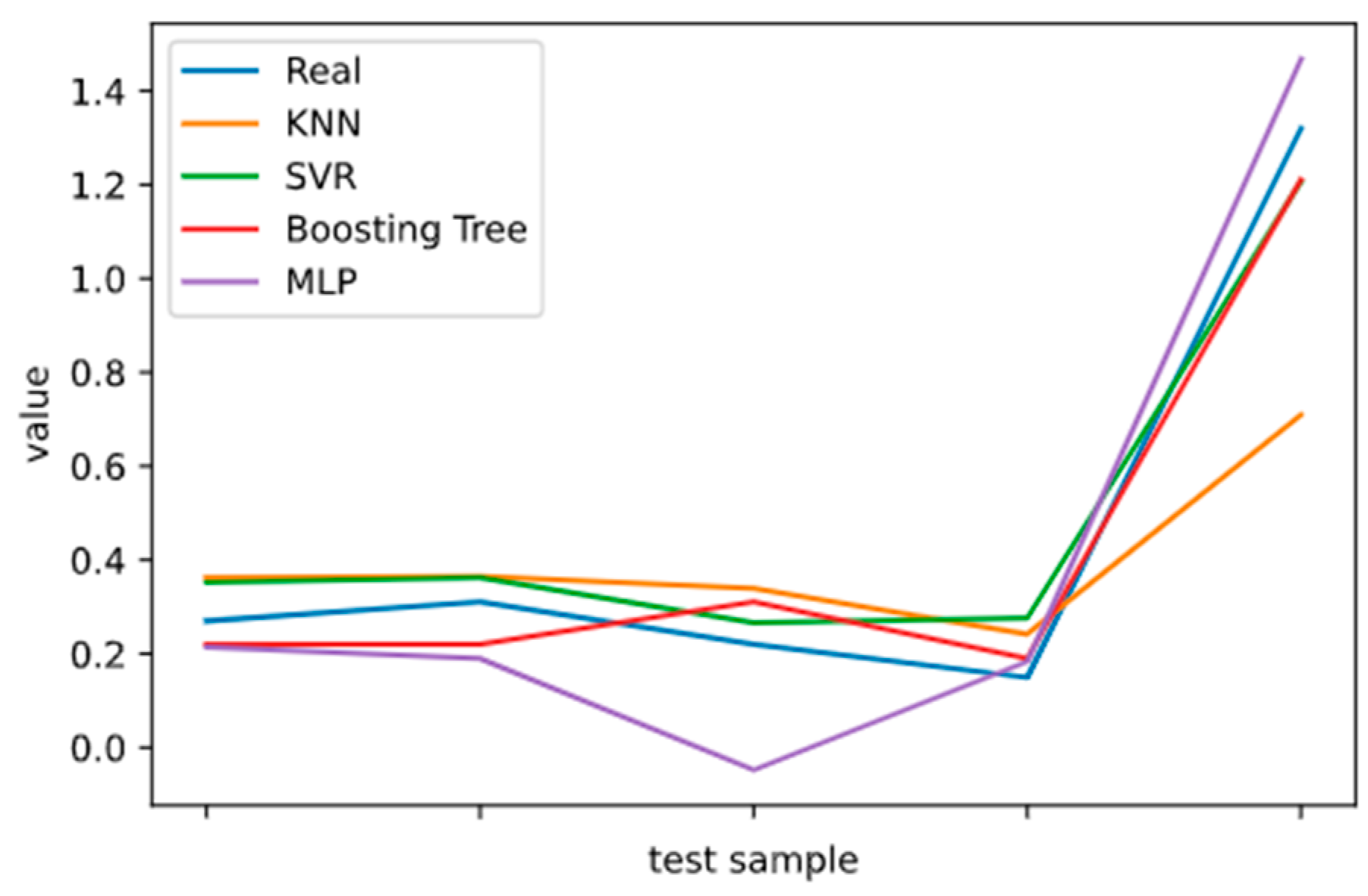
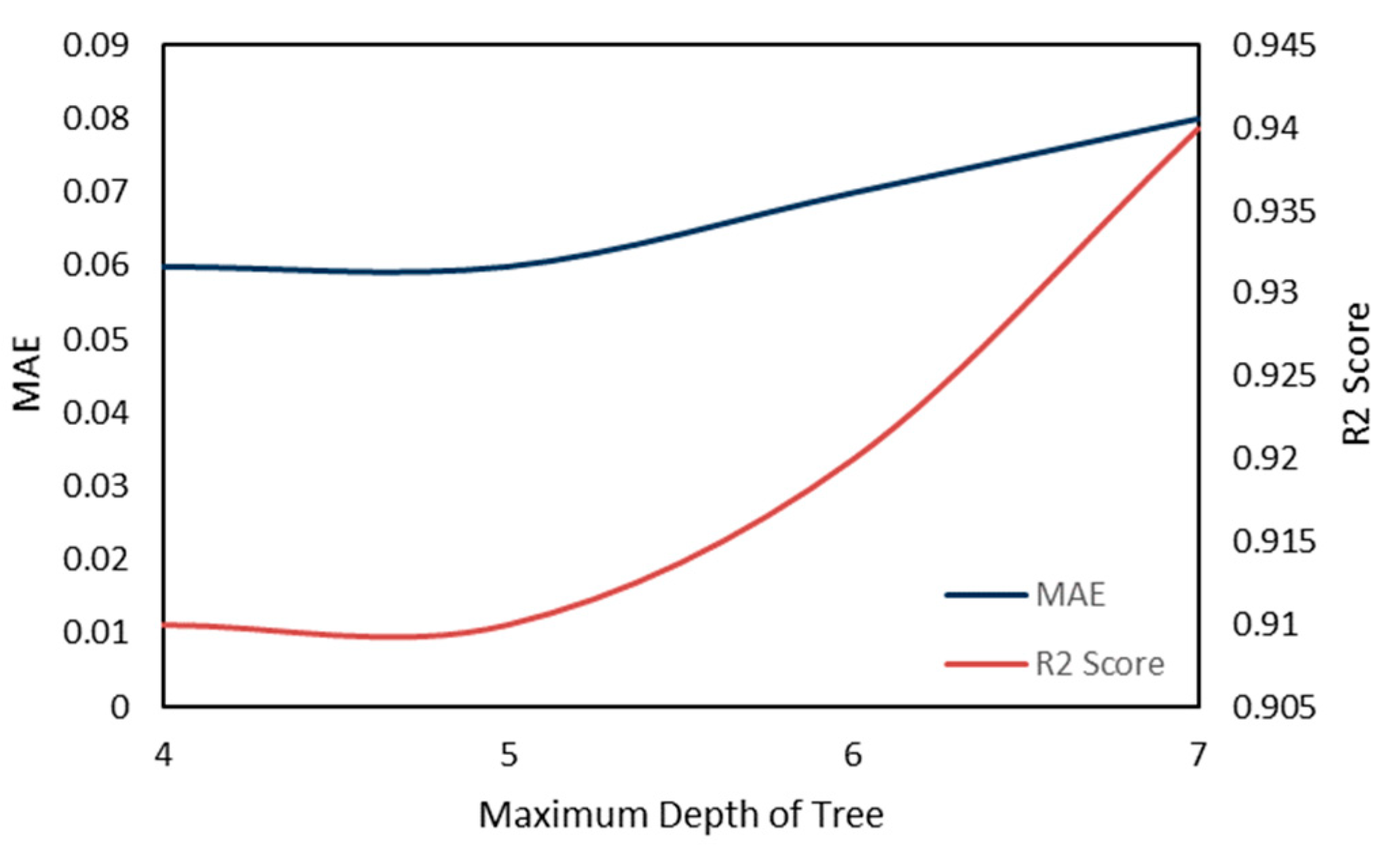
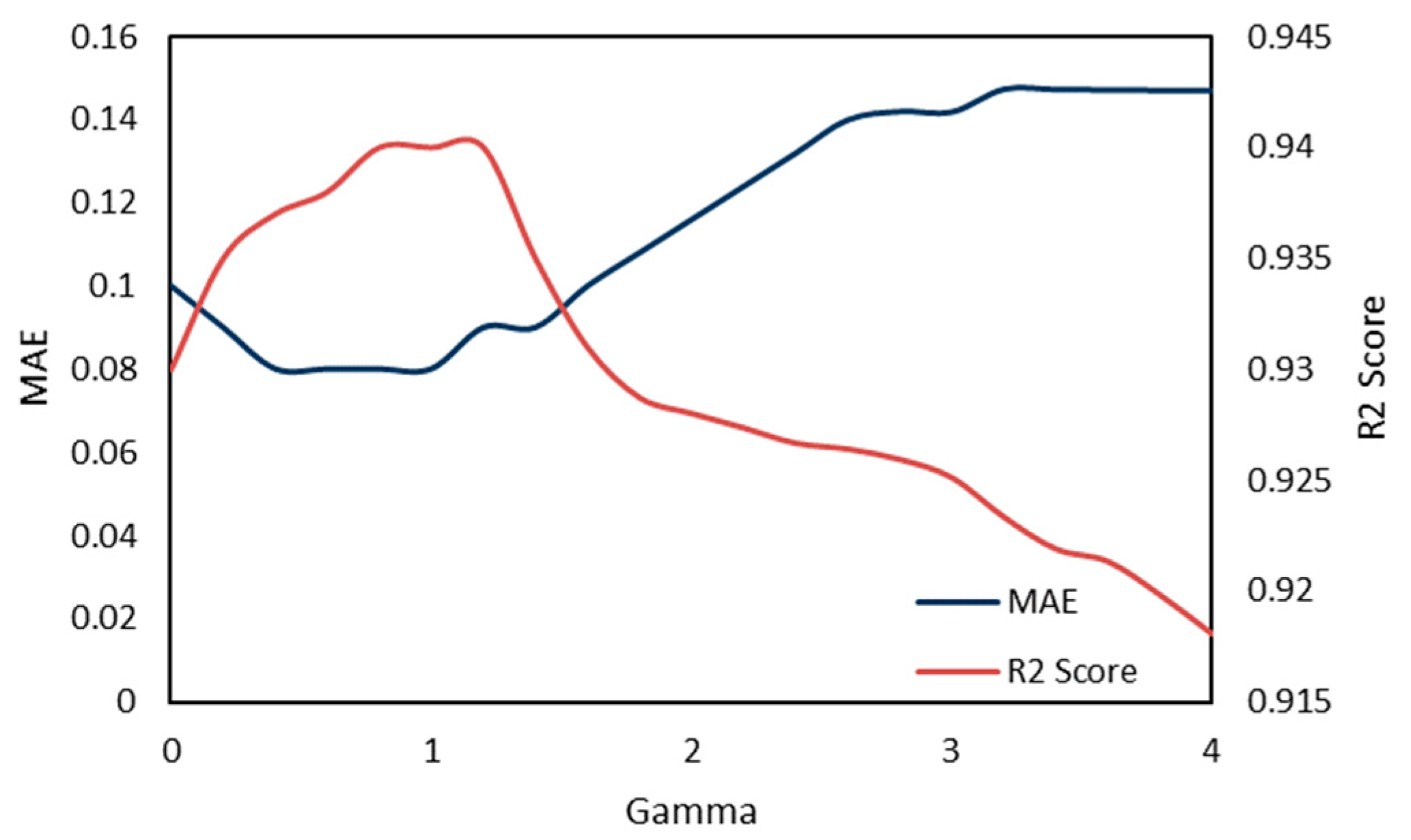
2.5. Model Performance Evaluation
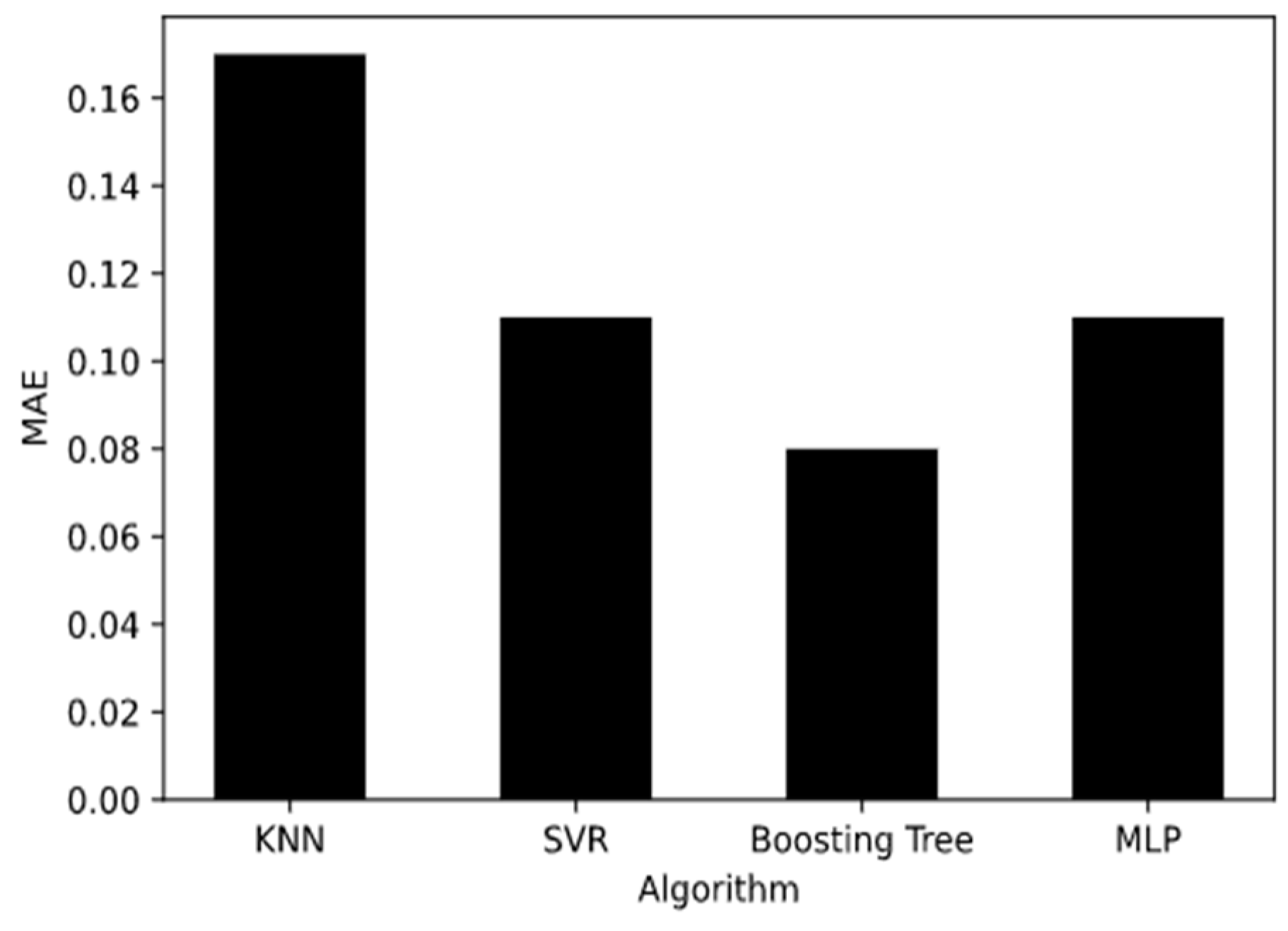
To evaluate machine learning models, several performance metrics are adopted and calculated to judge their performance. Based on case studies from other researchers, the mean absolute error (MAE) and coefficient of determination (R
2 Score) are often selected to evaluate the generalization ability of machine learning models [
67]. The MAE is the average difference between the true value and the predicted values, and it can be easily calculated and compared. Some researchers even used some revised forms of the MAE, such as the dynamic mean absolute error and mean absolute percentage error, to evaluate the accuracy of a prediction model [
