1. Introduction
Gas hydrates are clathrate compounds in which hydrogen bonding network of water trap typically non-polar gases, such as methane, ethane and propane [
1]. Gas hydrates form at high pressures and low temperatures. There are a number of similarities in the physical properties of ice and gas hydrates, as summarized by Sloan and Koh [
1].
Oil and gas pipelines commonly encounter hydrate-forming conditions, particularly in subsea pipelines. Gas hydrates, if formed, could block the gas pipelines and therefore poses considerable risks to flow assurance. Turner
et al. identified the existence of several distinct steps between the formation of gas hydrates and the blockage of gas pipelines; gas hydrate nucleation, gas hydrate crystal growth, gas hydrate particle cohesion and agglomeration, adhesion to pipeline walls and blockages [
2]. Then, prevention of any one of these steps could arrest gas hydrate formation from escalating to expensive and potentially catastrophic chain of events. Among these, cohesion and agglomeration properties of gas hydrate particles appear the key to hydrate management in flow assurance in natural gas pipelines.
One of the relevant outstanding questions is the presence of pre-melting layers on the surface of gas hydrates. A pre-melting layer, or surface melting layer, refers to a quasi-liquid layer that exists thermodynamically stably at the surface of a solid below the melting point [
3,
4]. This phenomenon has been observed for many solids since the days of Faraday [
3,
4,
5,
6,
7], and large amounts of efforts have been directed to the study of pre-melting of ice in particular, because of its wide-ranging environmental implications (see [
3,
4] for reviews). The presence of pre-melting layer, or lack thereof, greatly influences the adhesion/agglomeration properties. For example, when two blocks of ice cubes are brought together below the ice point, the pre-melting layers on each ice cube merge upon contact and form one liquid film that fill the gap between the two ice cubes which may have been present due to the surface roughness. Then, unlike the pre-melting layers on the outer surface, the merged water film is no longer exposed to the gas. It will freeze to form one large merged block of ice cube.
It has been known that the thickness of a pre-melting layer grows with the warming of the solid and eventually diverges at the melting point [
3,
4]. Pre-melting is responsible for the general lack of superheating of solids above the melting point, because the pre-melting layer provides the necessary nucleation sites for the emerging liquid phase upon melting.
Despite its importance, pre-melting has rarely been examined in relation to gas hydrates to date. In the literature, Aman
et al. experimentally inferred the presence of quasi liquid later on the surface of cyclopentane hydrates [
8]. Yang
et al. [
9] and Ding
et al. [
10] reported the presence of quasi liquid layer on the surface of gas hydrates from molecular dynamics simulation. In particular, Ding
et al. specifically reported the occurrence of surface melting on methane hydrate surfaces from molecular dynamics simulation [
10].
Unlike pre-melting of ice, in which the top-most layers of ice melts to form a thin film of liquid water below the ice point, pre-melting of gas hydrate, say methane hydrate, does not “melt” to form a thin film of liquid methane aqueous solution. The solubility of methane or other non-polar gases in water is so low that the dissociated gas would readily come out and join the surrounding gas. The net result of pre-melting of gas hydrates would then be the formation of a thin film of almost pure water. It is thus not immediately obvious if such a process is thermodynamically favorable. Below, we review the thermodynamic basis of pre-melting and apply surface thermodynamics to pre-melting of gas hydrate surfaces.
2. Thermodynamic Basis of Pre-Melting
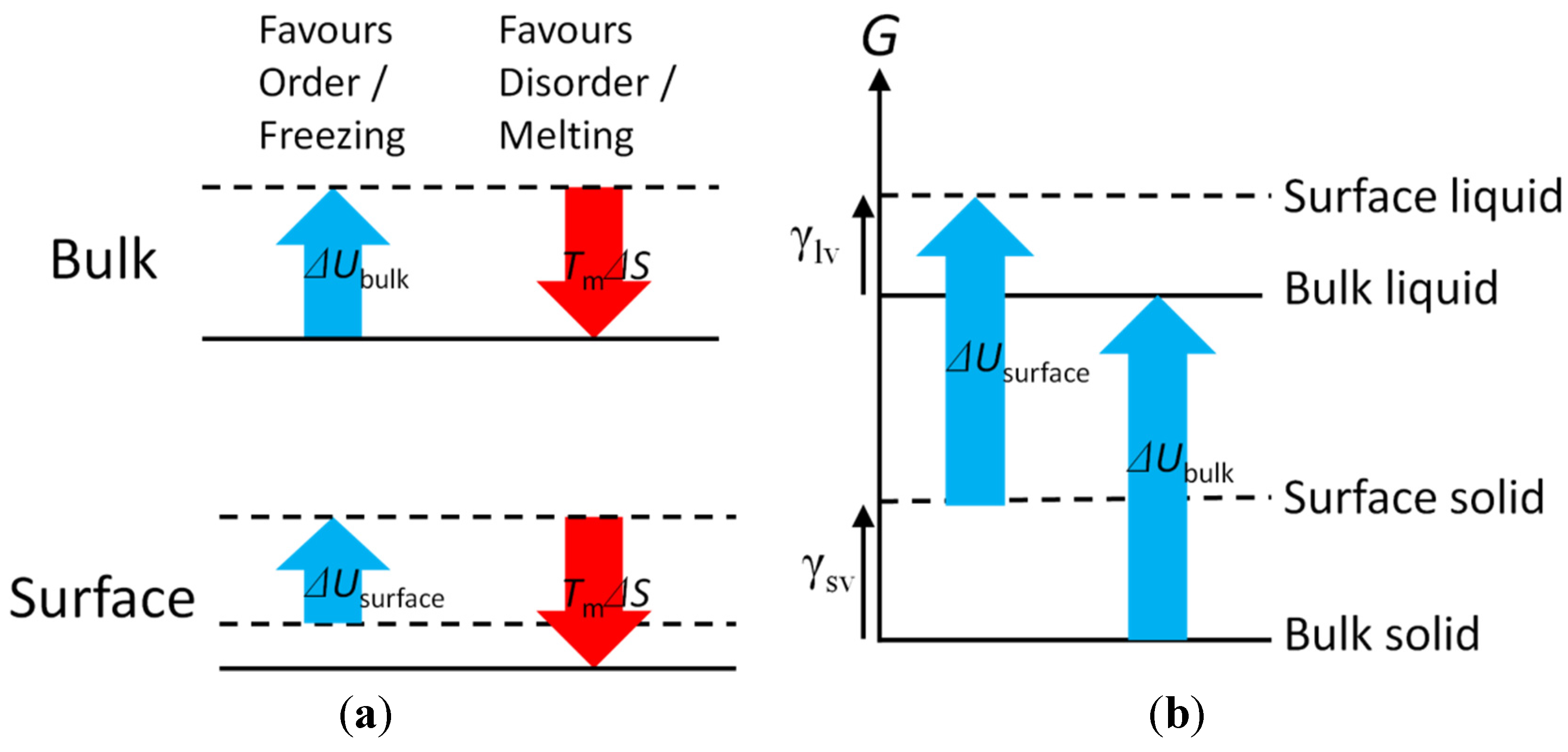
The internal energy, U, that arises from the bonding between atoms/molecules favours crystalline structures while the entropy favours disorder. Since the entropy (S) contribution to the Gibbs free energy, G, is −TS, the influence of the entropy term on G increases with temperature, T. Thus there is a point in T above which melting becomes favourable. This defines the melting point, Tm.
On a surface of a solid, the reduced numbers of bonds of atoms/molecules result in a higher U compared with that in the bulk at a given T. This lack of bonding of atoms/molecules at the surface, and the consequent increase in U, is the source of the interfacial free energy, γ, the energy required to bring atoms/molecules from inside of a bulk to a surface.
A liquid has reduced bonding density than an ordered, crystalline bulk solid of the same material. The extent depends on the number of chemical bonds per atom/molecule that would be broken upon melting. In a bulk, all the bonds will be lost upon melting in one go. In contrast, on a mathematically flat surface, an atom/molecule will lose half of its bonds when it comes to the surface from inside of the bulk. Upon melting, the remaining half will be broken. Therefore, more bonds will be broken upon melting in the bulk than on a surface, unless the molten liquid retains unusually high number densities of the bonds (high degrees of order). It follows that: (1) the change in
U upon melting on a surface is smaller than in the bulk (Δ
Usurface < Δ
Ubulk); (2) the surface tension of a solid is higher than that of the liquid of the same material (γ
lv < γ
sv); and (3) the crossover
T above which melting becomes favourable is lower on a surface than in the bulk (
Tsurface,m <
Tm). These relationships are schematically illustrated in
Figure 1.
