
Flexible Yttrium Coordination Geometry Inhibits “Bare-Metal” Guest Interactions in the Metal-Organic Framework Y(btc)

 ,
,
Abstract
:
1. Introduction
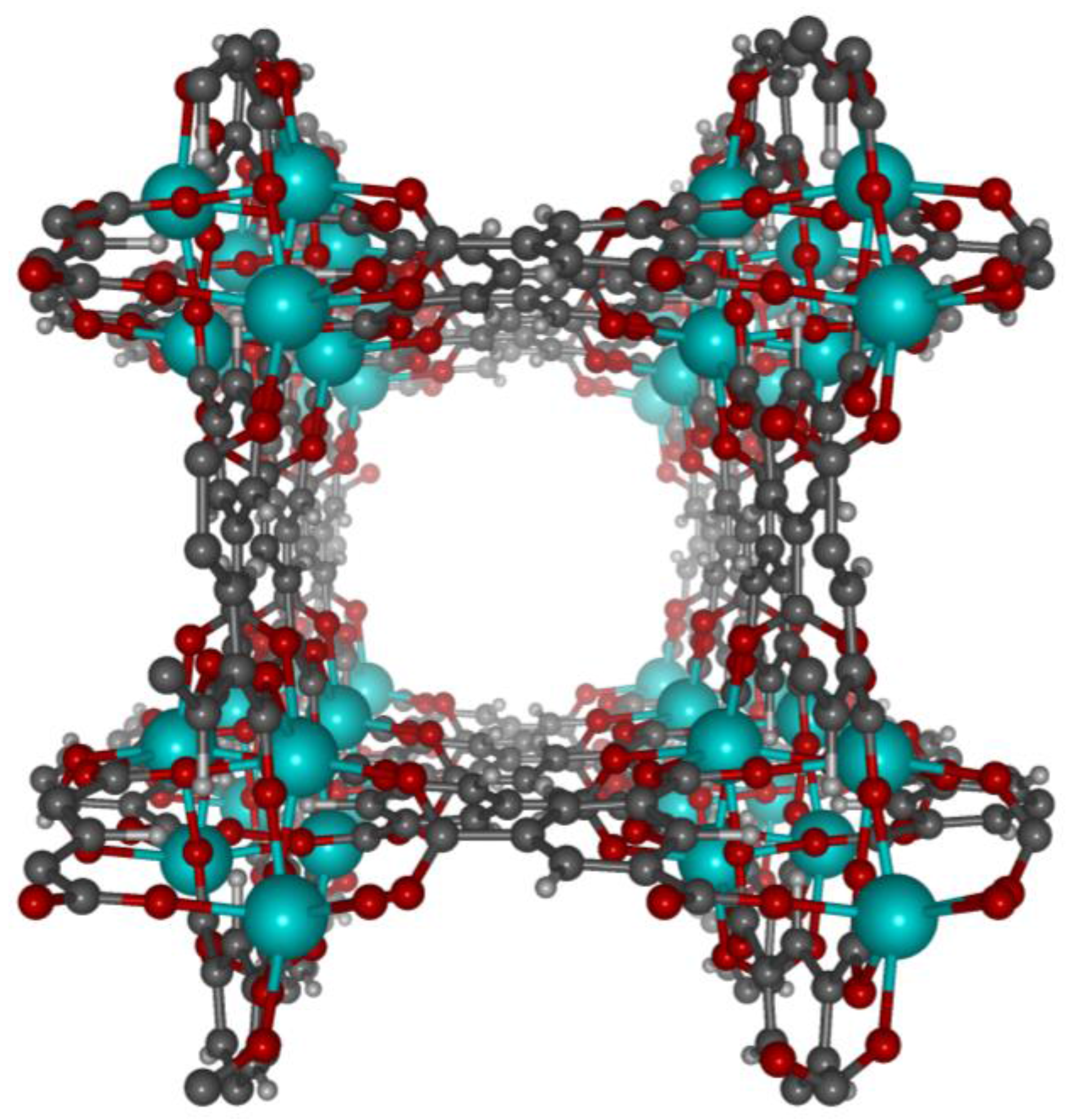
2. Results
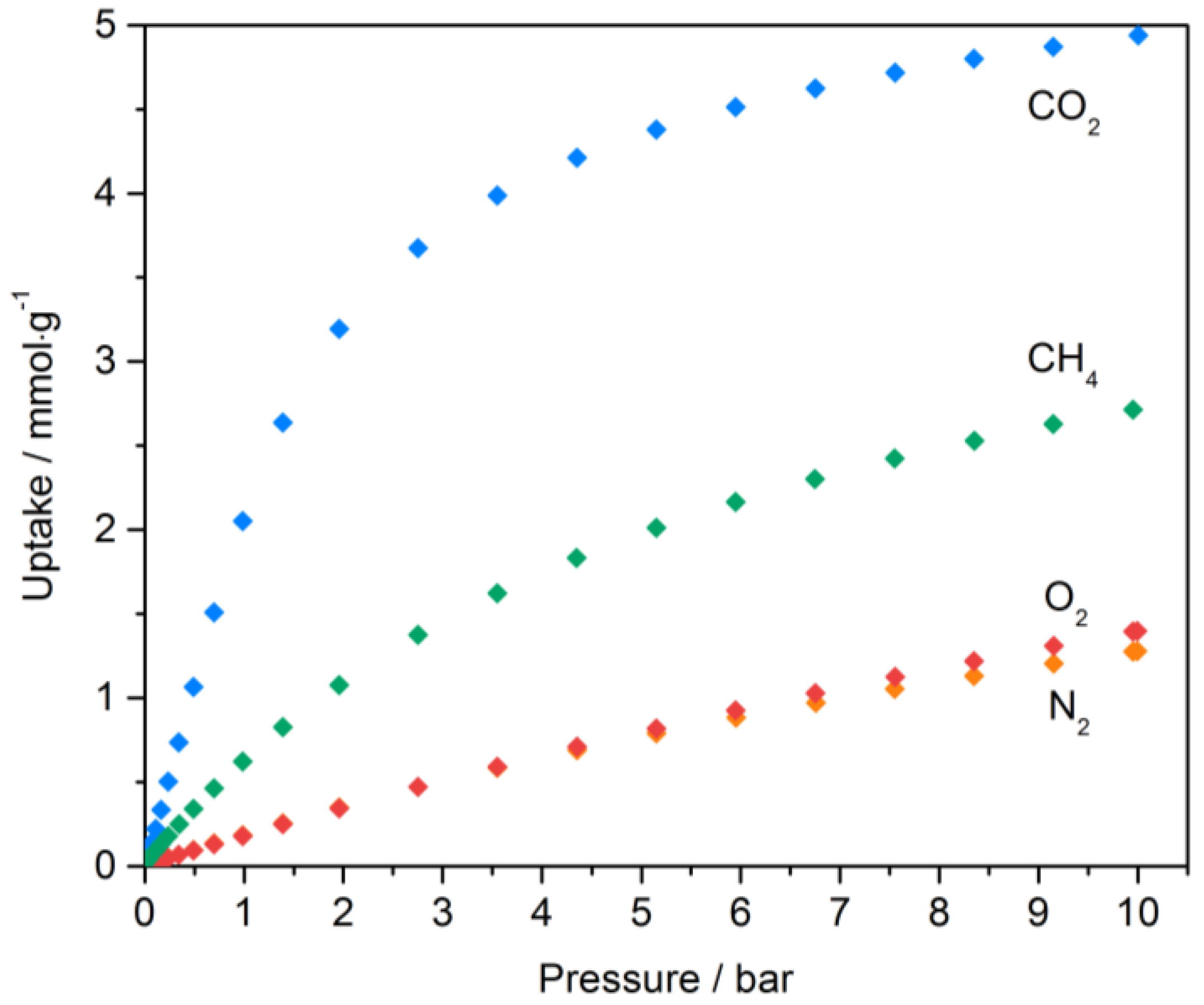
2.1. Carbon Dioxide Adsorption
2.1.1. Adsorption Isotherms
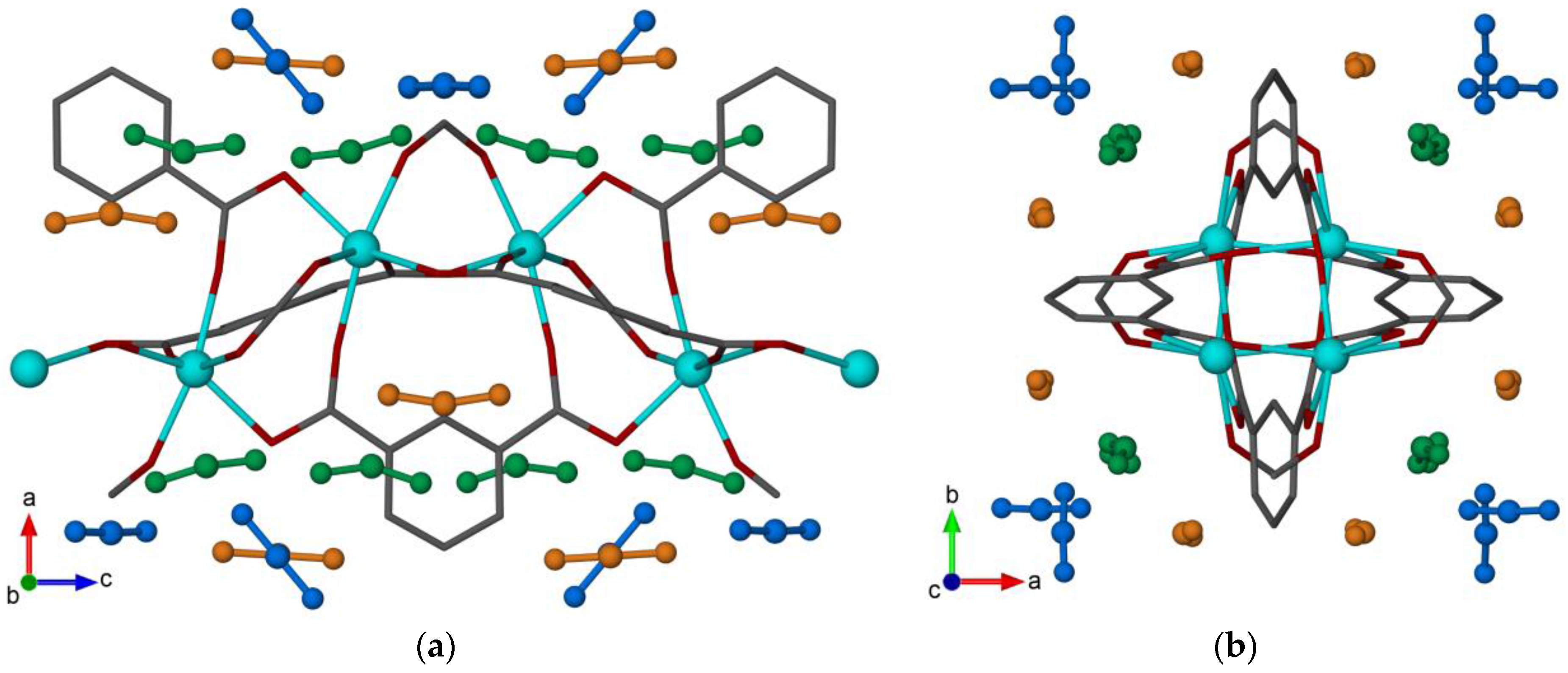
2.1.2. Binding Site Locations
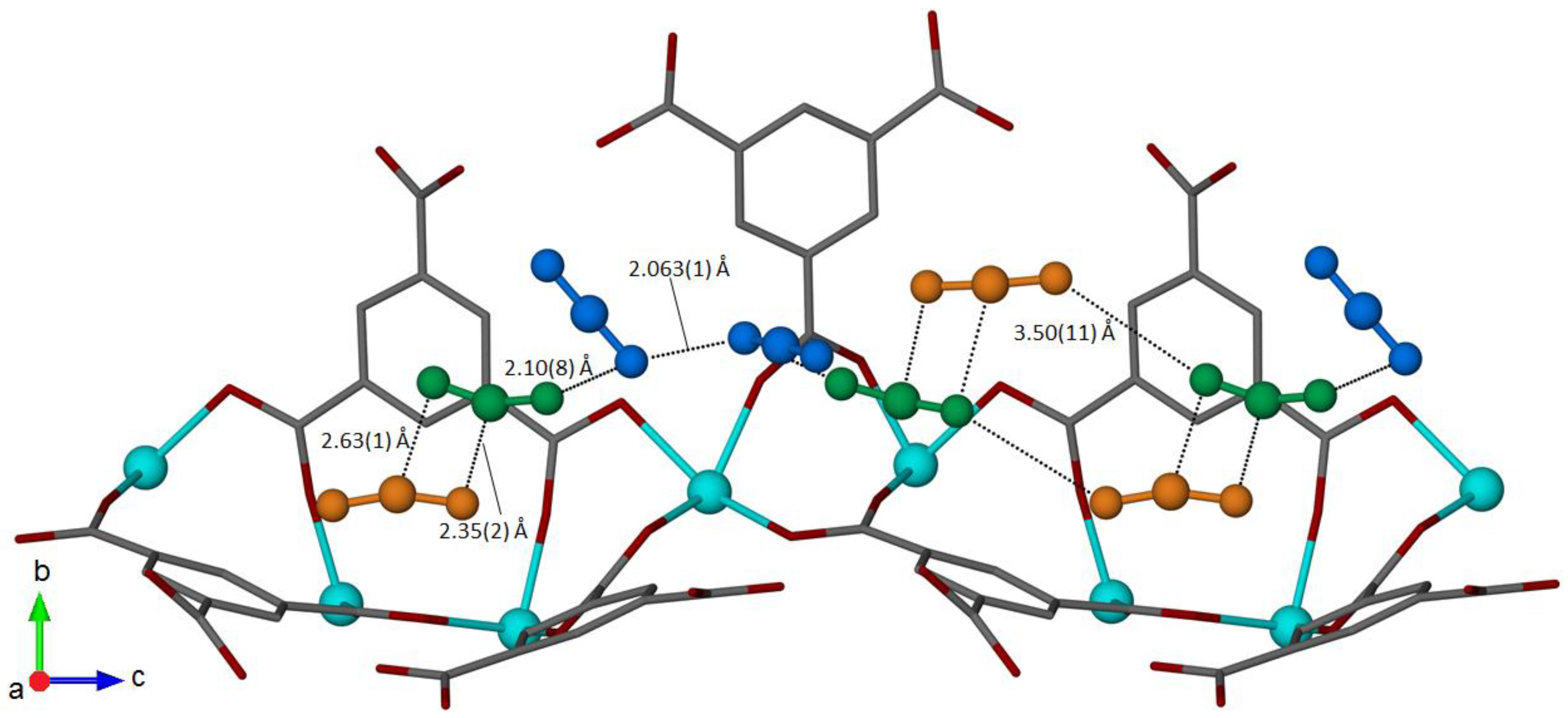
2.1.3. Intermolecular Interactions and Occupancy Restrictions
2.2. Methane and Oxygen Adsorption at High Dosage
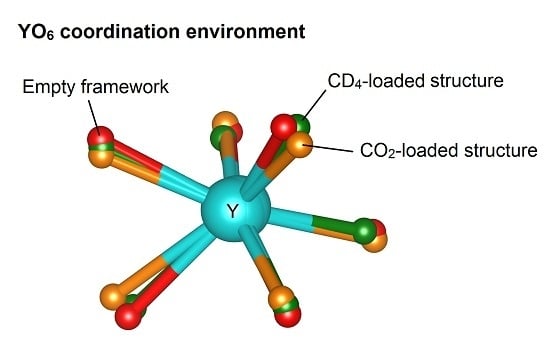
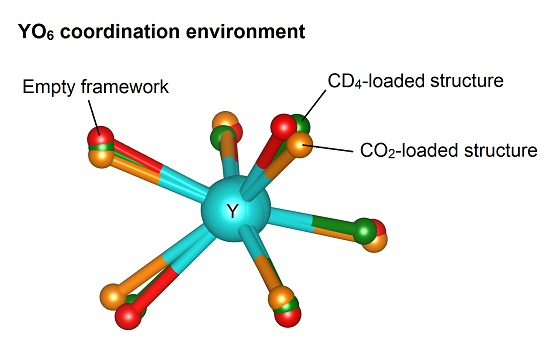
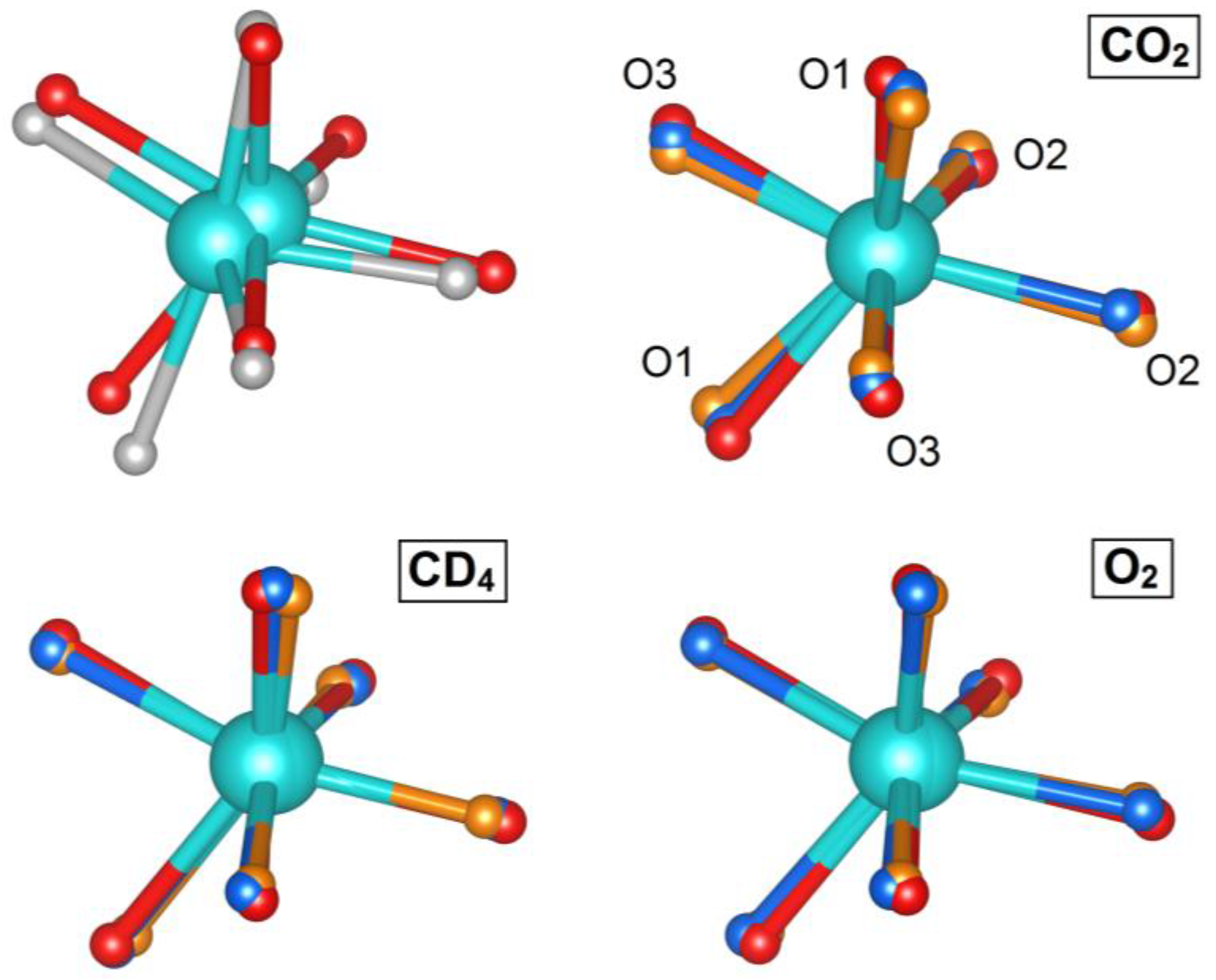
2.3. The Yttrium Coordination Sphere
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Isothermal Adsorption Measurements
4.3. Neutron Diffraction
