
Transport Mechanisms for CO2-CH4 Exchange and Safe CO2 Storage in Hydrate-Bearing Sandstone

Abstract
:1. Introduction

2. Experimental Description
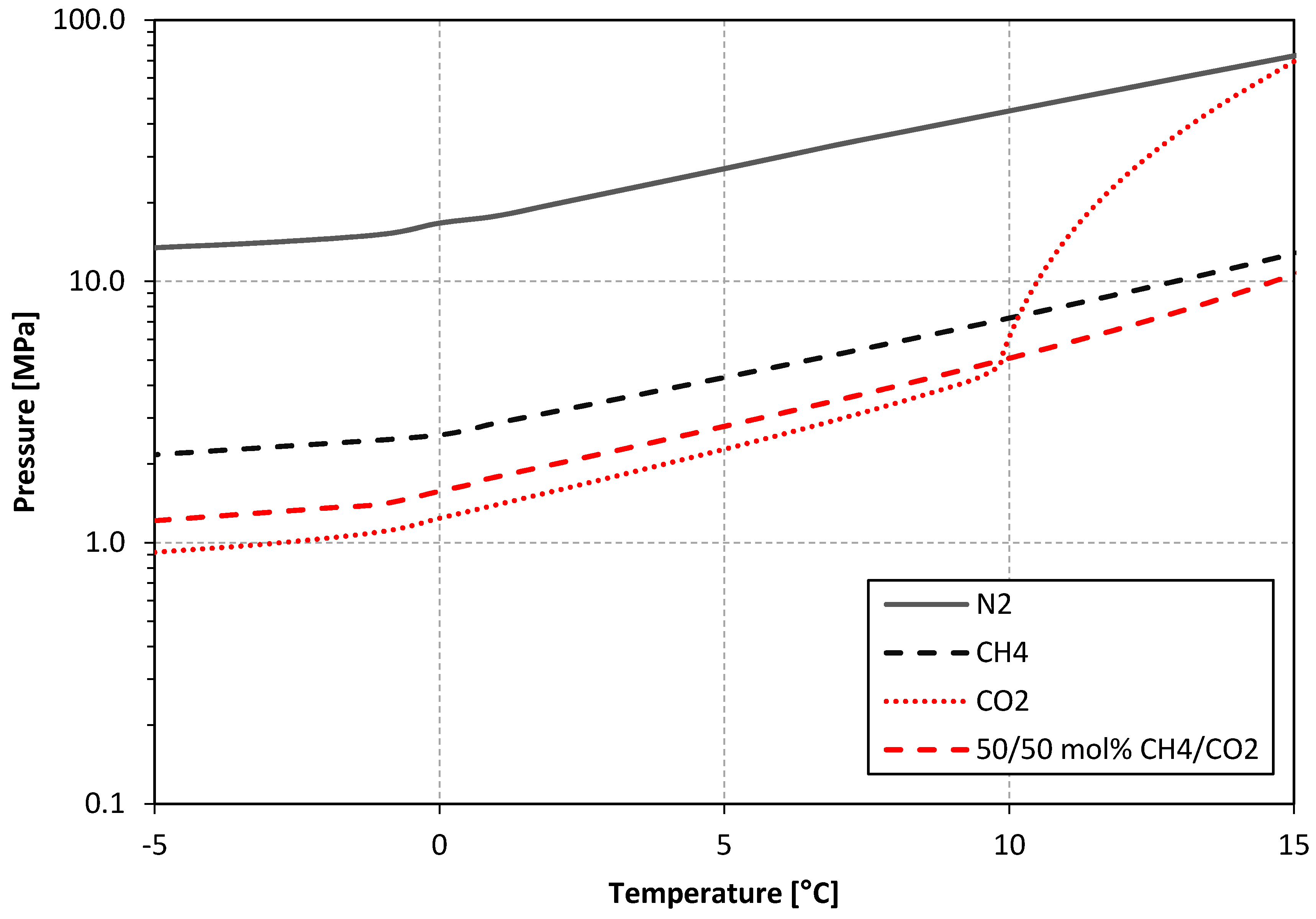
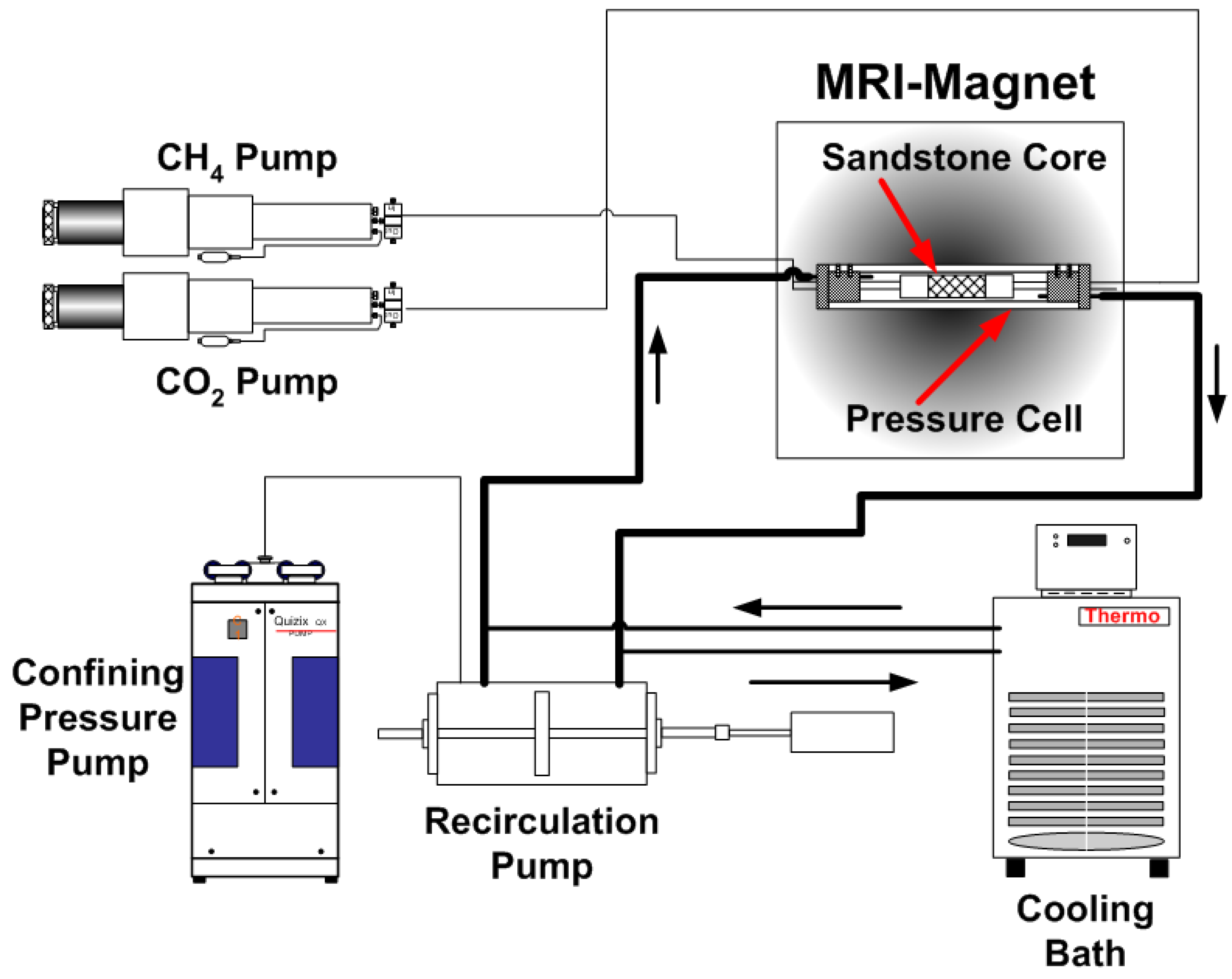
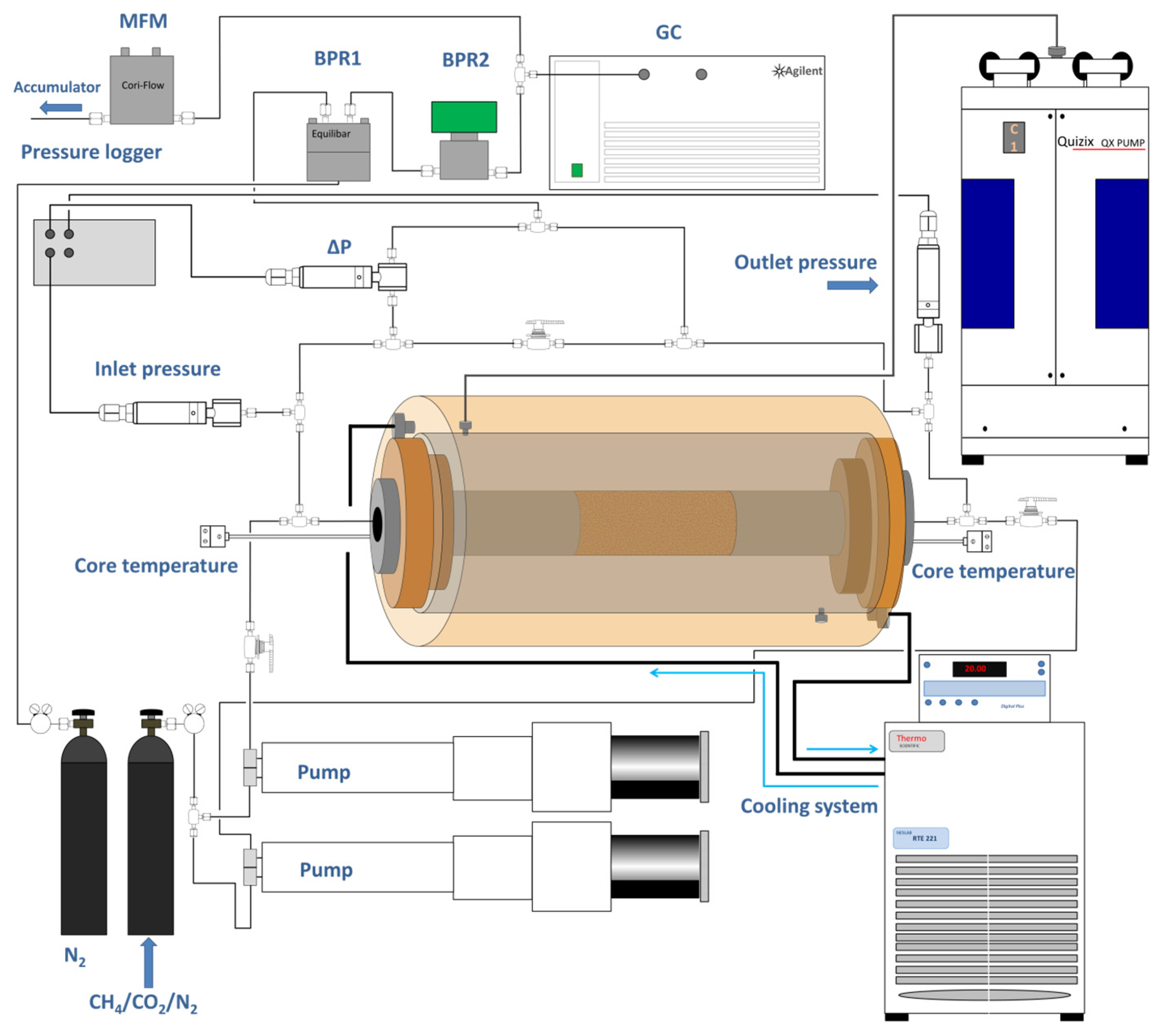
2.1. Experimental Design



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Paper name | NaCl concentration (wt%) | Swi (Frac) | Core length (cm) | Core diam. (cm) | Porosity (Frac) | Temp. (°C) |
|---|---|---|---|---|---|---|
| SS1 | 0.1 | 0.45 | 9.96 | 3.81 | 0.24 | 4.0 |
| DS1 | 3.0 | 0.50 | 10.08 | 3.81 | 0.24 | 4.0 |
| DS2 | 0.1 | 0.50 | 10.08 | 3.81 | 0.24 | 4.0 |
| c1 to c6 | 0.1 | 0.3–0.6 | 10.00 | 3.74 | 0.24 | 4.0 |
| w1 | 3.5 | 0.51 | 10.01 | 3.74 | 0.24 | 4.0 |
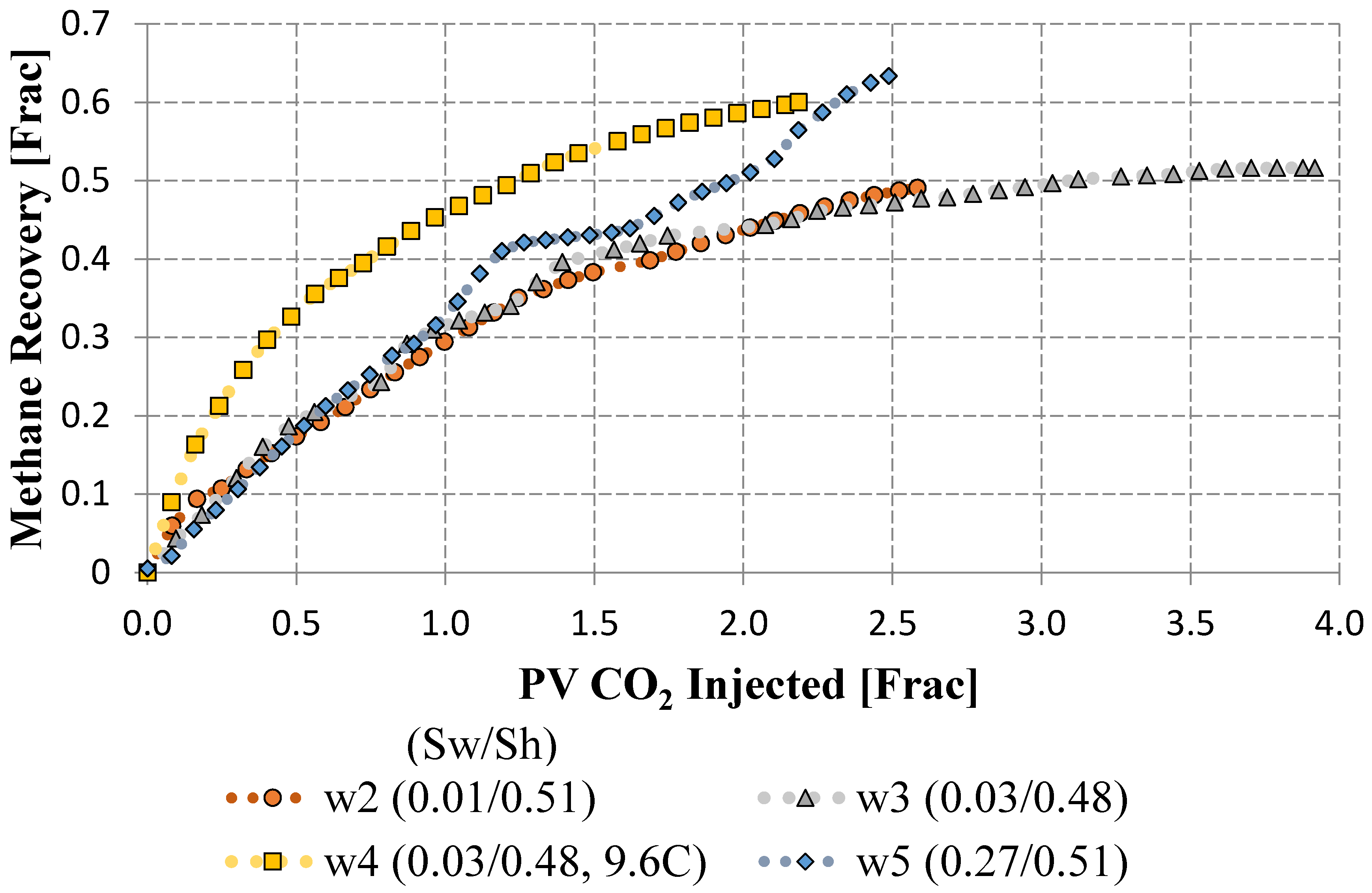
| w2 | 0.1 | 0.41 | 14.55 | 5.06 | 0.25 | 4.0 |
| w3 | 0.1 | 0.41 | 14.52 | 5.06 | 0.24 | 4.0 |
| w4 | 0.1 | 0.43 | 14.14 | 5.06 | 0.24 | 9.6 |
| w5 | 3.5 | 0.67 | 14.54 | 5.06 | 0.24 | 4.0 |
| w6 | 3.5 | 0.64 | 13.67 | 5.05 | 0.23 | 4.0 |

2.2. Experimental Procedure

3. Experimental Results and Discussion
3.1. Diffusion Driven Mass Transport–a Short Review of Previous Experimental Efforts
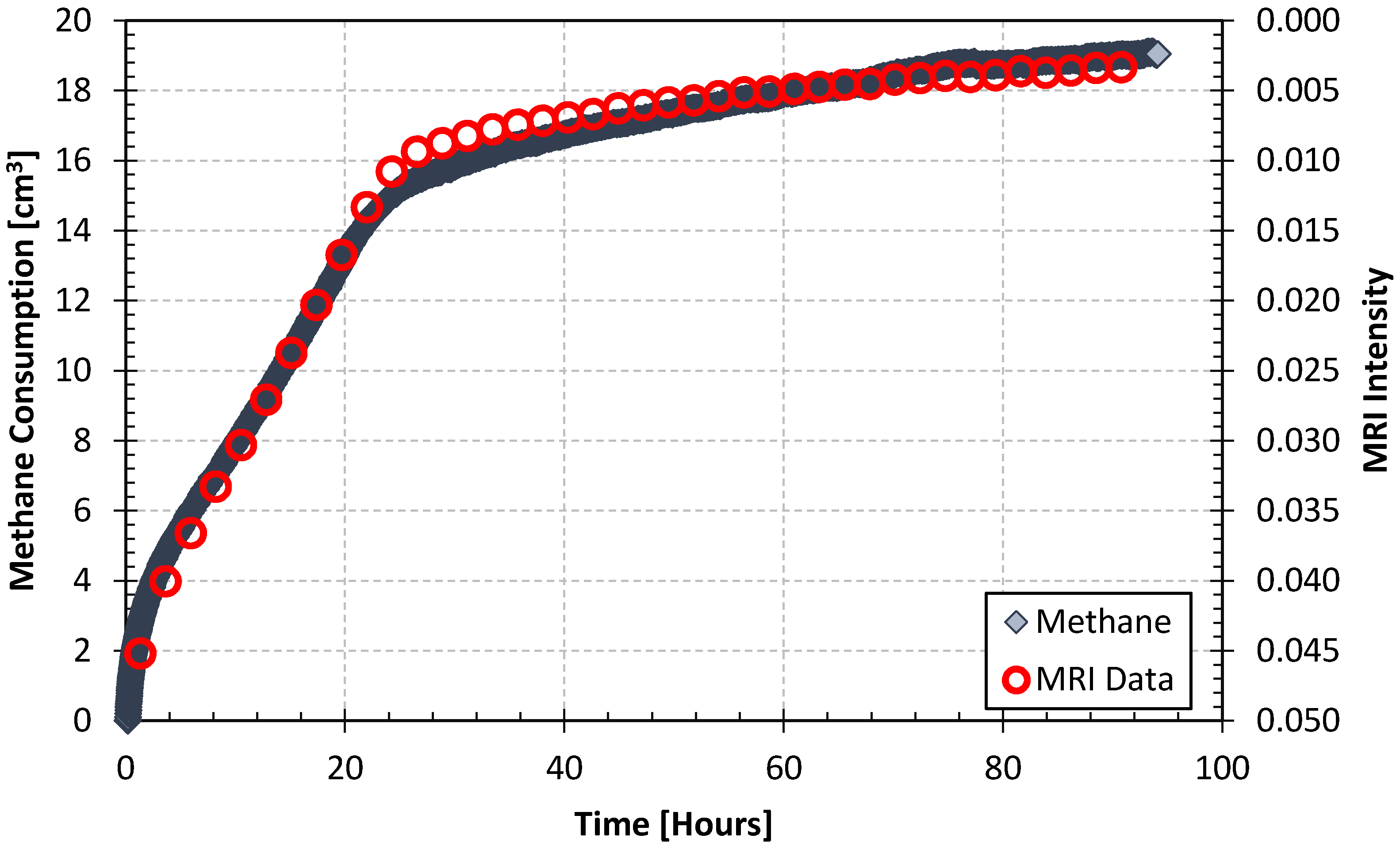
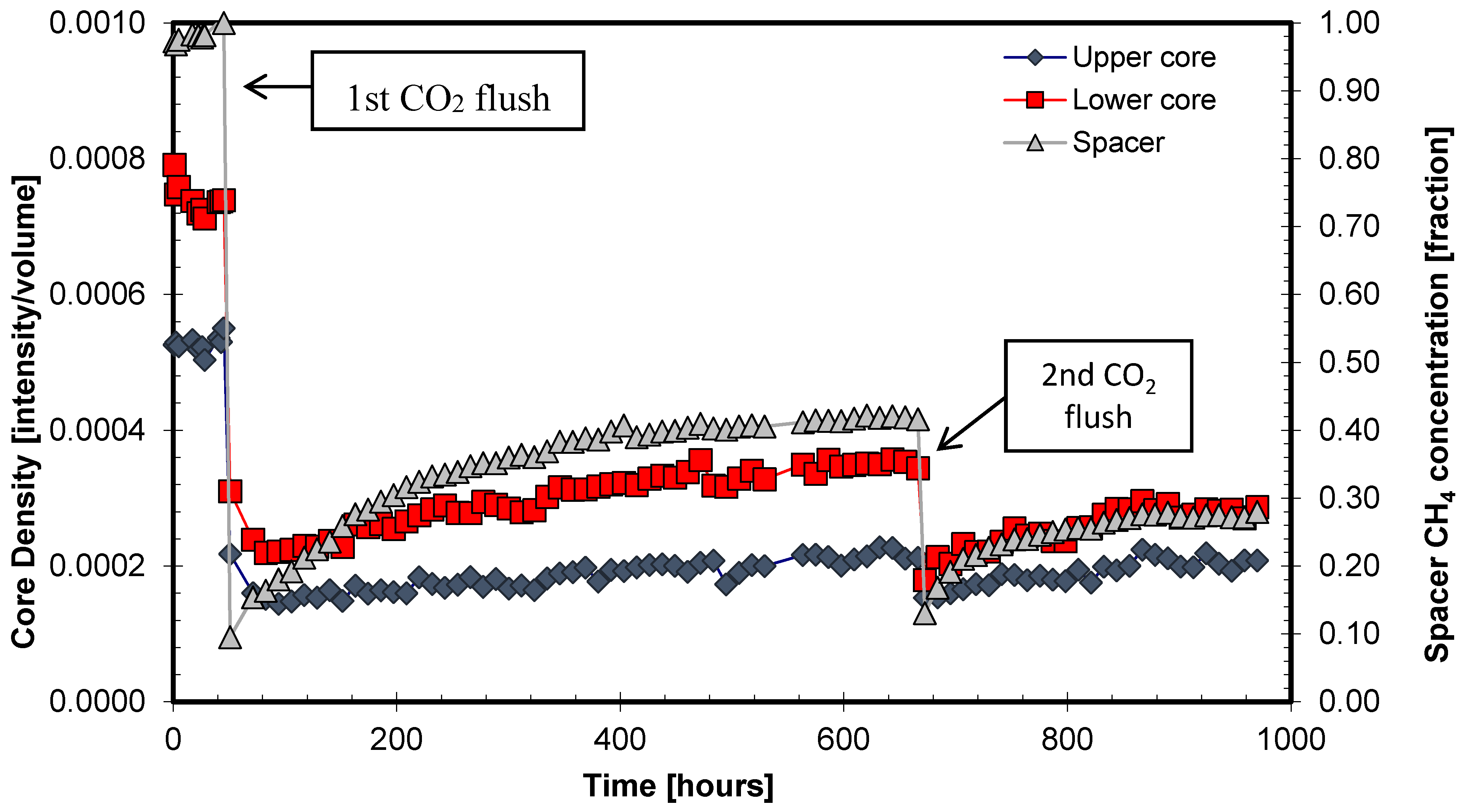
- Immediate production response was observed in the spacer volume after pure CO2 was injected into the spacer volume (injection occurred after 48 h and 670 h and lasted for 5 min before the system was shut in and the CH4 hydrate was soaked in the injected CO2). The production response is reflected by increasing MRI intensity (grey triangle) which indicates increased CH4 concentration in the spacer volume.
- Immediate intensity drops were observed in both core halves after CO2 was flushed through the spacer volume. Initial CO2 transportation is therefore not necessarily a slow process, but decreasing CO2 gas/liquid concentration during exchange may prevent mass transfer to radially more distant core segments. This will eventually affect the recovery unless CO2 is replenished. The majority of the injected CO2 will be channeled through the low resistance conduit (the spacer volume) during the 5 min flush. The advection-driven CO2 mass transport within the low-permeable CH4-hydrate saturated sediment was therefore not assumed to be significant, although some CO2-CH4 displacement was anticipated, especially at the core surfaces facing the open spacer volume. The fluid flow regime was strictly diffusion-driven after the shut-in.
- The monotonically increasing intensity trend in the core halves indicates exchange and release of CH4 with subsequent reduced exchange driving force. This is reflected in the spacer intensity curve shape, where the derivative of the curve trajectory approaches 0.

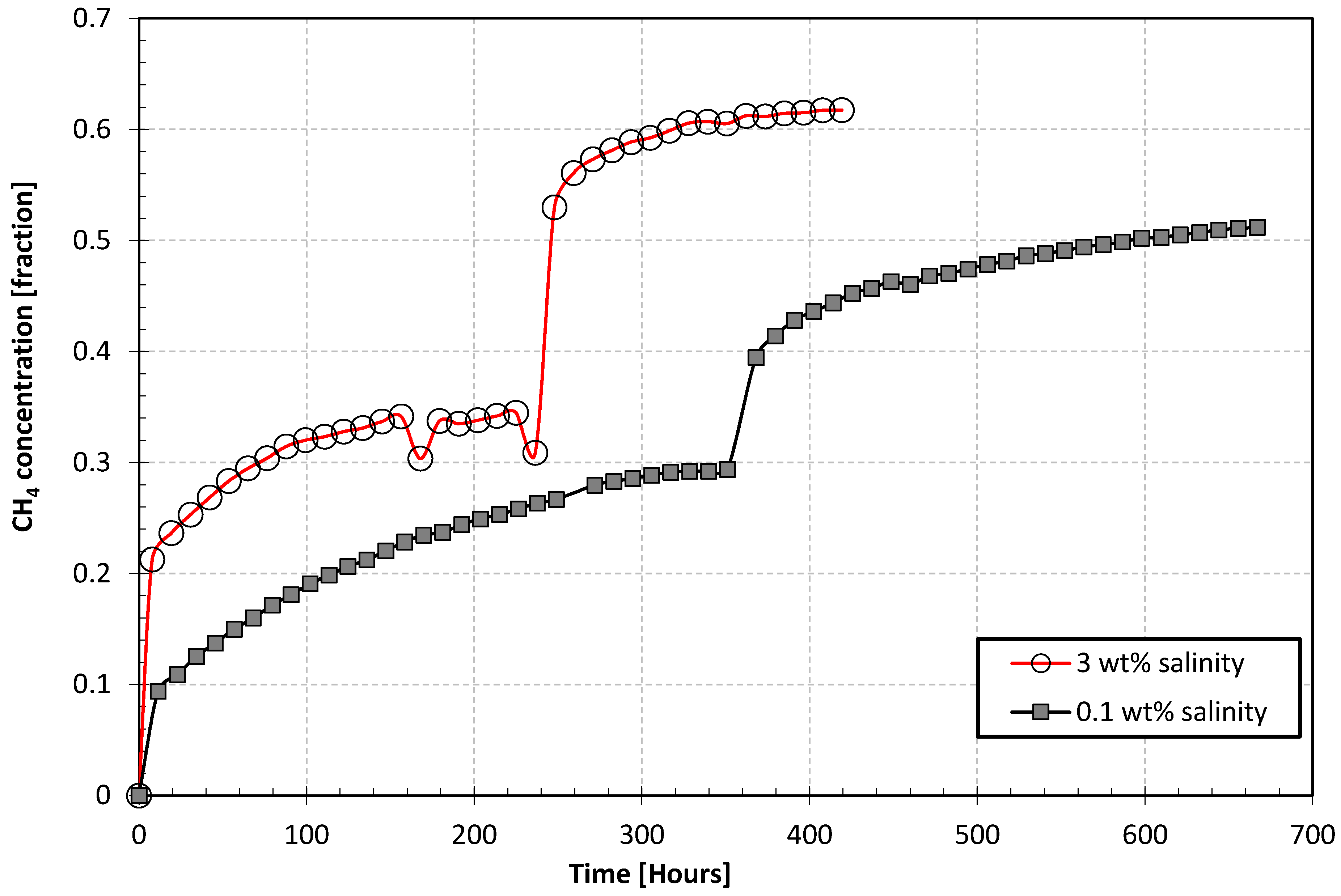
3.2. Salt Effects during Exchange


3.3. Maximizing Conversion Efficiency through Continuous CO2 Flow
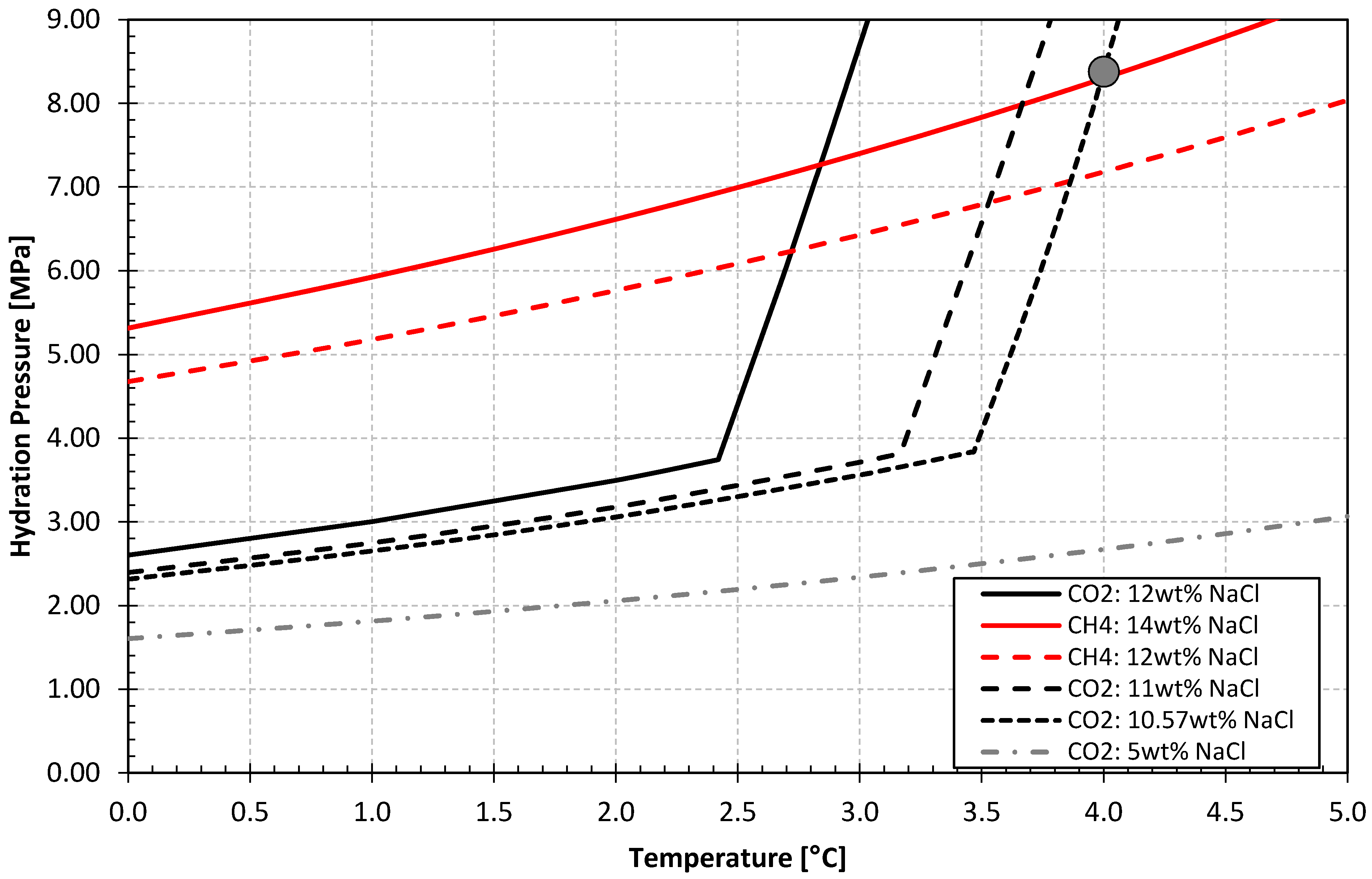
- Hydrate formation at 4 °C and 8.38 MPa.
- Injection of CO2 at 10 cm3/min to displace excess CH4 in the spacer volume after hydrate formation. MRI profiles (sagittal images of the spacer volume) were acquired during injection to confirm displacement.
- CO2 was injected at 0.033 cm3/min for several days (2–5) during the exchange process itself.
- Depressurization sequence to determine mixed hydrate composition.
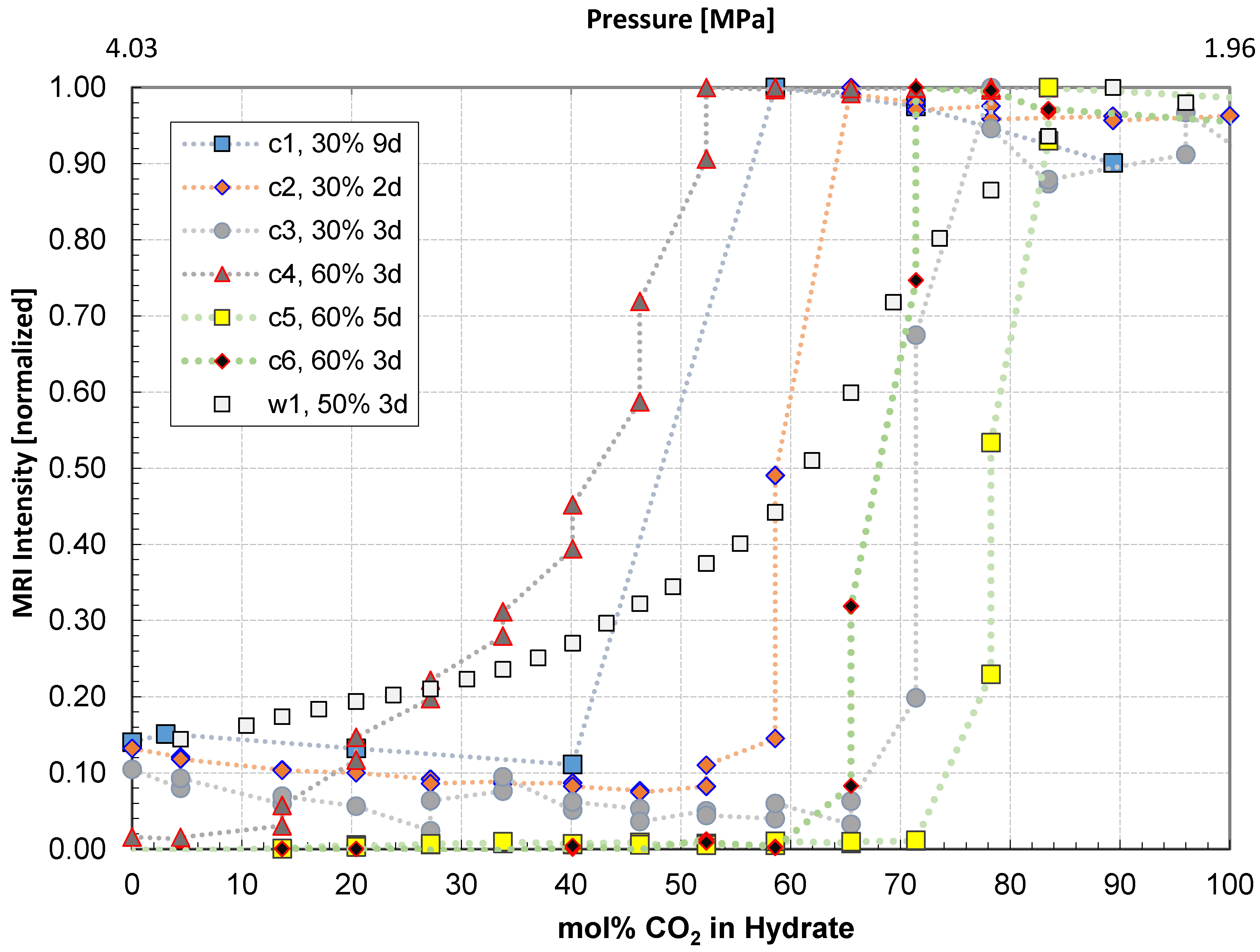
| Test | Saturation | Exposure time (days) | Conversion | Comment |
|---|---|---|---|---|
| c1 | 0.3 | 9 | 40%–60% | Huff and Puff. Non-uniform saturation distribution. Mass transport based on diffusion. |
| c2 | 0.3 | 2 | 58%–65% | Constant CO2 flow at 0.033 cm3/min. Uniform saturation. |
| c3 | 0.3 | 3 | 71%–78% | Constant CO2 flow at 0.033 cm3/min. Uniform saturation. |
| c4 | 0.6 | 3 | 14%–52% | Constant CO2 flow at 0.033 cm3/min. Non-uniform saturation, Sw exceeding 0.8 in some areas. |
| c5 | 0.6 | 5 | 71%–83% | Constant CO2 flow at 0.033 cm3/min. Uniform saturation. |
| c6 | 0.6 | 3 | 65%–71% | Constant CO2 flow at 0.033 cm3/min. Uniform saturation. |
| w1 | 0.5 | 3 | 16%–85% | Whole core. Constant CO2 flow at 0.033 cm3/min. Non-uniform saturation, plugging, significant residual water. |




3.4. Continuous CO2 Mass Transport in Non-Fractured Samples

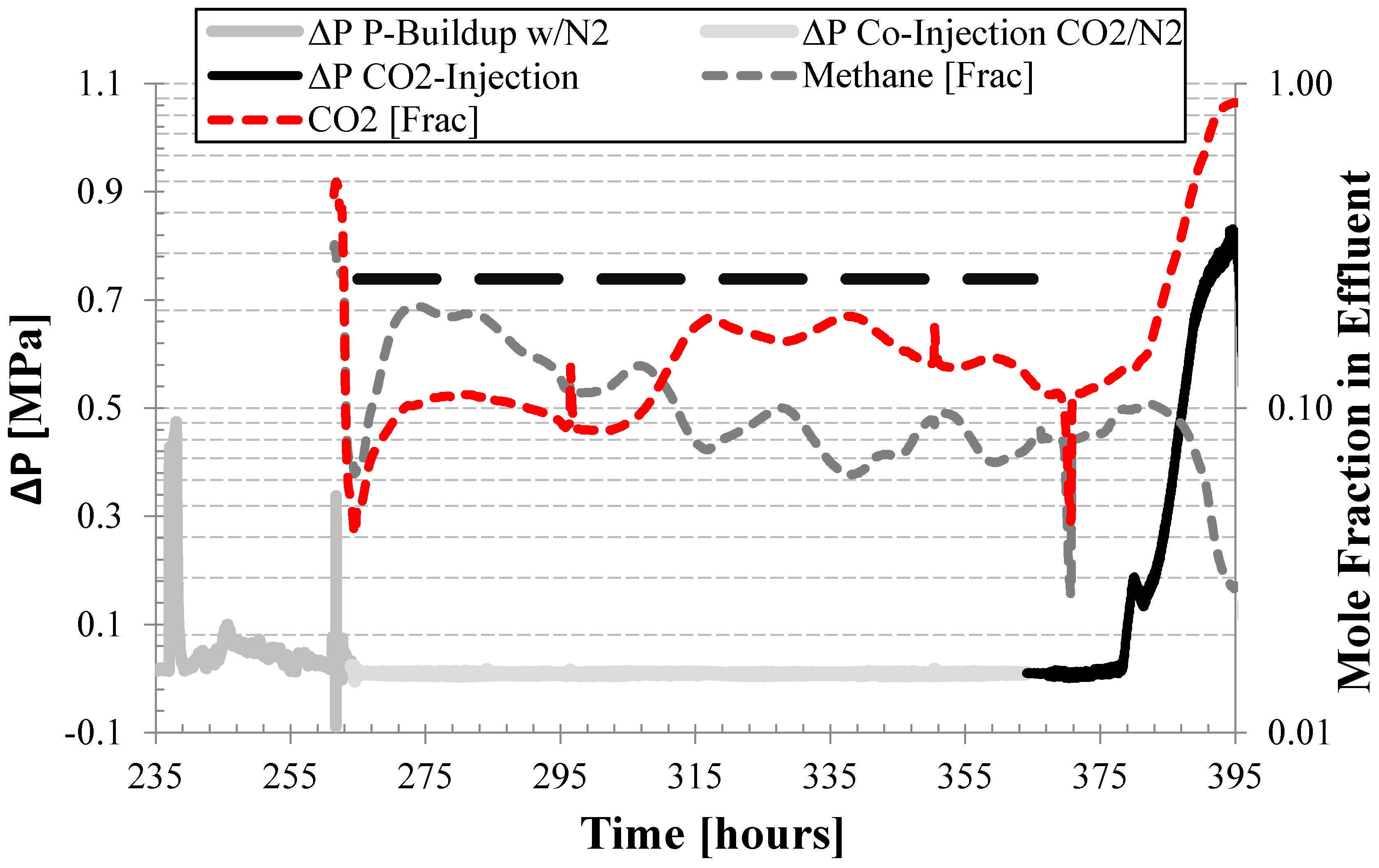
3.5. Flow Remediation through Binary Gas Injection

4. Conclusions
Acknowledgments
Author Contributions
Nomenclature
| Si | Saturation of fluid i |
| n | Measure of fluid amount in moles |
| m | Measure of fluid weight |
| Xi | Fraction of fluid i |
| Mi | Molar weight of fluid i |
| MDi | Molar Density of fluid i |
| υ | Hydration number |
| ΔP | Differential Pressure |
| PV | Pore Volume |
| Vi | Volume of fluid i |
| Ainterface | Interface Area |
| r | Radius |
| T | Temperature |
Supplementary Materials
Conflicts of Interest
References
- Kvenvolden, K.A. Methane hydrate—A major reservoir of carbon in the shallow geosphere? Chem. Geol. 1988, 71, 41–51. [Google Scholar] [CrossRef]
- Milkov, A.V. Global estimates of hydrate-bound gas in marine sediments: How much is really out there? Earth-Sci. Rev. 2004, 66, 183–197. [Google Scholar] [CrossRef]
- Klauda, J.B.; Sandler, S.I. Global distribution of methane hydrate in ocean sediment. Energy Fuels 2005, 19, 459–470. [Google Scholar] [CrossRef]
- Svandal, A.; Kuznetsova, T.; Kvamme, B. Thermodynamic properties and phase transitions in the H2O/CO2/CH4 system. Fluid Phase Equilib. 2006, 246, 177–184. [Google Scholar] [CrossRef]
- Anderson, G.K. Enthalpy of dissociation and hydration number of carbon dioxide hydrate from the clapeyron equation. J. Chem. Thermodyn. 2003, 35, 1171–1183. [Google Scholar] [CrossRef]
- Anderson, G.K. Enthalpy of dissociation and hydration number of methane hydrate from the clapeyron equation. J. Chem. Thermodyn. 2004, 36, 1119–1127. [Google Scholar] [CrossRef]
- Jung, J.W.; Espinoza, D.N.; Santamarina, J.C. Properties and phenomena relevant to CH4-CO2 replacement in hydrate-bearing sediments. J. Geophys. Res.: Solid Earth 2010, 115, B10102. [Google Scholar] [CrossRef]
- Kennett, J.P.; Cannariato, K.G.; Hendy, I.L.; Behl, R.J. Methane Hydrates in Quaternary Climate Change: The Clathrate Gun Hypothesis; American Geophysical Union: Washington, DC, USA, 2003. [Google Scholar]
- Lelieveld, J.; Crutzen, P.J.; Dentener, F.J. Changing concentration, lifetime and climate forcing of atmospheric methane. Tellus B 1998, 50, 128–150. [Google Scholar] [CrossRef]
- Birkedal, K.A.; Ersland, G.; Husebø, J.; Kvamme, B.; Graue, A. Geomechanical stability during CH4 production from hydrates—Depressurization or CO2 sequestration with CO2-CH4 exchange. In Proceedings of the 44th U.S. Rock Mechanics Symposium and 5th U.S.-Canada Rock Mechanics Symposium, Salt Lake City, UT, USA, 27–30 June 2010.
- Espinoza, D.N.; Santamarina, J.C. P-wave monitoring of hydrate-bearing sand during CH4–CO2 replacement. Int. J. Greenh. Gas Control 2011, 5, 1031–1038. [Google Scholar] [CrossRef]
- Tohidi, B.; Yang, J. CO2 hydrates could provide secondary safety factor in subsurface sequestration of CO2. Environ. Sci. Technol. 2010, 44, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Ballard, A.L.; Sloan, E.D., Jr. The next generation of hydrate prediction: An overview. J. Supramol. Chem. 2002, 2, 385–392. [Google Scholar] [CrossRef]
- Lee, H.; Seo, Y.; Seo, Y.-T.; Moudrakovski, I.L.; Ripmeester, J.A. Recovering methane from solid methane hydrate with carbon dioxide. Angew. Chem. 2003, 115, 5202–5205. [Google Scholar] [CrossRef]
- Lee, H.; Seo, Y.; Seo, Y.-T.; Kim, D.Y.; Moudrakovski, I.L.; Ripmeester, J.A.; Sang-Eon Park, J.-S.C.; Kyu-Wan, L. Replacement of methane hydrate by carbon dioxide: 13C NMR study for studying a limit to the degree of substitution. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2004; Volume 153, pp. 495–500. [Google Scholar]
- Park, Y.; Cha, M.; Cha, J.H.; Shin, K.; Lee, H.; Park, K.P.; Huh, D.G.; Lee, H.Y.; Kim, S.J.; Lee, J. Swapping carbon dioxide for complex gas hydrate structures. In Proceedings of the 6th International Conference on Gas Hydrates, Vancouver, BC, Canada, 6–10 July 2008; p. 6.