Figure 1.
Schematic illustration of free energy balance upon (a) freezing/melting and (b) surface vs. bulk of a material.
Figure 1.
Schematic illustration of free energy balance upon (a) freezing/melting and (b) surface vs. bulk of a material.
For these reasons, pre-melting is observed for many solids [
7]. However, there are a few important exceptions. Long chain n-alkanes are one group of such an example [
11]. Long chain n-alkanes form a rotator phase (each straight chain molecule retains rotational degree of freedom around its long axis) upon freezing and hence possess sufficient entropy in the solid form [
12,
13]. Thus the entropic gain upon melting the bulk rotator phase is much less than the case for which the rotational degree of freedom in the solid phase is suppressed (as in most other solids) [
12,
13,
14]. The end result is that, not only pre-melting is absent in the crystalline surface of long chain n-alkanes but also the surface of the liquid long chain n-alkanes remains frozen for a few Kelvins above
Tm [
12,
13,
15].
3. Can Pre-Melting Occur on the Surface of Gas Hydrates?
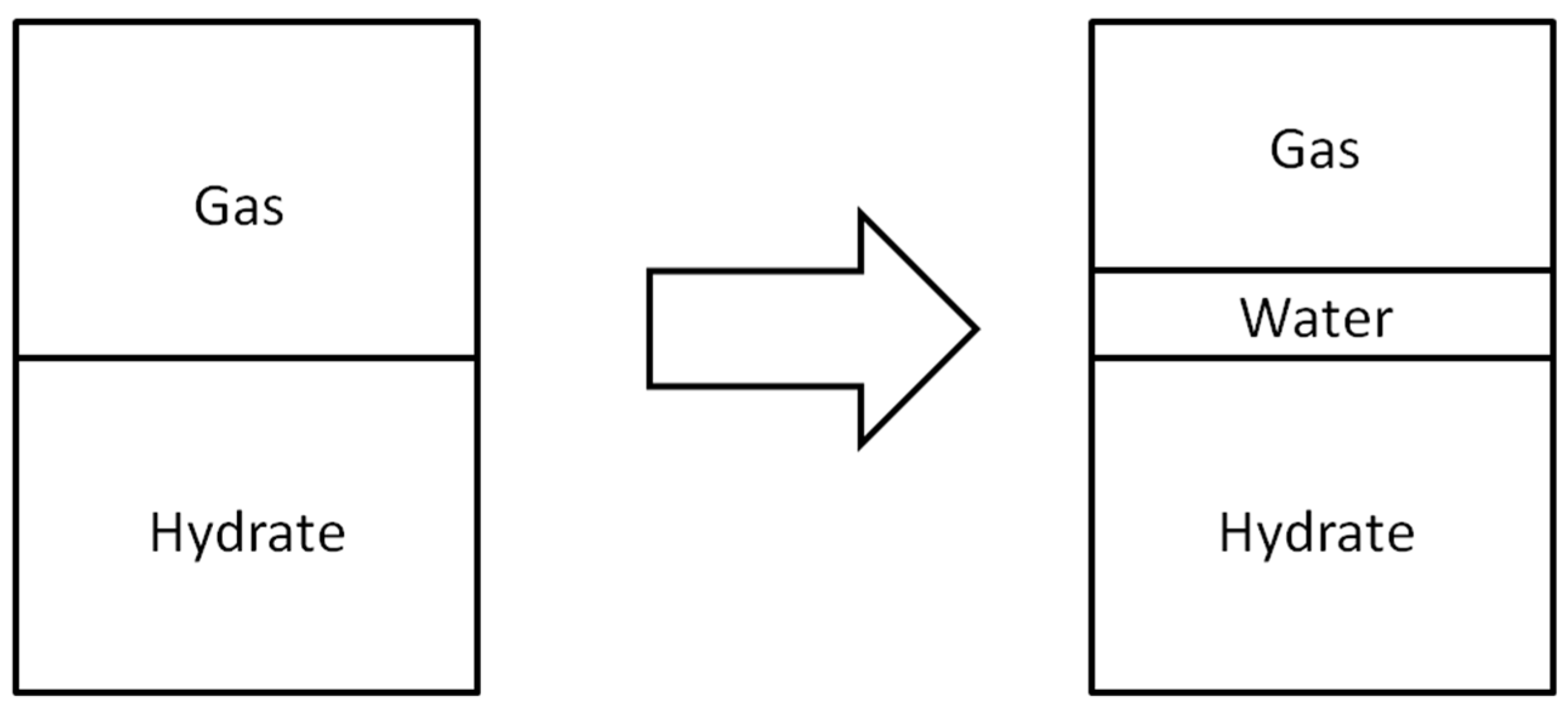
When a gas hydrate dissociates, it will become water and the guest gas that has been trapped in the clathrate structure. The guest gas will then become indistinguishable from the surrounding bulk gas medium, and the end result will be just a water film on the surface of the gas hydrate. The formation of such a water film newly introduces an additional interface, as shown in
Figure 2.