4.4. Structural Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liu, Y.; Kabbour, H.; Brown, C.M.; Neumann, D.A.; Ahn, C.C. Increasing the Density of Adsorbed Hydrogen with Coordinatively Unsaturated Metal Centers in Metal-Organic Frameworks. Langmuir 2008, 24, 4772–4777. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhong, C. Understanding Hydrogen Adsorption in Metal-Organic Frameworks with Open Metal Sites: A Computational Study. J. Phys. Chem. B 2006, 110, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Rowsell, J.L.C.; Yaghi, O.M. Effects of Functionalization, Catenation, and Variation of the Metal Oxide and Organic Linking Units on the Low-Pressure Hydrogen Adsorption Properties of Metal-Organic Frameworks. J. Am. Chem. Soc. 2006, 128, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhou, W.; Yildirim, T. High-Capacity Methane Storage in Metal-Organic Frameworks M-2(dhtp): The Important Role of Open Metal Sites. J. Am. Chem. Soc. 2009, 131, 4995–5000. [Google Scholar] [CrossRef] [PubMed]
- Rieth, A.J.; Tulchinsky, Y.; Dincă, M. High and Reversible Ammonia Uptake in Mesoporous Azolate Metal-Organic Frameworks with Open Mn, Co, and Ni Sites. J. Am. Chem. Soc. 2016, 138, 9401–9404. [Google Scholar] [CrossRef] [PubMed]
- Karra, J.R.; Walton, K.S. Effect of Open Metal Sites on Adsorption of Polar and Nonpolar Molecules in Metal-Organic Framework Cu-BTC. Langmuir 2008, 24, 8620–8626. [Google Scholar] [CrossRef] [PubMed]
- Ming, Y.; Purewal, J.; Yang, J.; Xu, C.C.; Soltis, R.; Warner, J.; Veenstra, M.; Gaab, M.; Muller, U.; Siegel, D.J. Kinetic Stability of MOF-5 in Humid Environments: Impact of Powder Densification, Humidity Level, and Exposure Time. Langmuir 2015, 31, 4988–4995. [Google Scholar] [CrossRef] [PubMed]
- DeCoste, J.B.; Peterson, G.W.; Schindler, B.J.; Killops, K.L.; Browe, M.A.; Mahle, J.J. The effect of water adsorption on the structure of the carboxylate containing metal-organic frameworks Cu-BTC, Mg-MOF-74, and UiO-66. J. Mater. Chem. A 2013, 1, 11922–11932. [Google Scholar] [CrossRef]
- Singh, M.P.; Dhumal, N.R.; Kim, H.J.; Kiefer, J.; Anderson, J.A. Influence of Water on the Chemistry and Structure of the Metal Organic Framework Cu-3(btc)(2). J. Phys. Chem. C 2016, 120, 17323–17333. [Google Scholar] [CrossRef]
- Duyker, S.G.; Halder, G.J.; Southon, P.D.; Price, D.J.; Edwards, A.J.; Peterson, V.K.; Kepert, C.J. Topotactic structural conversion and hydration-dependent thermal expansion in robust LnMIII(CN)6·nH2O and flexible ALnFeII(CN)6·nH2O frameworks (A = Li, Na, K; Ln = La–Lu, Y; M = Co, Fe; 0 ≤ n ≤ 5). Chem. Sci. 2014, 5, 3409–3417. [Google Scholar] [CrossRef]
- Duyker, S.G.; Peterson, V.K.; Kearley, G.J.; Ramirez-Cuesta, A.J.; Kepert, C.J. Negative Thermal Expansion in LnCo(CN)6 (Ln=La, Pr, Sm, Ho, Lu, Y): Mechanisms and Compositional Trends. Angew. Chem. Int. Ed. 2013, 52, 5266–5270. [Google Scholar] [CrossRef] [PubMed]
- Duyker, S.G.; Peterson, V.K.; Kearley, G.J.; Studer, A.J.; Kepert, C.J. Extreme compressibility in LnFe(CN)6 coordination framework materials via molecular gears and torsion springs. Nat. Chem. 2016, 8, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xu, H.; Liu, Y.; Zhao, Y.; Daemen, L.L.; Brown, C.; Timofeeva, T.V.; Ma, S.; Zhou, H.C. Hydrogen Adsorption in a Highly Stable Porous Rare-Earth Metal-Organic Framework: Sorption Properties and Neutron Diffraction Studies. J. Am. Chem. Soc. 2008, 130, 9626–9627. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, S.H.; Duyker, S.G.; Southon, P.D.; Peterson, V.K.; Kepert, C.J. Host-guest adsorption behavior of deuterated methane and molecular oxygen in a porous rare-earth metal-organic framework. Powder Diffr. 2014, 29, S96–S101. [Google Scholar] [CrossRef]
- Rosi, N.L.; Kim, J.; Eddaoudi, M.; Chen, B.L.; O'Keeffe, M.; Yaghi, O.M. Rod packings and metal-organic frameworks constructed from rod-shaped secondary building units. J. Am. Chem. Soc. 2005, 127, 1504–1518. [Google Scholar] [CrossRef] [PubMed]
- D'Alessandro, D.M.; Smit, B.; Long, J.R. Carbon Dioxide Capture: Prospects for New Materials. Angew. Chem. Int. Ed. 2010, 49, 6058–6082. [Google Scholar] [CrossRef] [PubMed]
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.-H.; Long, J.R. Carbon Dioxide Capture in Metal-Organic Frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Simmons, J.M.; Liu, Y.; Brown, C.M.; Wang, X.-S.; Ma, S.; Peterson, V.K.; Southon, P.D.; Kepert, C.J.; Zhou, H.C.; et al. Metal-Organic Frameworks with Exceptionally High Methane Uptake: Where and How is Methane Stored? Chem. Eur. J. 2010, 16, 5205–5214. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Li, L.; Lin, Z.; Song, L.; Wang, Z.H.; Chen, Q.; Yang, T.; Zhou, X.H.; Xiao, H.P.; Yin, X.J. Guest-induced reversible structural transitions and concomitant on/off luminescence switching of an Eu(III) metal-organic framework and its application in detecting picric acid. New J. Chem. 2015, 39, 2289–2295. [Google Scholar] [CrossRef]
- Li, J.R.; Kuppler, R.J.; Zhou, H.C. Selective gas adsorption and separation in metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chang, Z.; Song, W.C.; Song, H.; Song, H.B.; Hu, T.L.; Bu, X.H. A Controllable Gate Effect in Cobalt (II) Organic Frameworks by Reversible Structure Transformations. Angew. Chem. Int. Ed. 2013, 52, 11550–11553. [Google Scholar] [CrossRef] [PubMed]
- Liss, K.D.; Hunter, B.; