
An Experimental and Detailed Kinetics Modeling Study of Norbornadiene in Hydrogen and Methane Mixtures: Ignition Delay Time and Spectroscopic CO Measurements


Abstract
:1. Introduction
2. Experimental Methodology
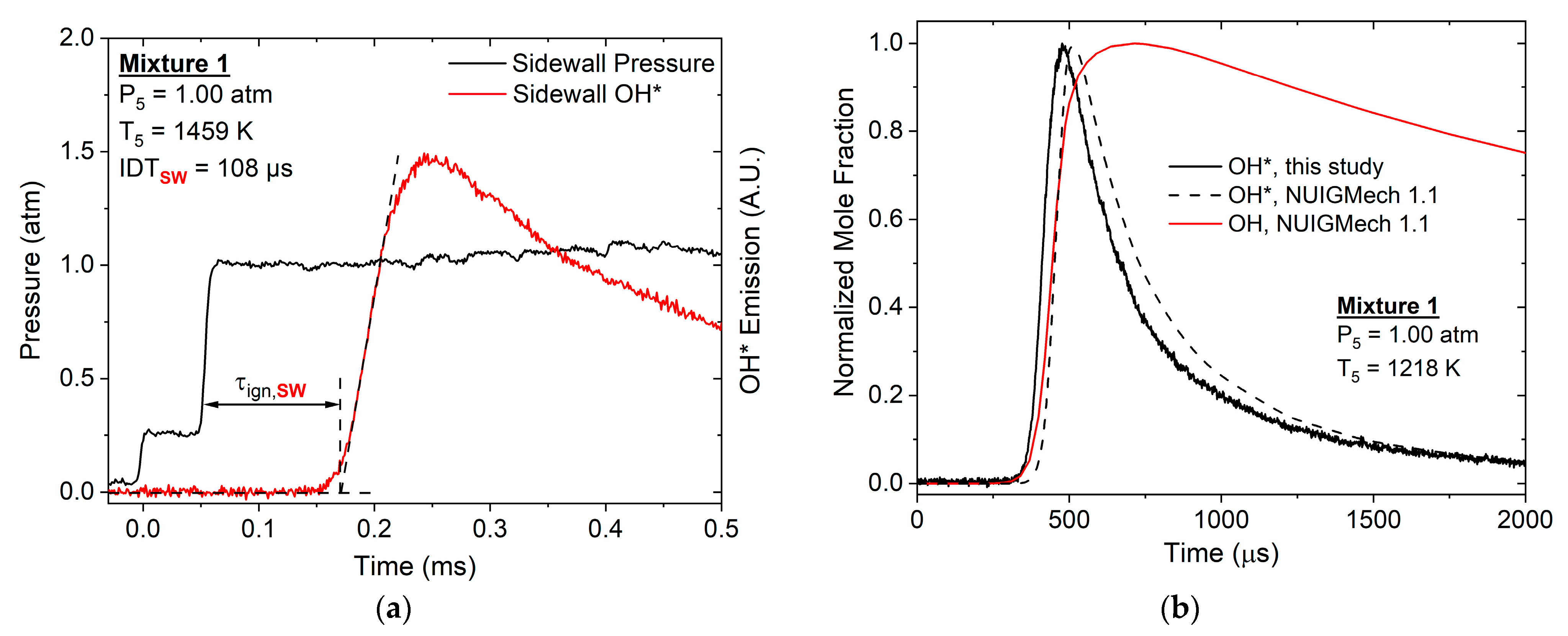
2.1. Shock-Tube Facilities
2.2. Chemiluminescnece Emission
2.3. CO Laser Diagnostic
2.4. Experimental Uncertainty
3. Detailed Kinetics Mechanism
- (1)
- Addition of the NBD decomposition reactions, namely NBD C6H5CH3 (toluene), NBD NCD (norcaradiene), and NBD C5H6 (cyclopentadiene) + C2H2 (acetylene).
- (2)
- As the chemistry of C6H5CH3, C5H6, and C2H2 is already included in NUIGMech 1.1, only the sub-mechanism of NCD was needed.