Figure 2.
Schematic illustration of change in the interfacial area upon pre-melting.
Figure 2.
Schematic illustration of change in the interfacial area upon pre-melting.
The change in the interfacial free energy component upon pre-melting per unit area is,
where the subscripts
g,
w and
h refer to gas, water and hydrate, respectively. Among the three relevant interfacial free energies, γ
gw is well known and about 72 mN/m, depending on the temperature and pressure [
16]. In contrast, γ
hw and γ
gh are difficult to measure. Still, some estimates have been made for the values of γ
hw. Uchida
et al. estimated CH
4, CO
2, and C
3H
8 hydrate-water interfacial tension values of 17 ± 3, 14 ± 3, and 25 ± 1 mN/m [
17]. Anderson
et al. estimated CH
4 and CO
2 hydrate-water interfacial tension values of 32 ± 3 and 30 ± 3 mN/m [
18]. Seo
et al. estimated C
2H
6 and C
3H
6 hydrate-water interfacial tension values of 39 ± 2 and 45 ± 1 mN/m [
19]. Using classical crystallization theory, Zhang
et al. reported the value of 9.3 mN/m for CO
2 hydrate-water interfacial tension [
20]. In contrast, Aman
et al. estimated the hydrate-water interfacial tension value of 0.32 ± 0.05 mN/m for liquid cyclopentane hydrate [
21].
For comparison, the interfacial tension between ice and water has been reported to be in the range of 28 to 33 mN/m [
22,
23,
24,
25]. Since gas hydrates are similar to ice in many aspects [
1], the similarity in the interfacial tension values is expected. The available estimates for the interfacial tension values between hydrates and water (and that between ice and water) may appear surprisingly large, given that both ice and hydrates are supposedly hydrophilic.
Surface energy of gas hydrates, γ
gh (hydrate-gas interface), is experimentally difficult to measure due to surface roughness and other complications. We note that the surface tension of ice has been reported to be around 130 mN/m [
26]. The similarity in γ
hw between ice and gas hydrates (see above) suggests that the number of the bonds that will be broken to create a unit area of new surface is similar between ice and gas hydrates. This similarity may be expected given that the guest molecules trapped in the gas hydrates are not chemically bonded and only the chemical and hydrogen bonds of water molecules would contribute to the surface and interfacial energies.
If we assume that γgh of gas hydrate surface is similar to that of ice (≈130 mN/m), and noting that the interfacial tension between ice and water is about 30 mN/m and that the surface tension of water is 72 mN/m, ΔGsurf ≈ −28 mN/m, which is negative. These calculations suggest that pre-melting will be favoured where ΔGsurf is large enough in negative to offset the free energy change upon dissociation of gas hydrate, ΔGfus. Since ΔGfus is proportional to the volume whereas ΔGsurf is proportional to the surface area, pre-melting is favoured for sufficiently thin pre-melting layers. In other words, pre-melting does occur on the surface of gas hydrates. The next question is; how thick can they be?
4. Thickness of the Pre-Melting Layers
The Δ
Gsurf gain is proportional to the surface area. In contrast, the Δ
Gfus penalty is proportional to the mass or volume of the water to be dissociated/melted. Then, for unit surface area, the Δ
Gfus becomes proportional to the thickness of the film. If we denote the thickness of the film as
h and the number density of water as ρ, then the volume of the film over unit surface area is
h and Δ
G per unit area is,
The enthalpy of fusion of ice is 334 J/g or 6.01 kJ/mol [
27]. In contrast, the enthalpy of dissociation of methane hydrate (to water and methane gas) is 56.9 kJ, per mol of methane [
1]. Given that there are 5.75 water molecules per methane molecule on average in methane hydrate, this value equates to 9.9 kJ/mol of water. These calculations suggest that the Δ
Gfus penalty of dissociating methane hydrates is about 40% greater than the Δ
Gfus penalty of melting ice (per mol of water). Thus, it may be expected that the thickness of the pre-melting layer of gas hydrates is smaller than that of ice.
The above estimate is for just below the respective melting/dissociation points. For greater subcoolings, the Δ
Gfus penalty becomes proportional to the subcooling of the system, Δ
T; Δ
Gfus(Δ
T) = (Δ
T/
Tm)Δ
Hfus = Δ
TΔ
Sfus. In contrast, the interfacial energy generally increases with cooling. However, the relevant interfacial energy values are not available, much less as functions of temperature. Nevertheless, we can still conclude that Δ
Gsurf is expected to be less sensitive to temperature because Δ
Gsurf is in the form of sum and difference of the three interfacial energy terms (see Equation (1)). For the first approximation, we ignore the temperature dependence on Δ
Gsurf. Then, Δ
G per unit area is,
From this, one may expect that the thickness of the pre-melting layer to decrease rapidly with subcooling because of the increase in the Δ
Gfus penalty with subcooling. Indeed, the thickness of the pre-melting layer of ice was found to decrease rapidly with subcooling [
3,
4]. With the about 40% greater Δ
Gfus penalty for gas hydrates than for water we found above, one may expect that the pre-melting layer of gas hydrates to thin out even more rapidly than that on ice with subcooling. Then, with the typically large subcoolings involved in gas hydrate systems (e.g., the subcooling for high pressure natural gases at the sea bed temperature is around 20 K), one may conclude that the surface of gas hydrates at such large subcoolings is effectively dry. However, this is not the end of the story.
5. Effects of Surface Forces
It has been known that the thickness of the pre-melting layers grows with warming of ice and eventually diverges at the melting point [
3,
4]. This is expected from the Δ
Gfus − Δ
Gsurf balance discussed above. What has been largely overlooked to date, however, is the effect of surface forces on such pre-melting layers. The central quantity that describes the stability of a thin liquid film on a solid surface is the disjoining pressure,
Π(
h) [
28]. The disjoining pressure is related to the difference in the chemical potential of a liquid in a film and that in a bulk liquid (there is a difference in the definition of disjoining pressure between the Western and the Russian conventions). Here we define
Π(
h)
vm ≡ µ
film − µ
bulk, where
vm is the molecular volume. Then, Δ
G(
h) per unit area becomes
For all liquid films, the disjoining pressure disappears as the film thickens and approaches bulk liquids; i.e., Π(h) → 0 as h → ∞. For stable films (µfilm < µbulk), Π(h) < 0 and hence dΠ(h)/dh > 0. For unstable films (µfilm > µbulk), Π(h) > 0 and hence dΠ(h)/dh < 0.
Multiple components exist in the disjoining pressure for a complex (polar and hydrogen bonding) liquid such as water [
28]. For simplicity, below we restrict our discussions on the implications of surface forces to the ubiquitous van der Waals component only.
It is worth recalling that water is denser than ice. Consequently, the refractive index of water is greater than that of ice. Sloan and Koh tabulated the refractive index of ice as 1.3082, sI gas hydrate as 1.346 and sII gas hydrate as 1.35 [
1]. For comparison, the refractive index of water is 1.33 [
27]. The theory of dispersion forces shows that a film which has an intermediate refractive index of that of the medium at either side of the film (
i.e., greater than that on the one side of the film but smaller than the other) is stable and can grow whereas a film which has a refractive index greater or smaller than that at either side of the film will be suppressed [
29,
30,
31]. Then, as we discussed in details previously [
11], the implication to the pre-melting of ice and gas hydrates is that the van der Waals forces for the ice/water/gas system must be attractive whereas the van der Waals forces for the clathrate/water/gas system must be repulsive. In other words, the pre-melting layers on ice were found to be thin, at least in part because the thickness was suppressed by the attractive van der Waals forces. No such thickness suppression by the surface forces exist for the pre-melting layers on gas hydrates. In other words, if we assume that the van der Waals component dominates the disjoining pressure, then
Π(
h) < 0 for the pre-melting layers on gas hydrates whereas
Π(
h) > 0 for the pre-melting layers on ice.