- Ota, M.; Morohashi, K.; Abe, Y.; Watanabe, M.; Smith, J.R.L.; Inomata, H. Replacement of CH4 in the hydrate by use of liquid CO2. Energy Convers. Manag. 2005, 46, 1680–1691. [Google Scholar] [CrossRef]
- Graue, A.; Kvamme, B.; Baldwin, B.; Stevens, J.; Howard, J.J.; Aspenes, E.; Ersland, G.; Husebo, J.; Zornes, D. Mri visualization of spontaneous methane production from hydrates in sandstone core plugs when exposed to CO2. SPE J. 2008, 13, 146–152. [Google Scholar] [CrossRef]
- Kvamme, B.; Graue, A.; Buanes, T.; Kuznetsova, T.; Ersland, G. Storage of CO2 in natural gas hydrate reservoirs and the effect of hydrate as an extra sealing in cold aquifers. Int. J. Greenh. Gas Control 2007, 1, 236–246. [Google Scholar] [CrossRef]
- Yuan, Q.; Sun, C.-Y.; Liu, B.; Wang, X.; Ma, Z.-W.; Ma, Q.-L.; Yang, L.-Y.; Chen, G.-J.; Li, Q.-P.; Li, S.; et al. Methane recovery from natural gas hydrate in porous sediment using pressurized liquid CO2. Energy Convers. Manag. 2013, 67, 257–264. [Google Scholar]
- Hester, K.C.; Stevens, J.C.; Howard, J.J. Composition studies to determine rate and extent of CO2 exchange in a hydrate-bearing core. In Preoceedings of the 7th International Conference on Gas Hydrates, Edinburgh, UK, 17–21 July 2011.
- Ersland, G.; Husebø, J.; Graue, A.; Baldwin, B.A.; Howard, J.; Stevens, J. Measuring gas hydrate formation and exchange with CO2 in bentheim sandstone using mri tomography. Chem. Eng. J. 2010, 158, 25–31. [Google Scholar] [CrossRef]
- Deusner, C.; Bigalke, N.; Kossel, E.; Haeckel, M. Methane production from gas hydrate deposits through injection of supercritical CO2. Energies 2012, 5, 2112–2140. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Pan, H.; Wang, X.; Li, F.; Sun, C.; Chen, G. Evaluation of different CH4-CO2 replacement processes in hydrate-bearing sediments by measuring P-wave velocity. Energies 2013, 6, 6242–6254. [Google Scholar] [CrossRef]
- Phale, H.A.; Zhu, T.; White, M.D.; McGrail, B.P. Simulation study on injection of CO2-microemulsion for methane recovery from gas-hydrate reservoirs. In SPE Gas Technology Symposium; Society of Petroleum Engineers: Calgary, AL, Canada, 2006. [Google Scholar]
- White, M.D.; Wurstner, S.K.; McGrail, B.P. Numerical studies of methane production from class 1 gas hydrate accumulations enhanced with carbon dioxide injection. Mar. Pet. Geol. 2011, 28, 546–560. [Google Scholar] [CrossRef]
- Schoderbek, D.; Boswell, R. Ignik sikumi #1, gas hydrate test well, successfully installed on the alaska north slope. Fire Ice 2011, 11, 1–5. [Google Scholar]
- Parshall, J. Production method for methane hydrate sees scientific success. J. Pet. Technol. 2012, 64, 50–51. [Google Scholar] [CrossRef]
- Thomas, W.J.; Adams, M.J. Measurement of the diffusion coefficients of carbon dioxide and nitrous oxide in water and aqueous solutions of glycerol. Trans. Faraday Soc. 1965, 61, 668–673. [Google Scholar] [CrossRef]
- Demurov, A.; Radhakrishnan, R.; Trout, B.L. Computations of diffusivities in ice and CO2 clathrate hydrates via molecular dynamics and Monte Carlo simulations. J. Chem. Phys. 2002, 116, 702–709. [Google Scholar] [CrossRef]
- Davies, S.R.; Lachance, J.W.; Sloan, E.D.; Koh, C.A. A novel approach to measuring methane diffusivity through a hydrate film using differential scanning calorimetry. In Preoceedings of the 6th International Conference on Gas Hydrates, Vancouver, BC, Canada, 6–10 July 2008.
- Husebø, J. Monitoring Depressurization and CO2-CH4 Exchange Production Scenarios for Natural Gas Hydrates; University of Bergen: Bergen, Norway, 2008. [Google Scholar]
- Birkedal, K.A. Empirical and Numerical Evaluation of Mechanisms in Gas Production from CH4-hydrates: Emphasis on Kinetics, Electrical Resistivity, Depressurization and CO2-CH4 Exchange; University of Bergen: Bergen, Norway, 2013. [Google Scholar]
- Circone, S.; Kirby, S.; Stern, L.A. Direct measurement of methane hydrate composition along the hydrate equilibrium boundary. J. Phys. Chem. 2005, 109, 9468–9475. [Google Scholar] [CrossRef]
- Petrich, C.; Eicken, H. Growth, structure and properties of sea ice. In Sea Ice; Wiley-Blackwell: Hoboken, NJ, USA, 2010; pp. 23–77. [Google Scholar]
- Duan, Z.; Sun, R. An improved model calculating CO2 solubility in pure water and aqueous NaCl solutions from 273 to 533 k and from 0 to 2000 bar. Chem. Geol. 2003, 193, 257–271. [Google Scholar] [CrossRef]
- Collett, T.S.; Boswell, R.; Lee, M.W.; Andersen, J.L.; Rose, K.; Lewis, K.A. Evaluation of long-term gas-hydrate-production testing locations on the Alaska North Slope. SPE Reserv. Eval. Eng. 2012, 15, 243–264. [Google Scholar] [CrossRef]
- Schoderbek, D.; Martin, K.L.; Howard, J.; Silpngarmlert, S.; Hester, K.C. North slope hydrate fieldtrial: CO2/CH4 exchange. In Proceedings of the Arctic Technology Conference, Houston, TX, USA, 3–5 December 2012.
- Kneafsey, T.J.; Nakagawa, S.; Borglin, S.E. Properties of Hydrate-bearing Sediments Subjected to Changing Gas Compositions; Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2013. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birkedal, K.A.; Hauge, L.P.; Graue, A.; Ersland, G. Transport Mechanisms for CO2-CH4 Exchange and Safe CO2 Storage in Hydrate-Bearing Sandstone. Energies 2015, 8, 4073-4095. https://doi.org/10.3390/en8054073
Birkedal KA, Hauge LP, Graue A, Ersland G. Transport Mechanisms for CO2-CH4 Exchange and Safe CO2 Storage in Hydrate-Bearing Sandstone. Energies. 2015; 8(5):4073-4095. https://doi.org/10.3390/en8054073
Chicago/Turabian StyleBirkedal, Knut Arne, Lars Petter Hauge, Arne Graue, and Geir Ersland. 2015. "Transport Mechanisms for CO2-CH4 Exchange and Safe CO2 Storage in Hydrate-Bearing Sandstone" Energies 8, no. 5: 4073-4095. https://doi.org/10.3390/en8054073
APA StyleBirkedal, K. A., Hauge, L. P., Graue, A., & Ersland, G. (2015). Transport Mechanisms for CO2-CH4 Exchange and Safe CO2 Storage in Hydrate-Bearing Sandstone. Energies, 8(5), 4073-4095. https://doi.org/10.3390/en8054073