- (3)
- NCD chemistry involves NCD CHT (1,3,5-cycloheptatriene), CHT CHTyl7 (cycloheptatrienyl) + H, and CHTyl7 + H(+M) CHT(+M).
- (4)
- The CHTyl7 sub-mechanism includes the reactions CHTyl7 C5H5 (cyclopentadienyl) + C2H2, eC7H6 (5-ethynyl-1,3-cyclopentadiene) + H CHTyl7, and eC7H6 + H C7H6 (fulvenallene) + H. The C5H5- and C7H6-related reactions can be found in NUIGMech 1.1.
- (5)
- Lastly, the cross-reactions between xylene (oC8H10, mC8H10, and pC8H10) and toluene were implemented, as well as the interactions between CHTyl7 with toluene and indene.
4. Results and Discussion
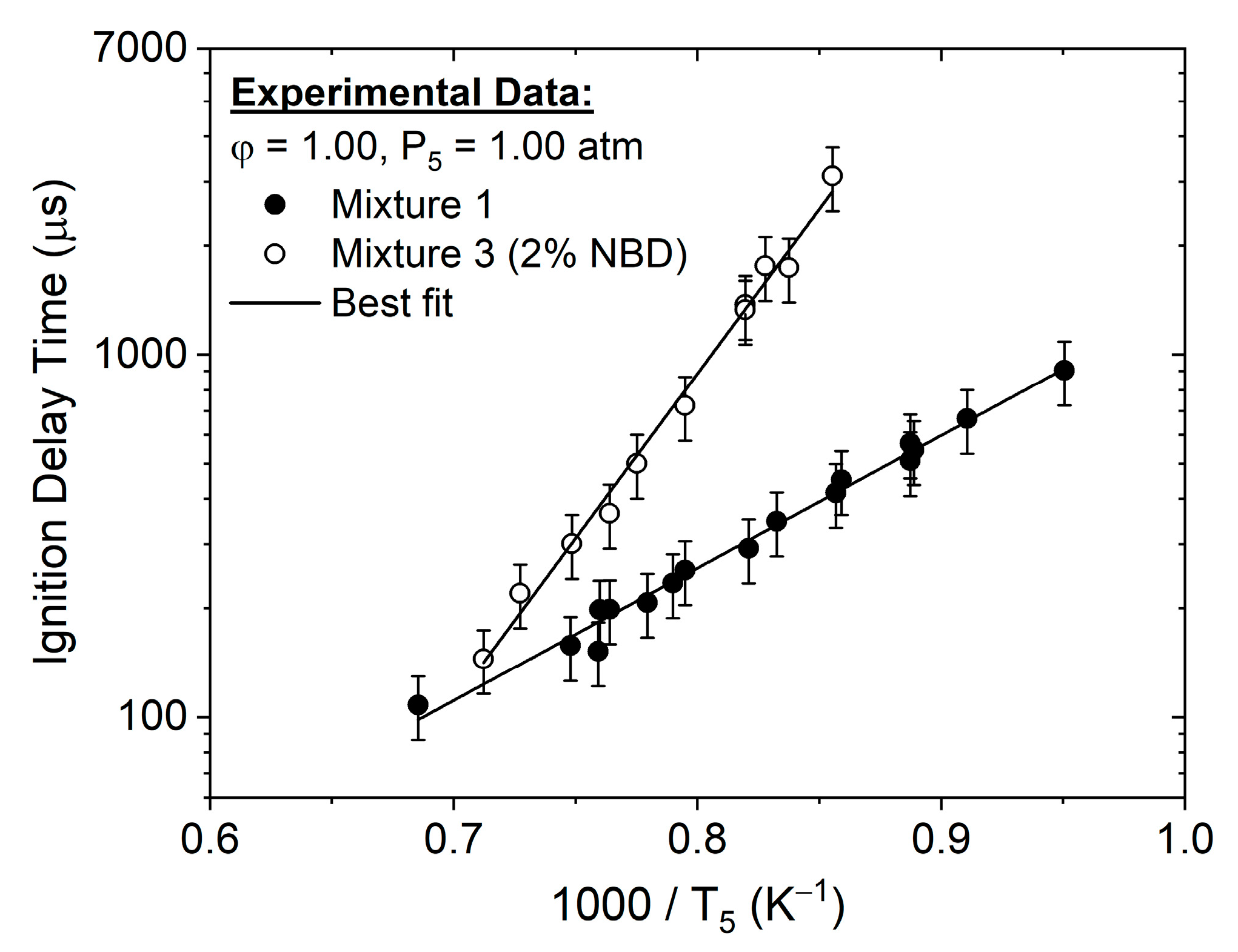
4.1. Ignition Delay Time Correlations
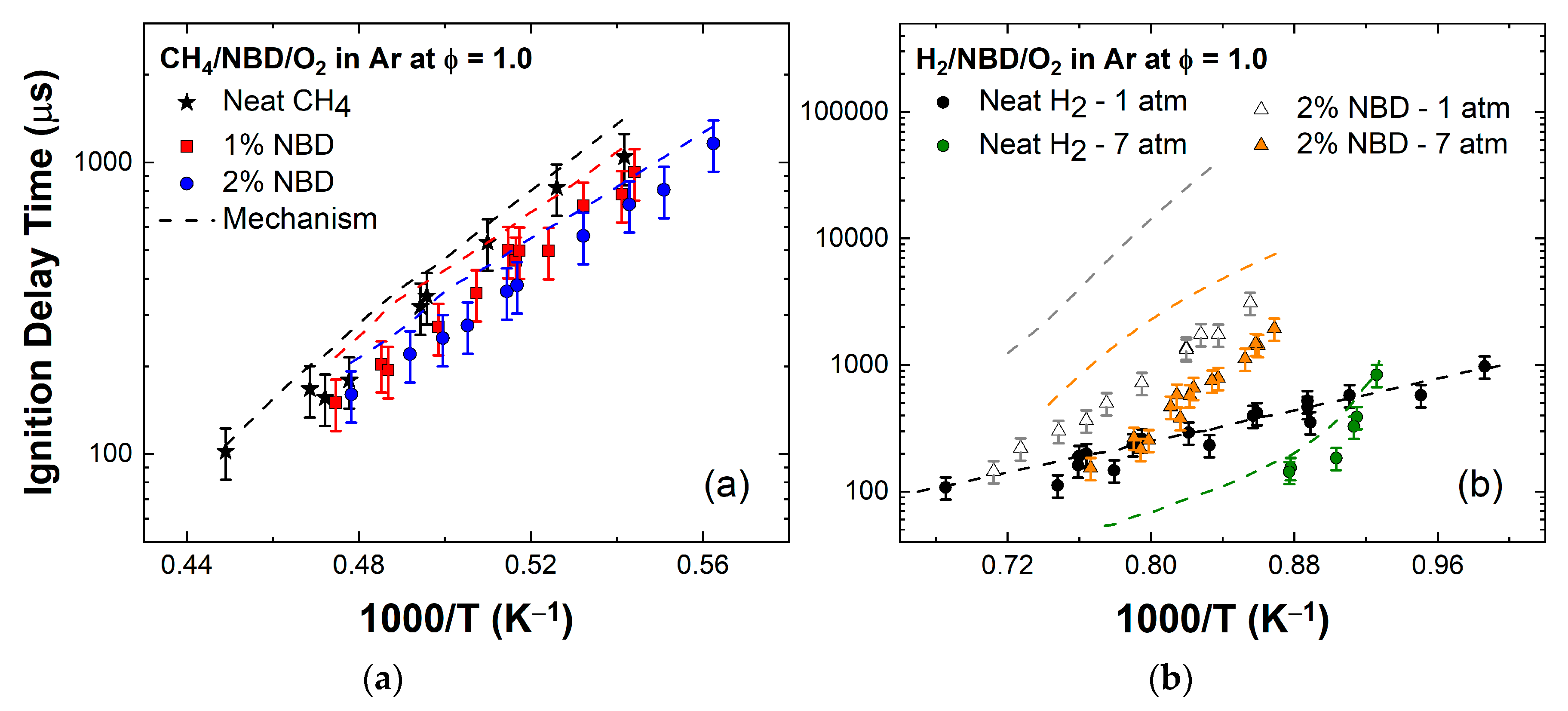
4.2. Hydrogen-Oxygen Mixtures
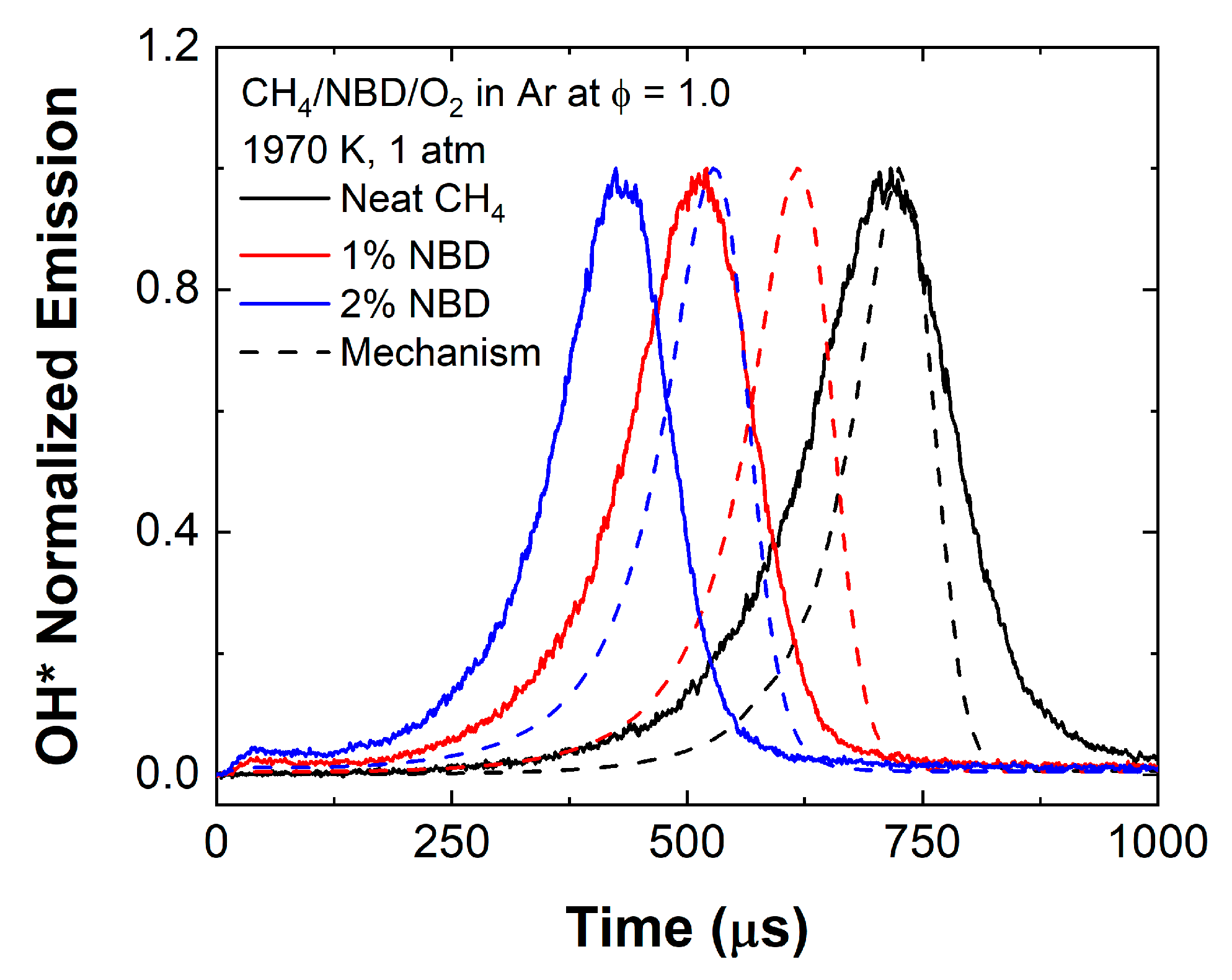
4.3. Methane–Oxygen Mixtures
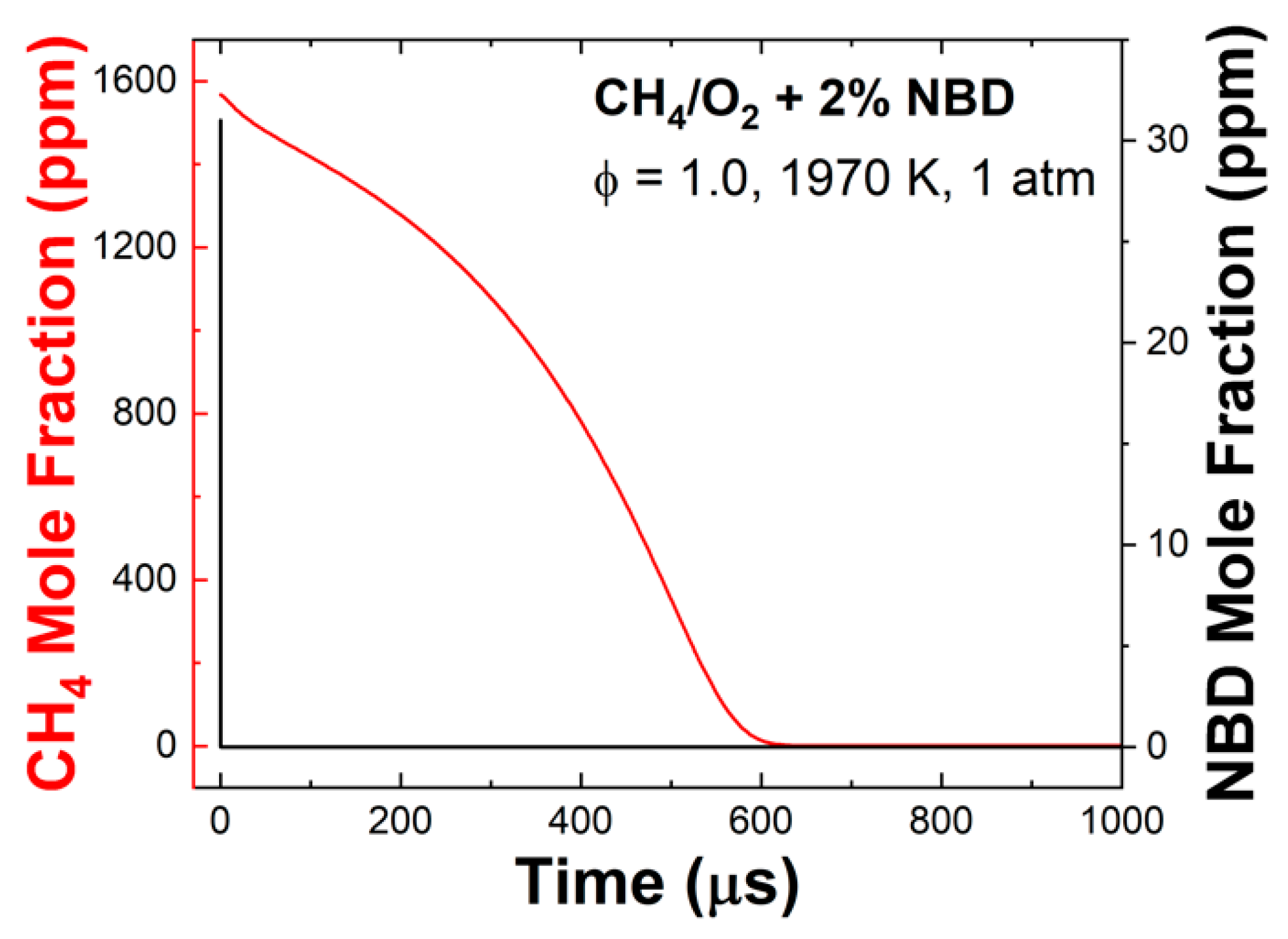
4.4. Chemical Kinetics Modeling