68,
69]. The more accurate the machine learning model is, the smaller the MAE will be. A perfect model causes the MAE to be close to 0. Let
and y denote the predicted value and the true value, respectively. Then, the MAE can be expressed as:
The MAE can intuitively measure the ‘gap’ between the predicted value and the real value. However, it is difficult to compare a model’s effect when the equivalence dimensions are different.
Since the dimensions of different datasets are different, it is difficult to compare them by using a simple difference calculation. The coefficient of determination (R
2 Score) can be adopted to evaluate the degree of coincidence of the predicted values and true values [
70]. Most recent research has proved the feasibility of using the R
2 Score to evaluate mixed-effect models, rather than just linear models [
71]. The calculation of the R
2 Score involves three important performance judgment terms—the sum of squares for regression (SSR), sum of squares for error (SSE), and sum of squares for total (SST) [
72]:
Based on the above relationship, the calculation method for the R
2 Score can be defined as [
73]:
where
is the mean square error between the predicted value
and the real value
;
is the variance of the real value
. Specifically, there are the following situations in the analysis of the R
2 Score:
If , the performance metric is 0, indicating that the predicted label value in the test sample is exactly the same as the true value. A perfect model has been built to predict all of the test samples.
If
, the numerator is equal to the denominator, indicating that our predicted values are all the mean values of the real values. In this situation, the prediction model explains none of the variability of the response data around its mean. Sánchez et al. (2019) also explained that, in this scenario, the inclusion of variables can be neglected, and the built prediction model is not adequate [
74].
If , the score is within the normal range. A value closer to 1 indicates a better fit, while a value closer to 0 indicates a worse fitting effect.
For a bad scenario in which , the numerator is greater than the denominator, that is, the error of the prediction data is greater than the variance of the real data. This indicates that the error of the predicted value is greater than the dispersion of the data, which indicates the misuse of a linear model for nonlinear data.





































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}