1. Introduction
Methane trapped in ocean and permafrost gas hydrate deposits is thought to be a fossil fuel reserve on the order of at least all conventional known reservoirs and may turn into a future energy source [
1,
2]. Published estimates of the last ten years vary, depending on the presumptions, between 0.2 × 10
15 m
3 STP (standard temperature pressure) and 120 × 10
15 m
3 STP CH
4 bonded in hydrate-bearing sediments [
3,
4]. Even the most conservative assumptions show an enormous energy potential when compared to conventional gas reserves of 0.15 × 10
15 m
3 STP [
5]. The gas production from hydrate-bearing sediments is, however, still a technical challenge. In order to release gas from hydrate-bearing sediments it is necessary to decompose the gas hydrate. In principle, this can be realized by distortion of the mechanical equilibrium (pressure reduction), the thermal equilibrium (heating) or the chemical equilibrium (e.g., injection of inhibitors or CO
2). The latter, coupling of controlled CH
4 gas production and CO
2 sequestration is often seen as an ideal solution to the demand for fossil fuel and has been proposed as an effective means of counteracting atmospheric CO
2 increase. The replacement of CH
4 in hydrate cages by CO
2 might also maintain the stability of a hydrate field to avoid potential slope slides and subsidence.
In theory, the exchange is simply possible because CO2 is a very good hydrate forming molecule and the resulting CO2 hydrate is stable at even higher temperatures at given pressure conditions compared to pure CH4 hydrate. However, in practice some other aspects come into play which have not been considered in the past.
First, pure CH
4 hydrates are very rare in natural environments [
6]; in fact, almost all natural gas hydrate deposits discovered so far contain minor proportions of higher hydrocarbons or other traces, such as CO
2 or H
2S. They form, depending on guest molecule concentrations, different crystallographic structures. Structure I (sI) gas hydrates are CH
4-rich, but may also include small fractions of CO
2 and H
2S. These hydrates predominantly originate from microbial CH
4 production and are the most abundant hydrate structure on earth [
7]. They have been reported for instance for many deposits in the Hydrate Ridge offshore Oregon or in the Blake Ridge [
8,
9]. Structure II (sII) and structure H (sH) hydrates form primarily from gas molecules of thermogenic origin and contain compounds from CH
4 up to butane and up to hexane for sII and sH, respectively. They have been found in the northern Gulf of Mexico and the northern Cascadia Margin [
10,
11].
Second, industrially emitted
pure CO
2 does not exist. The three methods prior to CO
2 capture are oxyfuel-, pre- and post-combustion, whereby only the latter is considered a well-developed technology [
12]. To separate and capture CO
2 from the gas stream a range of methods currently exist based on physical and chemical processes, including absorption, adsorption, membranes and cryogenics [
13]. Industrially emitted CO
2 includes typically further contaminants such as SO
2 and NO
2. SO
2 impurities are largely removed by gas scrubbing with alkaline solid or solution sorbents. NO
2 is mostly removed using catalytic reduction with NH
3, producing N
2 and H
2O. For a realistic benefit-cost ratio the cleaning process is barely complete and the resulting CO
2 still contains traces of impurities [
14].
Third, the replacement of CH
4 with CO
2 in the hydrate phase under lab conditions has been investigated by several groups. Depending on experimental conditions and setups, the reported exchange of CH
4 by means of CO
2 replacement in the hydrate phase varies widely: between 15% CH
4 within 800 h and 80% after only 15 h [
15,
16].
But the CO2 hydrate durability is only assured as far as the chemical environment does not change which is not the case in the presence of a proximate CH4 gas source. The compositional dis-equilibrium between hydrate phase and environment caused by the high CH4 concentration in the surrounding fluid—and with this a difference regarding the chemical potential in both phases—might induce a decomposition of the CO2 hydrate.
The follow-up scenario—the possible CO
2 hydrate dissolution and renewed CH
4 hydrate formation from the original source—has been investigated recently by Schicks
et al. [
17], demonstrating that conversion of CH
4 hydrate into CO
2 hydrate is reversible, when the CO
2 hydrate is exposed to a hydrocarbon atmosphere.
2. Results and Discussion
To converge on a real production scenario by means of CO2 sequestration into CH4 hydrate reservoirs, not only pure CH4 and CO2 have been used for this study. Mixed hydrates, formed from a CH4-C2H6 mixture (7% C2H6) and impure CO2 (containing 1% SO2) have been tested for the exchange process in both directions.
C
2H
6 is a common component of natural gas hydrates and causes, in concentrations used here, the formation of a sII hydrate [
18]. This study aims to examine whether structural changes occur during exchange reactions or the hydrate lattice structure stays unaffected by guest molecule exchange. Furthermore, the influence of potential structural changes on exchange efficiency can be determined.
The CO
2-impurities of 1% SO
2 shall be deemed to be industrially emitted CO
2. Previous experiments have shown that SO
2 is a good hydrate former and that mixed CO
2-SO
2 hydrates exceed the thermodynamic stability of a pure CO
2 hydrate in terms of
p-T stability [
19,
20]. Therefore, the influence of SO
2 on the exchange reactions has been studied too.
By using this variety of gas- and gas hydrate compositions, our work furthermore provides an insight into driving forces for guest molecule exchange.
2.1. Experiments
As shown in
Figure 1, the overall experimental procedure consists of three parts, where the first part comprises the hydrate formation. This part of the experiment, which is described in the Experimental section, usually takes two days. As it has no relevance for this study of exchange behavior in gas hydrates it is not considered in the following descriptions and diagrams. In all experiments the duration of the primary exchange has been scheduled for four days, the return exchange extends to about twice the time span. This experimental time scale has been chosen in order to approximate on real conditions where natural gas production from a gas hydrate deposit is a short event compared to the subsequent storage of the newly formed CO
2-rich hydrate in an environment which is potentially enriched in CH
4 dominated natural gas.
For this study we use the term “exchange” to describe the substitution/replacement of guest molecules in the hydrate phase with gas molecules of the adjacent gas phase and vice versa. It does not necessarily mean the replacement of gas molecules in one and the same hydrate cavity or, in other words, we assume that hydrate cavities have to be destroyed at least in parts to release gas molecules and to encase new guests. We also use the term “conversion” which here implies the general alteration of the hydrates with respect to their composition and structure. It comprises both the change from single hydrates to mixed hydrate as well as the change of the hydrate structure from sI to sII and vice versa. In similar context the terms “exchange or conversion reaction/process” are used. We also differentiate between hydrate-forming gas and exchange gas whereby hydrate forming gas is used during hydrate preparation and during return-conversion and exchange gas during the first conversion reaction. The term “reaction” should not suggest a chemical process but a response to a stimulus.
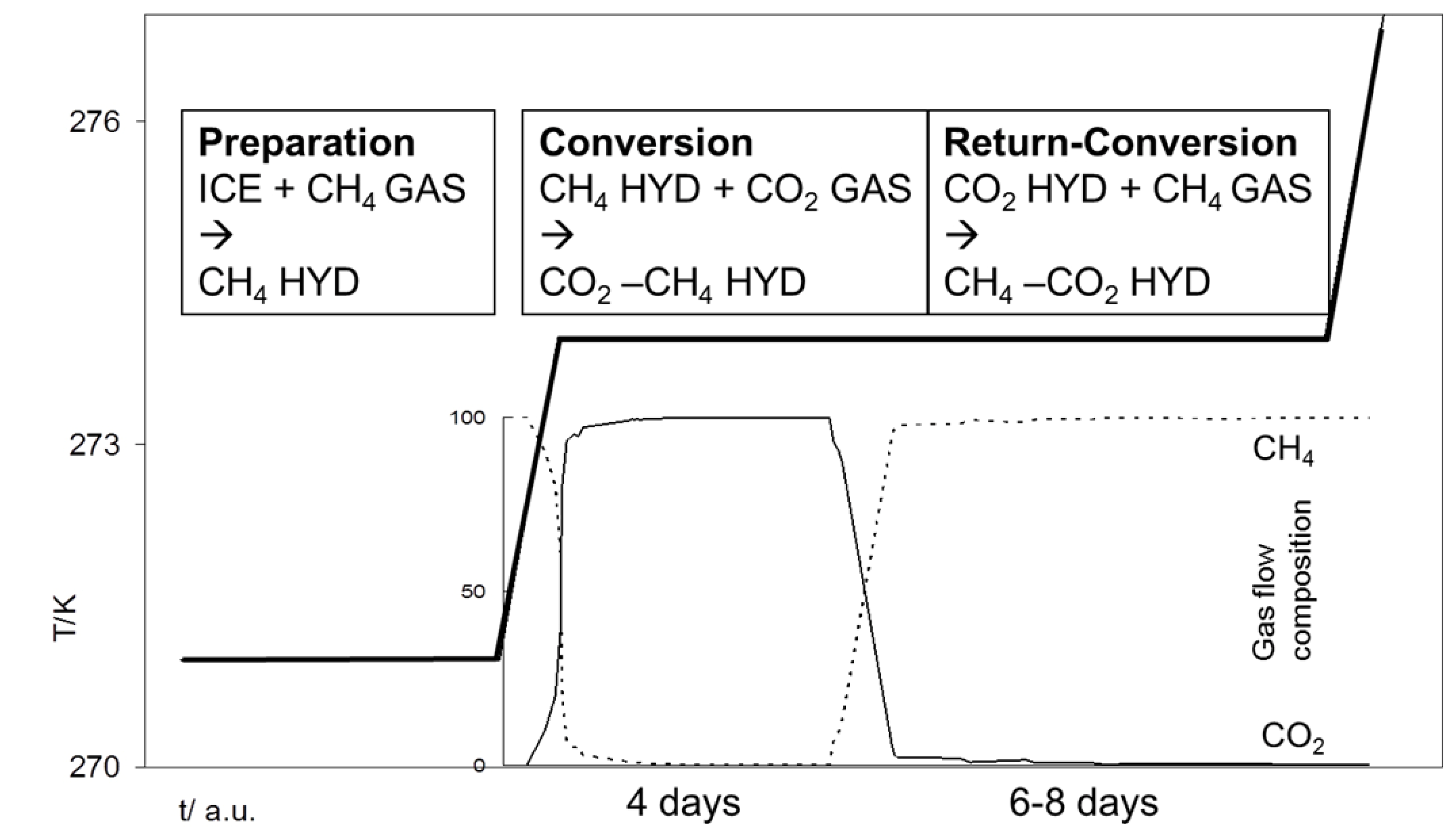
Figure 1.
Schematic illustration of the experimental process, subdivided into preparation, conversion and return-conversion. Temperatures versus time as well as gas composition (in %) are exemplarily shown for the CH4-CO2 exchange.
Figure 1.
Schematic illustration of the experimental process, subdivided into preparation, conversion and return-conversion. Temperatures versus time as well as gas composition (in %) are exemplarily shown for the CH4-CO2 exchange.
2.1.1. The CH4-CO2 Exchange
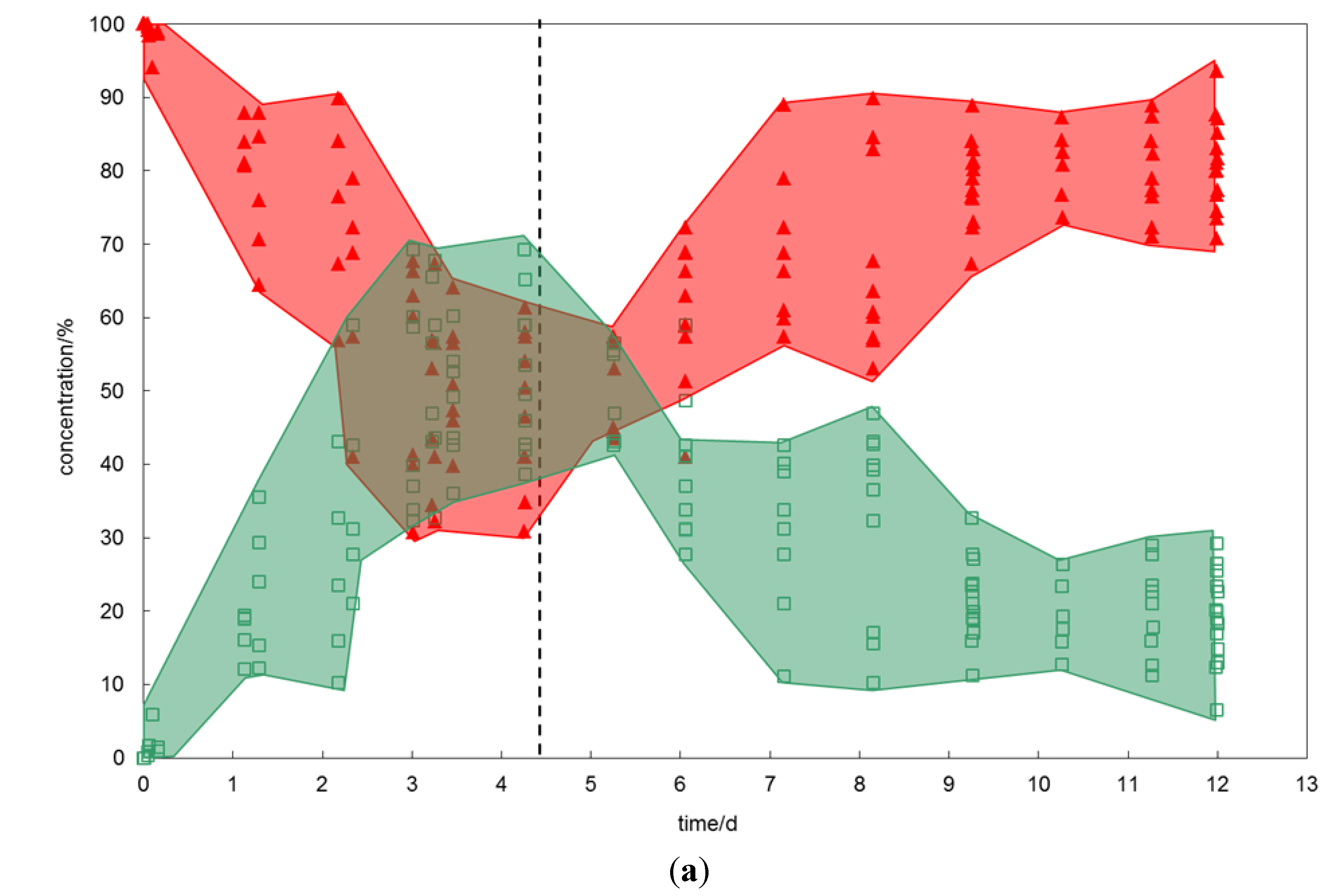
In the reference experiment pure substances have been used (
Figure 2a). The experiment started with the preparation of a pure sI CH
4 hydrate. The exchange process has been induced by offering pure CO
2 gas. For that, the gas flow has been changed from CH
4 to CO
2. After about four days of exposing the hydrate phase to CO
2 gas, the reaction ceased and results in a mixed CH
4-CO
2 hydrate with relative CO
2 concentrations between 40% and 70% in the hydrate phase.
On the reverse exchange, offering CH4 again to the newly formed mixed CH4-CO2 hydrate, no complete removal of the CO2 has been observed within eight days. In fact, the conversion reactivity slowed down after about five days and exhibited a stationary state with between 10% and 30% CO2 in the mixed CH4-CO2 hydrate within the following days.
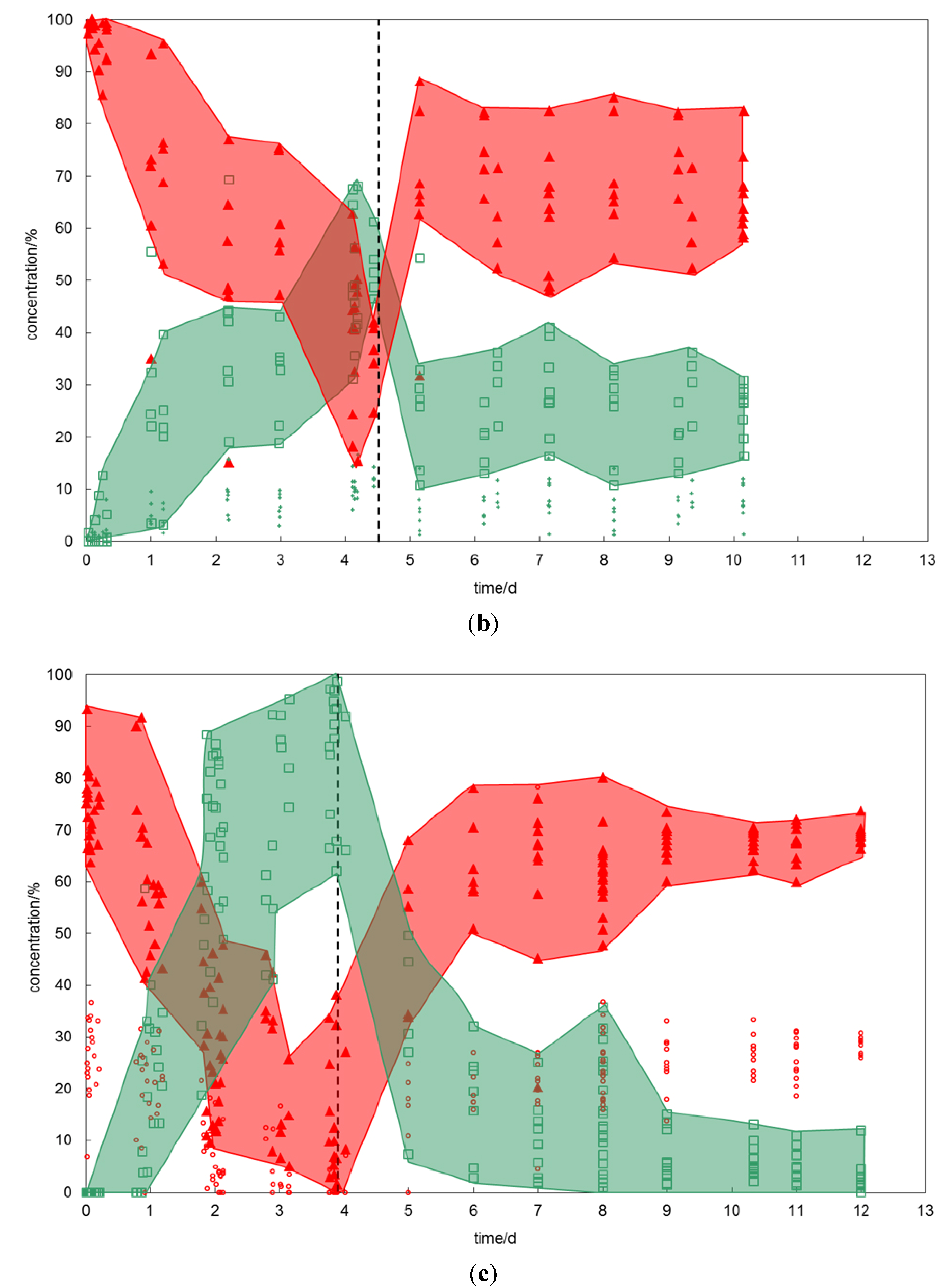
2.1.2. The CH4–CO2-SO2 Exchange
On the second sequence of experiments, simple CH
4 hydrate has been exposed to a CO
2-SO
2 gas mixture (
Figure 2b). After four days, the hydrate composition reached values of up to 15% SO
2 and between 45% and 70% CO
2. The supplier gas stream was then changed back to pure CH
4 and the relative concentrations of the CH
4-CO
2-SO
2 hydrate determined for about six days. Already after one day, the relative concentrations leveled out to values of 12% to 40% CO
2 and 2% to 12% SO
2. The respective CH
4 concentrations varied between 50 and 85%. Because SO
2 is a good hydrate former it became strongly enriched in the hydrate phase. While only one percent was offered by the gas phase, up to 15% have been detected in the hydrate phase. This was a result of the continuous gas flow during the experimental procedure which allowed a permanent supply with feed gas and accordingly enrichment in the hydrate phase.
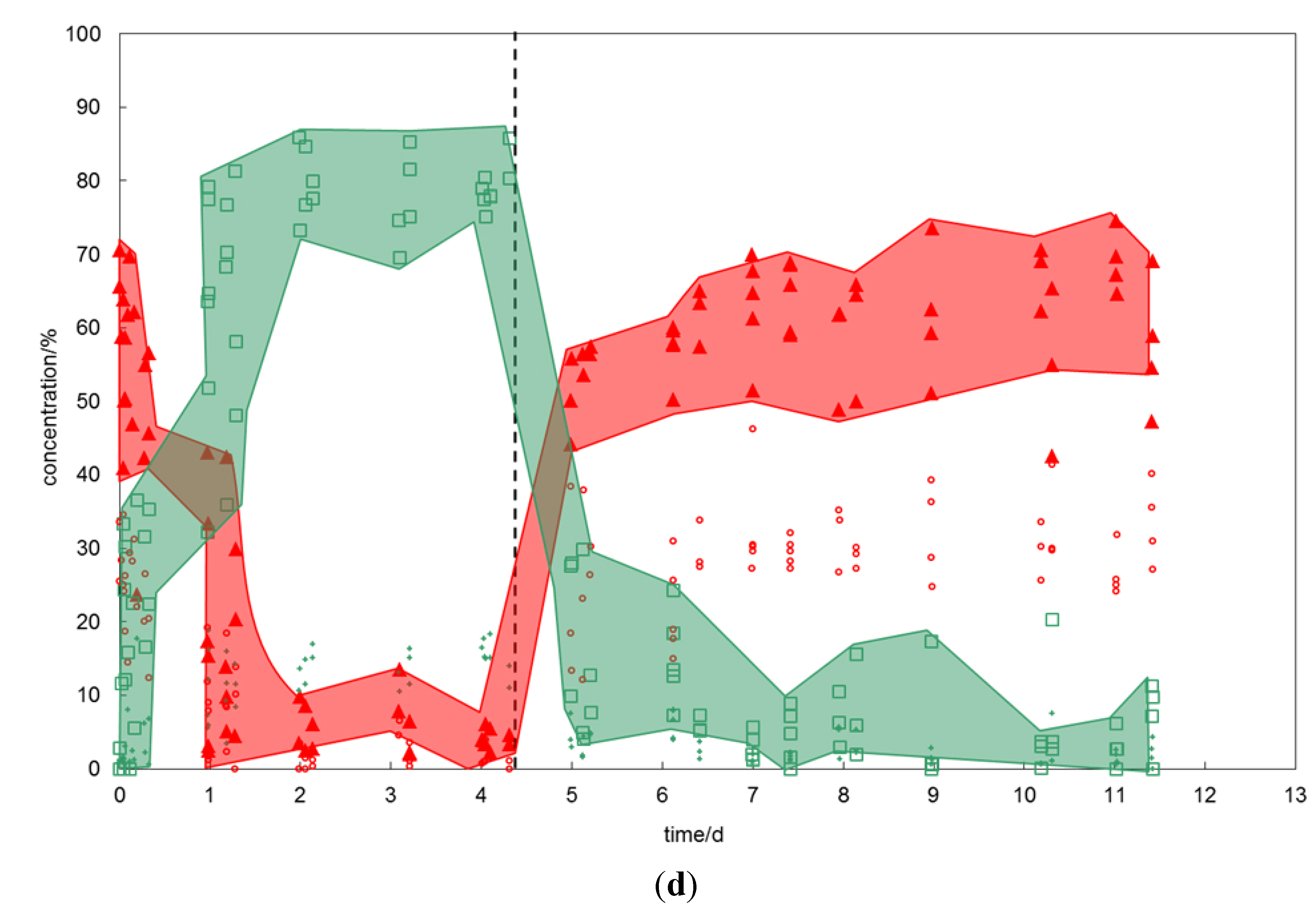
Figure 2.
(a) Changes in hydrate composition (red—CH4, green—CO2) with time when CH4 hydrate is exposed to CO2 and vice versa. The red and green areas emphasize the heterogeneity of the hydrate phase. At the starting point (time zero) the prepared CH4 hydrate was exposed to exchange gas, the dashed line marks the time when the gas composition has been changed back to the initial hydrate forming gas. (b) Changes in hydrate composition (red—CH4, green—CO2, small green diamonds—SO2) with time when CH4 hydrate is exposed to CO2-SO2 gas and vice versa. (c) Changes in hydrate composition (red—CH4, small red circles—C2H6, green—CO2) with time when CH4-C2H6 hydrate is exposed to CO2 gas and vice versa. (d) Changes in hydrate composition (red—CH4, small red circles—C2H6, small green diamonds—CO2, plus—SO2) with time when CH4-C2H6 hydrate is exposed to CO2-SO2 gas and vice versa.
Figure 2.
(a) Changes in hydrate composition (red—CH4, green—CO2) with time when CH4 hydrate is exposed to CO2 and vice versa. The red and green areas emphasize the heterogeneity of the hydrate phase. At the starting point (time zero) the prepared CH4 hydrate was exposed to exchange gas, the dashed line marks the time when the gas composition has been changed back to the initial hydrate forming gas. (b) Changes in hydrate composition (red—CH4, green—CO2, small green diamonds—SO2) with time when CH4 hydrate is exposed to CO2-SO2 gas and vice versa. (c) Changes in hydrate composition (red—CH4, small red circles—C2H6, green—CO2) with time when CH4-C2H6 hydrate is exposed to CO2 gas and vice versa. (d) Changes in hydrate composition (red—CH4, small red circles—C2H6, small green diamonds—CO2, plus—SO2) with time when CH4-C2H6 hydrate is exposed to CO2-SO2 gas and vice versa.
2.1.3. The CH4-C2H6–CO2 Exchange
For this experiment, pure CO
2 gas has been provided to a mixed CH
4-C
2H
6 hydrate (
Figure 2c). The starting composition of the CH
4-C
2H
6 hydrate showed a strong enrichment of C
2H
6 in the hydrate lattice. Although the feed gas has offered only 7% C
2H
6, up to 35% have been incorporated into the hydrate lattice. The initial CH
4-C
2H
6 hydrates were sII hydrates, evident from the peak positions of the Raman bands (see explanation and Raman spectra below).
On offering pure CO2, the magnitude of the exchange reaction exceeded that of previously discussed experiments using simple CH4 hydrate. The newly formed mixed CH4-CO2 hydrates showed a CO2 content of more than 60%. Pure CO2 hydrates were also detected. The concentration of C2H6 decreased to values below detection limit. In the course of the conversion, the initial sII hydrate has been transformed into sI hydrate marked by the change in Raman peak position.
During the reverse exchange, the CO2 concentration in the hydrate decreased rapidly and the hydrate composition reached, already after one day, a level of 60% to 80% CH4, 20% to 30% C2H6 and only 0 to 15% residual CO2. These values remained constant until the end of the experiment. Furthermore, the Raman spectra indicated a reformation to sII hydrate.
2.1.4. The CH4-C2H6–CO2-SO2 Exchange
Here impure variations of gas and hydrate have been combined (
Figure 2d). The results are similar to that of the previous experiment but the effects are even more pronounced. The initial mixed CH
4-C
2H
6 hydrate contained 20% to 35% C
2H
6 and exhibited sII characteristics on the Raman band positions.
On offering CO2-SO2 gas, the molecule exchange occurred rapidly and the final (after four days) hydrate composition showed CH4 and C2H6 concentrations of less than 10%, up to 90% CO2 and up to 20% SO2 in the hydrate structure. The hydrate has converted into sI.
When offering the CH4-C2H6 gas to the newly formed hydrate, the structure changed again into a sII hydrate. The conversion rate was high for the first day but for the remaining six days the system reached a stationary state with a relatively constant hydrate composition comprising 50% to 75% CH4, 25% to 40% C2H6, 0% to 15% CO2 and up to 5% SO2.
In
Figure 2a–d it is noticeable that the compositional data of the exchange reaction are inhomogeneous. If the replacement reaction is considered a surface process, this scattering mirrors different stages of the replacement process, whereby unreacted or less reacted material occurs in the subsurface region [
21]. Despite much effort to apply the similar procedure when focusing on a hydrate crystal, we cannot exclude variations caused by different focus depth, especially given the fact that the hydrate crystal morphology varies highly.
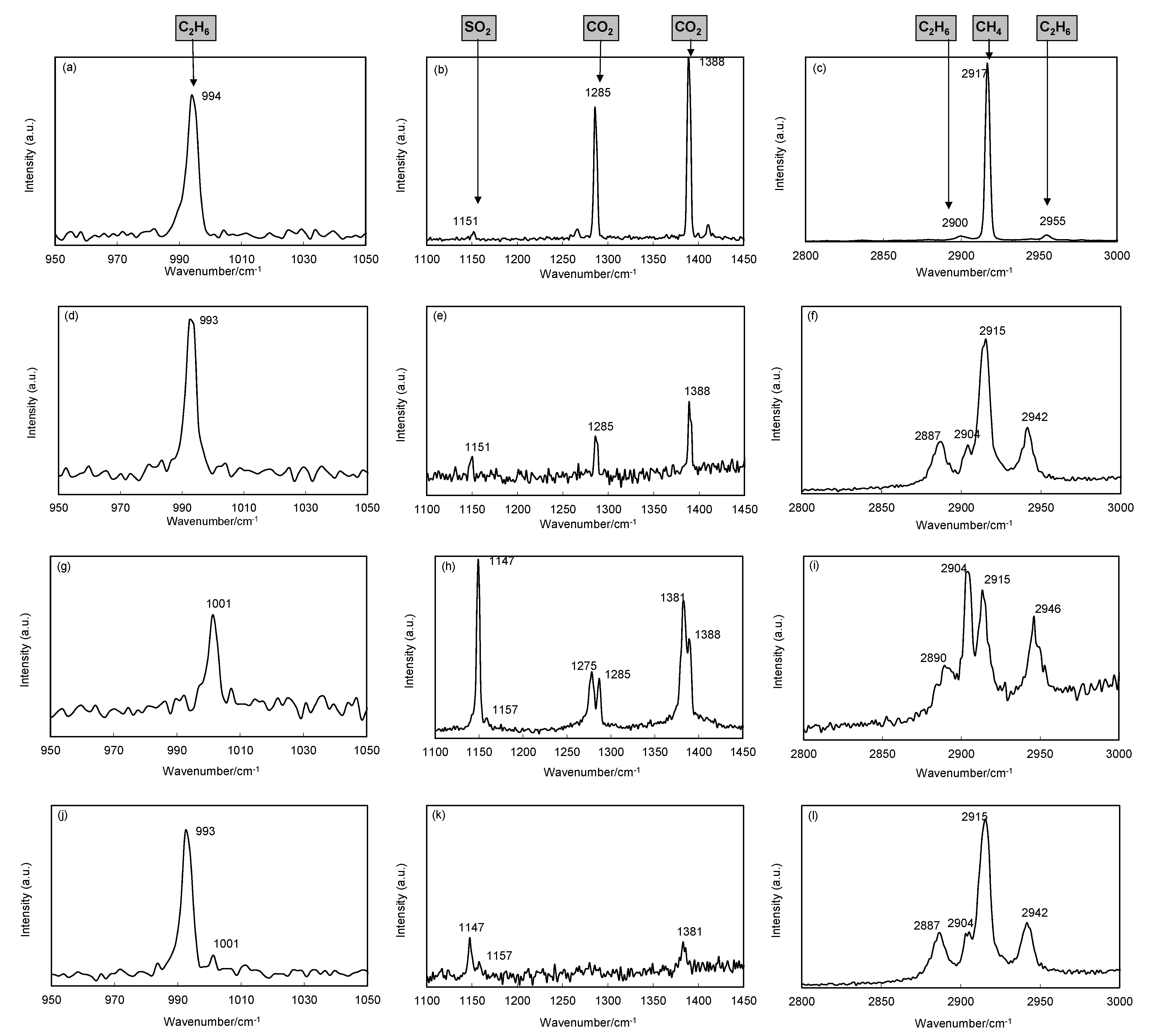
2.2. The Raman Spectra
Exemplarily, typical spectra of the investigated guest molecules at different experimental stages of the CH
4-C
2H
6–CO
2-SO
2 exchange are summarized in
Table 1 and shown in
Figure 3.
Table 1.
Summary of Raman shifts for molecules measured in this study.
Table 1.
Summary of Raman shifts for molecules measured in this study.
| Molecule | Vibrational Mode | Raman Peak Positions (cm−1) |
|---|
| gas | sI hydrate | sII hydrate |
|---|
| 512 | 51262 | 512 | 51264 |
|---|
| Methane | υ1 | C-H sym. stretch | 2917 | 2915 | 2904 | 2915 | 2904 |
| Ethane | υ3 | C-C stretch | 994 | | 1001 | | 993 |
| υ1 | CH3 sym. stretch | 2900 | | 2890 | | 2887 |
| 2υ11 | CH3 d-stretch | 2955 | | 2946 | | 2942 |
| Carbon dioxide | υ1 | sym. stretch (O–C–O) | 1285 | | 1275 | | 1275 |
| 2υ2 | bending overtone | 1388 | | 1381 | | 1381 |
| Sulphur dioxide | υ1 | S-O sym. stretch | 1151 | 1157 | 1147 | | 1147 |
For completeness, the typical Raman spectra of the investigated molecules in the gaseous state are shown
Figure 3a–c. The Raman signal of C
2H
6 can be determined at 994 cm
−1, 2900 cm
−1 and 2955 cm
−1 and the CH
4 peak position has been detected at 2917 cm
−1. For CO
2 the two Fermi diads occur at 1285 cm
−1 and 1388 cm
−1, the SO
2 Raman band appears at 1151 cm
−1. At the beginning of the exchange experiment (
Figure 3d–f; 4 min after offering the exchange gas CO
2-SO
2), the mixed CH
4-C
2H
6 hydrate is predominant and occurs in form of sII hydrate. The structure determination is based on the C
2H
6 vibrational modes at 993 cm
−1, 2887 cm
−1 and 2942 cm
−1 which can be assigned to C
2H
6 encased in the large cavities of sII as well as the position of the CH
4 bands at 2904 cm
−1 and 2915 cm
−1 which can be assigned to CH
4 in the large and small cavities, with a strong dominance of the latter. These observations are in good agreement with Hester
et al. [
22]. At this stage, the Raman bands of SO
2 at 1151 cm
−1 and CO
2 at 1285 cm
−1 and 1388 cm
−1 are small and indicative of gas phase appearance only.
Figure 3.
Typical Raman bands of the exchange reaction of CH
4-C
2H
6 against CO
2-SO
2.
Figure 3a–c shows the typical spectra of investigated molecules in the gas phase.
Figure 3d–f shows the Raman spectra during the starting situation with sII mixed CH
4-C
2H
6 hydrate and CO
2-SO
2 gas just arrived.
Figure 3g–i shows the spectra of the converted sI mixed hydrate after about two days.
Figure 3j–l shows the Raman spectra of the return-conversed hydrate at the end of the exchange experiment.
Figure 3.
Typical Raman bands of the exchange reaction of CH
4-C
2H
6 against CO
2-SO
2.
Figure 3a–c shows the typical spectra of investigated molecules in the gas phase.
Figure 3d–f shows the Raman spectra during the starting situation with sII mixed CH
4-C
2H
6 hydrate and CO
2-SO
2 gas just arrived.
Figure 3g–i shows the spectra of the converted sI mixed hydrate after about two days.
Figure 3j–l shows the Raman spectra of the return-conversed hydrate at the end of the exchange experiment.
Three days later (
Figure 3g–i) the conversion to sI hydrate is evident by the shift in peak positions of C
2H
6 to 1001 cm
−1, 2890 cm
−1 and 2946 cm
−1. The observation is in agreement with the appearance of the CH
4 Raman bands, where the band at 2904 cm
−1, indicative for large cage occupation, exceeds that of small cages at 2915 cm
−1. This is in agreement with findings of Schicks
et al. [
23] and characterizes the typical dominance of large cages in sI structure. The Raman band of gaseous SO
2 has split into two bands at wavenumbers of 1147 cm
−1 and 1157 cm
−1 similar to observations of Beeskow-Strauch
et al. [
19], the CO
2 Raman bands appear with two bands at 1275 cm
−1 and 1381 cm
−1 which both indicate the incorporation of CO
2 into the hydrate lattice. Additionally, two smaller peaks with band positions similar to gaseous CO
2 are present. Due to the fact that Raman analysis is not able to distinguish between CO
2 molecules encased in different types of clathrate cavities [
24], these bands were assigned as gaseous artifacts. Following the change of gas composition back to the initial hydrate forming gas mixture of CH
4-C
2H
6, a renewed conversion of the hydrate structure from sI to sII is observable by the return of band positions typically for sII hydrates (
Figure 3j–l). Contrary to the initially recorded sII Raman signature, relicts of the former sI hydrate structure remain, shown by Raman bands of ethane indicative for sI.
It is not possible to quantify the degree of structural conversion and return-conversion. Based on Raman band intensities and the disappearance of sII Raman bands of ethane, we conclude a complete conversion from sII into sI. This is in good agreement with studies based on PXRD measurements on the conversion of CH
4-C
2H
6 hydrates to CO
2-rich mixed hydrate, which also indicate a complete conversion of sII into sI [
17]. The Raman spectra show as well, that the reverse conversion from sI to sII, remains incomplete, which suggests the coexistence of sI, and sII hydrate in the final hydrate.
For all Raman measurements the O-H stretching has been detected in the range from 3000 cm−1 to 3800 cm−1. Overall, the O-H Raman feature did not change in peak shape or size to suggest the presence of a liquid water phase. However, the structural changes in the course of guest molecule exchange and hence hydrate conversion from sI to sII and vice versa induced a dissociation and reformation of hydrates which likely caused a temporary and spatially limited appearance of free water molecules.
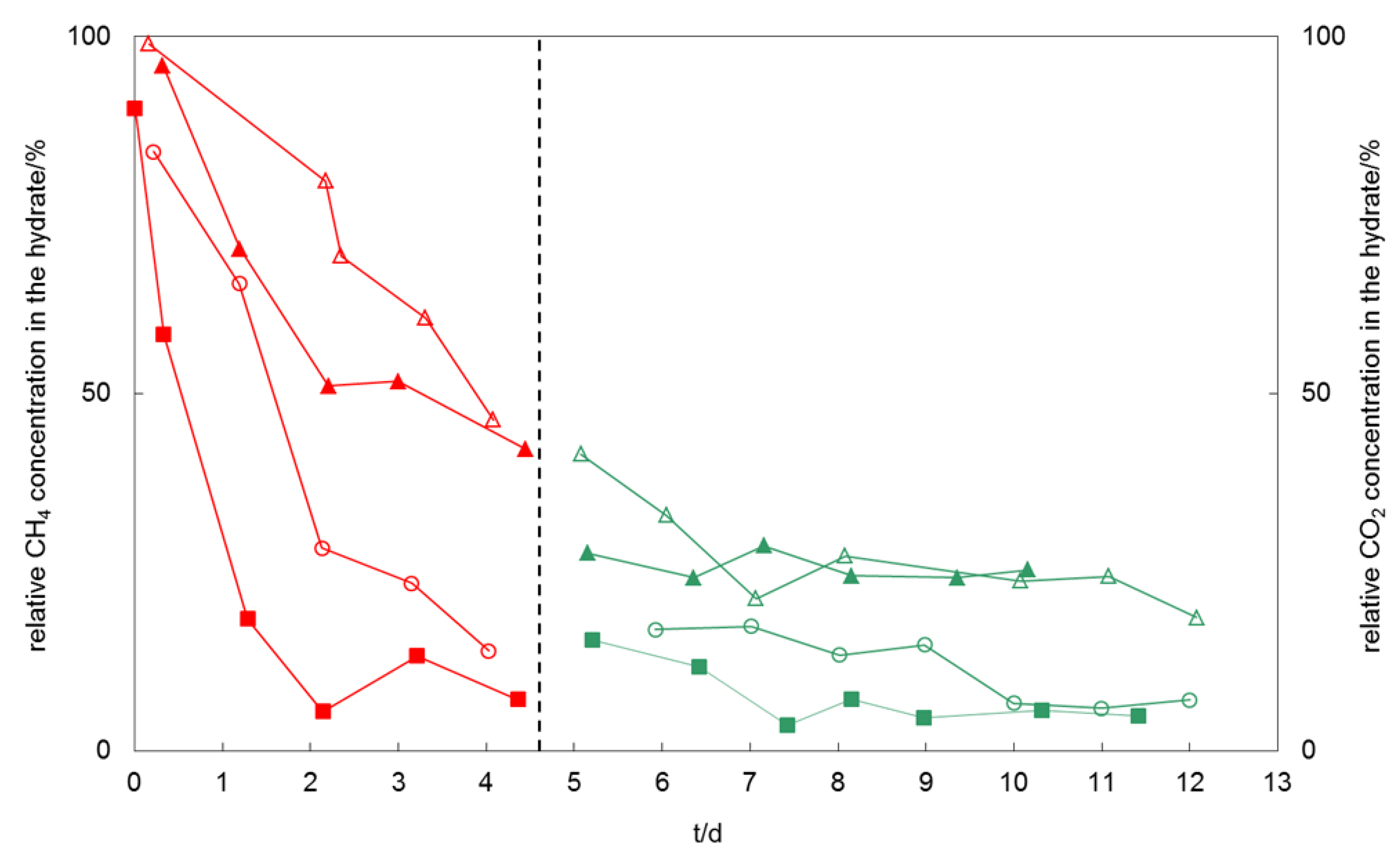
2.3. Discussion of Results
Toward a scenario of CH
4 production and CO
2 storage in gas hydrates we visualized the effectiveness and rate of molecule exchange depending on impurities by plotting the mean values of the relative concentration of CH
4 and CO
2versus time (
Figure 4). The data show that impurities in gas or hydrate do increase CH
4 recovery rates and effectiveness but also decrease the ability to retain CO
2 in the hydrate lattice when it comes to a renewed exchange. In particular the addition of C
2H
6 causes an increase in exchange effectiveness. While after four days of pure CH
4-CO
2 exchange about 50% CH
4 remain in the hydrate lattice, less than 10% of CH
4-remnants have been detected in the presence of C
2H
6 impurities. The CO
2 retention is highest for the pure CH
4-CO
2 exchange and decreases with impurity content whereby C
2H
6 impurities result in the strongest CO
2 release. SO
2 impurities in CO
2 cause an increase in molecule exchange rates during the initial phase of the exchange reaction, but result in no significant difference to the final hydrate compositions.
The results of our study indicate three influencing factors for the effectiveness and intensity of the exchange reaction.
Figure 4.
Mean values of the relative concentration of CH4 (left part of the diagram) and CO2 (right part) in the hydrate phase versus time. These visualize the effectiveness and rate of molecule exchange with respect to methane release and CO2 retention, respectively. (open triangles: exchange of CH4–CO2; full triangle: exchange of CH4–CO2-SO2; open circles: CH4-C2H6–CO2; full squares: exchange of CH4-C2H6–CO2-SO2). The dashed line indicates the change of gas flow from initially CO2-rich to finally CH4-rich.
Figure 4.
Mean values of the relative concentration of CH4 (left part of the diagram) and CO2 (right part) in the hydrate phase versus time. These visualize the effectiveness and rate of molecule exchange with respect to methane release and CO2 retention, respectively. (open triangles: exchange of CH4–CO2; full triangle: exchange of CH4–CO2-SO2; open circles: CH4-C2H6–CO2; full squares: exchange of CH4-C2H6–CO2-SO2). The dashed line indicates the change of gas flow from initially CO2-rich to finally CH4-rich.
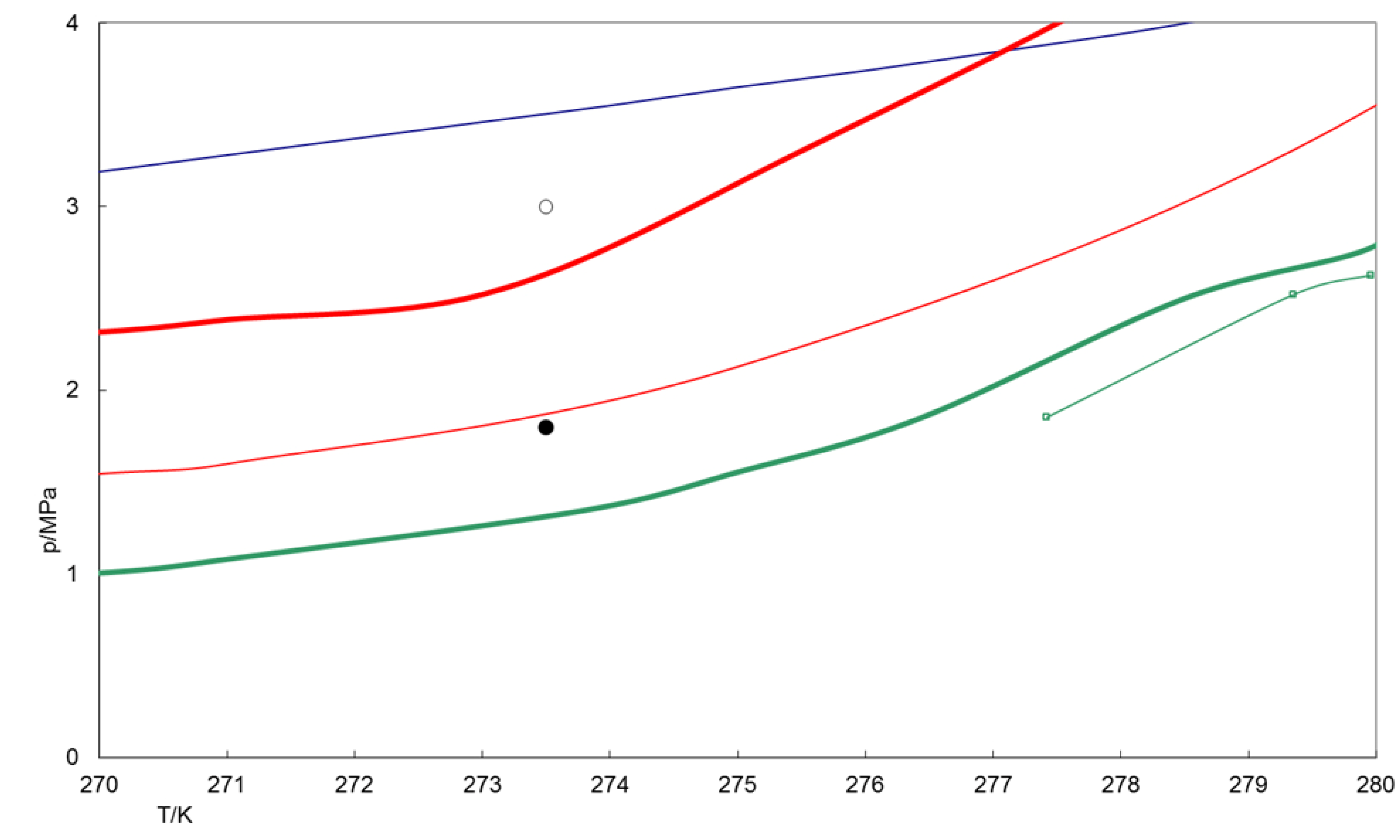
2.3.1. Thermodynamic p-T Stability
By comparison of the
p-T conditions of the phase boundaries, CO
2 hydrate is stable at higher temperatures at given pressure compared to simple CH
4 hydrate [
25]. Accordingly, the phase boundaries of mixed CO
2-CH
4 hydrates fall in between the two phase boundary lines for simple CO
2 and CH
4 hydrates (
Figure 5).
Impurities of 1% SO2 in the gas phase increase the p-T stability field of the resulting CO2-rich mixed hydrate, compared to simple CO2 hydrate. This may lead to the assumption that, if the aim for thermodynamic stability in terms of p-T conditions is the key driving force for the exchange reaction, the exchange effectiveness will be intensified in presence of SO2. In fact, we observed a comparatively rapid conversion rate at the beginning of the exchange experiment but the overall exchange effectiveness remains unaffected. This suggests that a larger p-T stability area might be relevant for the initiation of the exchange reaction.
The mixed CH4-C2H6 hydrate is also stable at higher temperatures at a given pressure compared to simple CH4 hydrate. The stability conditions for the chosen CH4-C2H6 hydrate with respect to temperature and pressure are close to that of pure CO2 hydrate. This finding may imply a lower exchange rate. However, the exchange rate of mixed CH4-C2H6 hydrates to CO2-rich hydrate exceeds that of pure CH4 hydrates. This indicates that the driving force is the aim for chemical equilibrium between all phases. The conversion of CH4-C2H6-hydrate to CO2-rich hydrate and vice versa is coupled to structural conversion (sI-sII).
Figure 5.
Stability fields of simple CO
2 hydrates (bold green line), mixed CO
2-SO
2 hydrate (fine green line) formed from CO
2(99%)-SO
2(1%) gas mixture, CH
4 hydrate (bold red line) and CH
4(93%)-C
2H
6(7%) hydrates (fine red line). Also shown are the CO
2 vapor-liquid boundary (blue line) and experimental p-T conditions (dots). Data according CSMHyd [
25], data of CO
2-SO
2 hydrate stability taken from Beeskow-Strauch [
19].
Figure 5.
Stability fields of simple CO
2 hydrates (bold green line), mixed CO
2-SO
2 hydrate (fine green line) formed from CO
2(99%)-SO
2(1%) gas mixture, CH
4 hydrate (bold red line) and CH
4(93%)-C
2H
6(7%) hydrates (fine red line). Also shown are the CO
2 vapor-liquid boundary (blue line) and experimental p-T conditions (dots). Data according CSMHyd [
25], data of CO
2-SO
2 hydrate stability taken from Beeskow-Strauch [
19].
2.3.2. Guest-to-Cavity-Ratio
A unit cell of sI comprises two small cavities (5
12) and six large cavities (5
126
2), whereas a unit cell of sII consists of sixteen small cavities (5
12) and eight large cavities (5
126
4). Although both CH
4 and C
2H
6 are individually sI hydrate formers, the binary system of CH
4-C
2H
6 with a ratio of 93/7 is typically observed to form sII hydrates. In fact, CH
4-C
2H
6 mixtures containing 75% to 99% CH
4 form sII hydrates. SII is the preferred structure due to the relative stability of the molecules in the hydrate cages (guest-to-cavity-ratio) together with their ratio of large to small cavities in a unit cell of sII hydrates [
18,
26]. Hendriks
et al. [
27] explain it invoking the abundance of small cavities in sII hydrates which are stabilized exclusively by CH
4. Therefore, the competition between CH
4 and C
2H
6 for the occupation of large cavities in the hydrate is reduced compared to sI hydrates.
When CO2 or a mixture of CO2 and SO2 gas are offered to the CH4-C2H6 mixed sII hydrate, a conversion to sI hydrate occurs. The reason is the large size of both these newly offered molecules which makes them preferentially suitable for large cages. Consequently, the aim to maximize the number of large cages enforces a structural conversion of sII to sI during the exchange reaction. During the return-exchange the number of CH4 molecules increases which induces a reverse structural conversion of sI to sII. This conversion, however, remains incomplete resulting in the coexistence of sI and sII structures in the final hydrate. We assume that the ability of CH4 to occupy large cages of sI attenuates the demand to increase the ratio of small to large cages. It shows that an optimal guest-to-cavity-ratio not necessarily equates with complete structural conversion.
Generally, offering new guest molecules to existing hydrates might cause the impulse for a structural rearrangement to optimize guest-to-cavity-ratios. This restructuring process is attended by a strong molecular disorder which results in a large interface between gas and hydrate phase. In the course of an exchange reaction, it supports an intense molecule exchange between hydrate phase and adjacent gas phase.
2.3.3. Chemical Equilibrium
Guest molecule exchange as well as structural conversion between sI and sII occurs in both directions. This basic finding indicates that the compositional disequilibrium, induced by changes of the surrounding gas phase composition, is the key driving force for the exchange process in gas hydrates. The dominant influence of obtaining an equilibrium of the chemical potential in all phases is especially evident when investigating the return-exchange of the simple CO2-CH4 exchange experiment. This reaction can neither be driven due to a “higher stability” of the resulting hydrate phase in terms of p-T conditions, nor does CH4 offer a better guest-to-cavity ratio in large cages. Nevertheless the exchange reaction occurs, with the driving force being the chemical disequilibrium between hydrate and surrounding environment.
3. Experimental Section
The experiments were performed using ready-made certified class 1 gases (gas certification ISO 9001) of pure CO
2, CO
2 + 1% SO
2, pure CH
4 and CH
4 + 7% C
2H
6 supplied by Linde AG. The experiments have been performed by varying gas compositions as shown in
Table 2 together with
p-T conditions of the respective experiments.
Table 2.
List of performed experiments that shows the combination of used gas mixtures and respective p-T conditions.
Table 2.
List of performed experiments that shows the combination of used gas mixtures and respective p-T conditions.
| Experiment | Combination of Gas Mixtures | Experimental Conditions |
|---|
| Preparation: hydrate-forming gas | Conversion: offered exchange gas | Return-conversion: offered exchange gas | p/MPa | T/K |
|---|
| (a) | 100% CH4 | 100% CO2 | 100% CH4 | 3 | 273.5 |
| (b) | 100% CH4 | 99% CO2 + 1% SO2 | 100% CH4 | 3 | 273.5 |
| (c) | 93% CH4+7% C2H6 | 100% CO2 | 93% CH4 + 7% C2H6 | 1.8 | 273.5 |
| (d) | 93% CH4+7% C2H6 | 99% CO2 + 1% SO2 | 93% CH4 + 7% C2H6 | 1.8 | 273.5 |
3.1. Experimental Setup
Hydrate clathrate formation and exchange experiments were performed in a small-scale (0.4 cm3 volume) Hastelloy pressure cell covered with a quartz window. The cell was mounted to an Olympus microscope and attached to a Raman spectroscope (LabRam, Jobin Yvon, 532 DPSS laser, 100 mW). This setup allows both the visual observation of the hydrate behavior together with semi-quantitative analysis (relative concentration of compounds in gas phase and hydrate phase) based on Raman spectra. The non-destructive technique of confocal laser Raman spectroscopy is particularly suited to analyze individual selected phases. Several Raman spectra at different areas of the sample were taken at certain time slots during the experiment.
The pressure cell is designed to operate in continuous gas flow mode via gas in- and outlet ports. The inflow conduit passes through the cell on a long way to cool down the gas to working temperature prior to entering the sample cell. Temperature control is realized by a thermostat and the temperature is determined with a precision of ±0.1 K. A pressure controller regulates the sample pressure with a precision of ±2% rel. The system pressure was measured with a P3MB pressure transducer (Hottinger Baldwin Messtechnik) with a precision of ±0.01% rel. Prior to the experiment start, air was completely flushed out of the cell system by passing the experimental gas through the entire gas flow system.
Laser Raman spectra of mixed systems can be used to determine the relative molar proportions of the components as described in Beeskow-Strauch
et al. [
19] based on the procedures and the cross section factors published by Burke and Schrötter [
28,
29]. The integrated band intensities of the specimen are proportional to the number of molecules present in the irradiated volume of the sample. Therefore, comparison of the relative corrected integrated band intensities of the species permits their relative proportions to be determined [
30].
3.2. Experimental Process
The experimental process has been subdivided into three parts: (1) the production of the initial hydrate sample, (2) the hydrate conversion using exchange gas injection and (3) the reverse-conversion using the preliminary hydrate-forming gas (see
Figure 1,
Table 2).
The initial hydrate sample has been produced using fine grained ice, prepared in a cryo-mill and transferred into the pre-cooled pressure cell (<273 K). SEM investigation of the ice indicates a particle size between 10 µm and 30 µm (
Figure 6). The pressure cell was sealed and exposed to the chosen gas in a continuous gas stream of 1 mL/min. All experiments were run with a pressure of 3 MPa and 1.8 MPa when using pure CH
4 gas and CH
4-C
2H
6 gas, respectively. Subsequent slow heating to about 274.5 K assured the melting of all ice and the ultimately remaining of only hydrate crystals. At this stage, slight cooling (to 273.5 K) of the system enables hydrate growth close to equilibrium conditions. Raman spectra of the growing clathrates were recorded on a regular basis to observe possible changes in clathrate structure or composition. This part of the experiment lasted several hours until no further changes in hydrate composition were recorded and the system reached a stationary state. The
p-T conditions of the experiments were chosen to retain the gaseous state of aggregation of the test gas and to be within the stability fields of the hydrate phases (see
Figure 5).
Figure 6.
SEM picture of the prepared fine grained ice. The surface of the ice particle is covered with frost and includes fissures and cracks indicating a large surface area.
Figure 6.
SEM picture of the prepared fine grained ice. The surface of the ice particle is covered with frost and includes fissures and cracks indicating a large surface area.
(2) The hydrate conversion process has been initiated by changing the gas composition from the hydrate-forming gas to the exchange gas (in the simplest case: changing from CH4 to CO2 gas). For that, the gas inflow route has been isolated from the pressure cell and completely flushed with the exchange gas. The pressure and temperature in the hydrate-containing pressure cell was kept constant. The re-opening of the inflow conduit instantaneously supplies the hydrate sample with the exchange gas and therefore coincides with the start of hydrate conversion. This technique prevents the mixing of hydrate-forming gas and exchange gas in the conduits which would result in a sluggish and undetermined conversion start.
The exchange reaction was running for four days. Regular measurements by laser Raman spectroscopy of gas phase and hydrate phase document the changes in relative guest molecule concentration. Also, structural changes in the hydrate phase could be detected by determining the Raman band position and the rate of cage occupancy. Thus, the Raman measurements also indicate the slow-down of the conversion process when no significant changes in the composition in the newly formed mixed hydrate occurred.
(3) After finishing the first conversion process, the durability of the newly formed CO2-rich mixed hydrate has been tested by offering the initial hydrate-forming gas to this mixed hydrate. For that, the gas inflow route has been isolated again from the pressure cell and completely flushed with the hydrate-forming gas before re-opening of the inflow conduit. In similar manner, the rates of the return-conversion have been documented by laser Raman spectroscopy. The reaction proceeded for six to eight days.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