4.4.1. OH* Emission Profiles
4.4.2. CO Time-History Profiles
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Walsh, R.; Wells, J.M. The enthalpy of formation of bicyclo[2,2,1]hepta-2,5-diene thermodynamic functions of bicyclo[2,2,1]heptane and bicyclo[2,2,1]hepta-2,5-diene. J. Chem. Thermodyn. 1975, 7, 149–154. [Google Scholar] [CrossRef]
- Zou, J.-J.; Zhang, M.-Y.; Zhu, B.; Wang, L.; Zhang, X.; Mi, Z. Isomerization of norbornadiene to quadricyclane using Ti-containing MCM-41 as photocatalysts. Catal. Lett. 2008, 124, 139–145. [Google Scholar] [CrossRef]
- Pan, L.; Zou, J.-J.; Zhang, X.; Wang, L. Photoisomerization of norbornadiene to quadricyclane using transition metal doped TiO2. Ind. Eng. Chem. Res. 2010, 49, 8526–8531. [Google Scholar] [CrossRef]
- Bach, R.D.; Schilke, I.L.; Schlegel, H.B. The energetics of valence isomerization in the norbornadiene−quadricyclane system. J. Org. Chem. 1996, 61, 4845–4847. [Google Scholar] [CrossRef]
- Fan, H.-F.; Chin, T.-L.; Lin, K.-C. Kinetics of catalytic isomerization of quadricyclane to norbornadiene using near infrared absorption spectroscopy: Conversion rate and diffusion motion in heterogeneous reaction. J. Phys. Chem. B 2004, 108, 9364–9370. [Google Scholar] [CrossRef]
- Walsh, R.; Wells, J.M. The kinetics of the Diels–Alder addition of cyclopentadiene to acetylene and the decomposition of norbornadiene. Int. J. Chem. Kinet. 1975, 7, 319–329. [Google Scholar] [CrossRef]
- Li, Z.; Anderson, S.L. Pyrolysis and isomerization of quadricyclane, norbornadiene, and toluene. J. Phys. Chem. A 1998, 102, 9202–9212. [Google Scholar] [CrossRef]
- Jin, K.-R.; Zheng, Z.-H.; Wu, L.-N.; Xu, Q.; Liu, B.-Z.; Wang, Z.-D.; Tian, Z.-Y. Pyrolysis of norbornadiene: An experimental and kinetic modeling study. Combust. Flame 2022, 242, 112155. [Google Scholar] [CrossRef]