As such, we will be able to predict the thickness of pre-melting layers if the interfacial energy values (ΔGsurf) and the disjoining pressure are known.
6. Implications to Cohesion and Friction Forces between Gas Hydrate Particles
As we have seen, pre-melting is thermodynamically favorable for both ice and gas hydrates. However, there is an important difference in terms of implications such pre-melting layers have on the cohesion and frictional forces.
When two blocks of ice cubes are brought together below the ice point, the pre-melting layers on each ice cube merge upon contact and form one liquid film and fill the gap between the two ice cubes that may have been present due to the surface roughness. Then, unlike the pre-melting layers on the outer surface, the merged film is no longer exposed to the gas. It will freeze to form one large merged block of ice cube.
In contrast, pre-melting of gas hydrate does not “melt” to form a thin film of a gas aqueous solution of the same concentration. The solubility of non-polar gases in water is simply too low to maintain the same composition as that of gas hydrate. Then the dissociated gas would readily come out of the aqueous phase and merge with the surrounding gas. The net result of pre-melting of gas hydrates would then be the formation of a thin film of almost pure water.
Then, when two blocks of gas hydrate cubes are brought together below the thermodynamic equilibrium dissociation temperature of the gas hydrate, the almost pure water film on either surface of the gas hydrate will merge and fill the gap between the two blocks. Unlike for ice, however, the almost pure water cannot readily re-form gas hydrate, because there is not sufficient amount of guest gas available. The diffusion rate of gas from outside of the joined block into the merged film is generally a slow process that depends on the surface roughness of the original blocks.
The presence of pre-melting layer will increase the cohesive force between gas hydrate particles due to capillary bridging [
32]. In contrast, the presence of pre-melting layer will render frictional forces between gas hydrate particles small. The reason is that if each gas hydrate particle is coated with pre-melting layer of almost pure water and if such layer remains liquid when trapped between two gas hydrate particles, then the trapped water will act as an excellent lubricant. Our recent study indeed showed that the cohesion hysteresis between cyclopentane hydrate particles in liquid cyclopentane was very small (below the detection limit of the instrument) [
33].
7. Conclusions
As we search through the relevant literature, it became clear that many fundamental data that are central to the surface and interfacial properties of gas hydrates are lacking. For example, very few water—gas hydrate interfacial energy values are available in the literature, let alone their temperature dependence, and none for the gas—gas hydrate interfacial energy values. Future experimental effort should be directed to the measurements of these important fundamental quantities.
Despite the lack of relevant data, surface thermodynamic analyses provide a few powerful predictions. The presence of pre-melting layers is thermodynamically favourable for gas hydrates. Unfortunately, there are not sufficient data to predict whether such pre-melting layers are thicker or thinner than those on ice, at a given subcooling. There are two opposing factors that influence the thickness of the pre-melting layers on gas hydrates. One is the enthalpy of fusion below the thermodynamic dissociation temperature, which is unfavourable for the growth of the pre-melting layer and the cost is greater than that for ice. The other is the repulsive van der Waals forces which favour thick pre-melting layers on gas hydrates, as opposed to the attractive van der Waals forces which suppress the thickness of pre-melting layers on ice.
As for the effect of the pre-melting layers on cohesive and frictional forces, the effect for gas hydrates is expected to be vastly different from that for ice. When two blocks of ice are brought together, the pre-melting layers will join the two blocks and transform them into a single block of ice. In contrast, when two blocks of gas hydrate particles are brought together, the pre-melting layers will remain liquid between the two blocks in the absence of fast gas diffusion. Such trapped liquid will cause increased cohesive forces due to capillary action and reduced friction forces because it will act as a lubricant.


{kind=link}
{kind=link}