- Chen, J.; Liu, M.; Zhu, Y.; Jin, K.; Tian, Z.; Yang, L.; Zhou, C.-W. Oxidation of norbornadiene: Theoretical investigation on H-atom abstraction and related radical decomposition reactions. Propuls. Power Res. 2023, 12, 104–113. [Google Scholar] [CrossRef]
- Petersen, E.L. Interpreting endwall and sidewall measurements in shock-tube ignition studies. Combust. Sci. Technol. 2009, 181, 1123–1144. [Google Scholar] [CrossRef]
- Gaydon, A.; Hurle, I. The shock tube in high-temperature chemical physics. Reinhold 1963. [Google Scholar]
- Petersen, E.L.; Rickard, M.J.A.; Crofton, M.W.; Abbey, E.D.; Traum, M.J.; Kalitan, D.M. A facility for gas- and condensed-phase measurements behind shock waves. Meas. Sci. Technol. 2005, 16, 1716. [Google Scholar] [CrossRef]
- Cooper, S.P.; Mathieu, O.; Schoegl, I.; Petersen, E.L. High-pressure ignition delay time measurements of a four-component gasoline surrogate and its high-level blends with ethanol and methyl acetate. Fuel 2020, 275, 118016. [Google Scholar] [CrossRef]
- Mulvihill, C.R.; Petersen, E.L. Concerning shock-tube ignition delay times: An experimental investigation of impurities in the H2/O2 system and beyond. Proc. Combust. Inst. 2019, 37, 259–266. [Google Scholar] [CrossRef]
- Hall, J.M.; Petersen, E.L. An optimized kinetics model for OH chemiluminescence at high temperatures and atmospheric pressures. Int. J. Chem. Kinet. 2006, 38, 714–724. [Google Scholar] [CrossRef]
- Smith, G.P.; Golden, D.M.; Frenklach, M.; Moriarty, N.W.; Eiteneer, B.; Goldenberg, M.; Bowman, C.T.; Hanson, R.K.; Song, S.; Gardiner, W.C.; et al. GRI-Mech 3.0. 1999. Available online: http://www.me.berkeley.edu/gri_mech/ (accessed on 10 September 2023).
- Spearrin, R.M.; Goldenstein, C.S.; Jeffries, J.B.; Hanson, R.K. Quantum cascade laser absorption sensor for carbon monoxide in high-pressure gases using wavelength modulation spectroscopy. Appl. Opt. 2014, 53, 1938–1946. [Google Scholar] [CrossRef]
- Mathieu, O.; Mulvihill, C.R.; Petersen, E.L. Assessment of modern detailed kinetics mechanisms to predict CO formation from methane combustion using shock-tube laser-absorption measurements. Fuel 2019, 236, 1164–1180. [Google Scholar] [CrossRef]
- Hanson, R.K.; Spearrin, R.M.; Goldenstein, C.S. Spectroscopy and Optical Diagnostics for Gases; Springer: Cham, Switzerland, 2016. [Google Scholar]
- He, D.; Nativel, D.; Herzler, J.; Jeffries, J.B.; Fikri, M.; Schulz, C. Laser-based CO concentration and temperature measurements in high-pressure shock-tube studies of n-heptane partial oxidation. Appl. Phys. B 2020, 126, 142. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, B.; Gong, S.; Wang, L.; Zhang, X.; Liu, G. Experimental and modeling studies of quadricyclane and 2-ethylnorbornane pyrolysis from atmospheric to high pressure. Combust. Flame 2021, 226, 163–181. [Google Scholar] [CrossRef]
- El-Sabor Mohamed, A.A.; Panigrahy, S.; Sahu, A.B.; Bourque, G.; Curran, H.J. An experimental and kinetic modeling study of the auto-ignition of natural gas blends containing C1–C7 alkanes. Proc. Combust. Inst. 2021, 38, 365–373. [Google Scholar] [CrossRef]
- Petersen, E.L.; Kalitan, D.M.; Rickard, M.J. Reflected shock ignition of SiH4/H2/O2/Ar and SiH4/CH4/O2/Ar mixtures. J. Propuls. Power 2004, 20, 665–674. [Google Scholar] [CrossRef]
- Petersen, E.L.; Röhrig, M.; Davidson, D.F.; Hanson, R.K.; Bowman, C.T. High-pressure methane oxidation behind reflected shock waves. In Symposium (International) on Combustion; Elsevier: Amsterdam, The Netherlands, 1996; Volume 26, pp. 799–806. [Google Scholar] [CrossRef]
- Baigmohammadi, M.; Patel, V.; Martinez, S.; Panigrahy, S.; Ramalingam, A.; Burke, U.; Somers, K.P.; Heufer, K.A.; Pekalski, A.; Curran, H.J. A comprehensive experimental and simulation study of ignition delay time characteristics of single fuel C1–C2 hydrocarbons over a wide range of temperatures, pressures, equivalence ratios, and dilutions. Energ. Fuel 2020, 34, 3755–3771. [Google Scholar] [CrossRef]
- Krejci, M.C.; Mathieu, O.; Vissotski, A.J.; Ravi, S.; Sikes, T.G.; Petersen, E.L.; Kérmonès, A.; Metcalfe, W.; Curran, H.J. Laminar flame speed and ignition delay time data for the kinetic modeling of hydrogen and syngas fuel blends. J. Eng. Gas Turbines Power 2013, 135, 021503. [Google Scholar] [CrossRef]
- Glassman, I. Combustion, 2nd ed.; Academic Press: Cambridge, MA, USA, 1987. [Google Scholar]
- Petersen, E.L. A Shock Tube and Diagnostics for Chemistry Measurements at Elevated Pressures with Application to Methane Ignition; Stanford University: Stanford, CA, USA, 1998. [Google Scholar]
- He, Y.; Wang, Y.; Grégoire, C.; Niedzielska, U.; Mével, R.; Shepherd, J.E. Ignition characteristics of dual-fuel methane-n-hexane-oxygen-diluent mixtures in a rapid compression machine and a shock tube. Fuel 2019, 249, 379–391. [Google Scholar] [CrossRef]
- Grégoire, C.M.; Cooper, S.P.; Petersen, E.L. Chemical kinetics investigation of toluene combustion in a shock tube using spectroscopic CO and H2O laser absorption. Fuel 2023, 332, 126234. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mixture | Fuel Blend | XNBD | XH2 | XCH4 | XO2 | XAr | ϕ |
|---|---|---|---|---|---|---|---|
| 1 | 100% H2 | - | 0.029993 | - | 0.014997 | 0.955010 | 1.00 |
| 2 | 100% CH4 | - | - | 0.001687 | 0.003418 | 0.994895 | 0.99 |
| 3 | 98/2% H2/NBD | 0.000529 | 0.026457 | - | 0.018005 | 0.955009 | 1.00 |
| 4 | 99/1% CH4/NBD | 0.000016 | - | 0.001625 | 0.003366 | 0.994993 | 1.01 |
| 5 | 98/2% CH4/NBD | 0.000031 | - | 0.001567 | 0.003408 | 0.994994 | 1.00 |
| Mixture | Temperature (K) | Pressure (atm) | Diagnostics |
|---|---|---|---|
| 1 | 1014–1459 | 0.93–1.87 | OH* |
| 1080–1140 | 6.95–7.51 | ||
| 2 | 1846–2316 | 0.90–1.05 | CO and OH* |
| 3 | 1169–1404 | 0.96–1.24 | OH* |
| 1151–1305 | 6.32–7.08 | ||
| 4 | 1838–2174 | 0.95–1.05 | CO and OH* |
| 5 | 1778–2091 | 0.97–1.08 | CO and OH* |
| Mixture | A | Ea (kcal/mol) |
|---|---|---|
| 1 | 0.56 | 15.2 |
| 2 | 5.23 10−4 | 53.6 |
| 3 | 4.87 10−5 | 41.5 |
| 4 | 6.51 10−4 | 51.7 |
| 5 | 2.14 10−3 | 46.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandberg, M.G.; Grégoire, C.M.; Mohr, D.J.; Mathieu, O.; Petersen, E.L. An Experimental and Detailed Kinetics Modeling Study of Norbornadiene in Hydrogen and Methane Mixtures: Ignition Delay Time and Spectroscopic CO Measurements. Energies 2023, 16, 7278. https://doi.org/10.3390/en16217278
Sandberg MG, Grégoire CM, Mohr DJ, Mathieu O, Petersen EL. An Experimental and Detailed Kinetics Modeling Study of Norbornadiene in Hydrogen and Methane Mixtures: Ignition Delay Time and Spectroscopic CO Measurements. Energies. 2023; 16(21):7278. https://doi.org/10.3390/en16217278
Chicago/Turabian StyleSandberg, Matthew G., Claire M. Grégoire, Darryl J. Mohr, Olivier Mathieu, and Eric L. Petersen. 2023. "An Experimental and Detailed Kinetics Modeling Study of Norbornadiene in Hydrogen and Methane Mixtures: Ignition Delay Time and Spectroscopic CO Measurements" Energies 16, no. 21: 7278. https://doi.org/10.3390/en16217278
APA StyleSandberg, M. G., Grégoire, C. M., Mohr, D. J., Mathieu, O., & Petersen, E. L. (2023). An Experimental and Detailed Kinetics Modeling Study of Norbornadiene in Hydrogen and Methane Mixtures: Ignition Delay Time and Spectroscopic CO Measurements. Energies, 16(21), 7278. https://doi.org/10.3390/en16217278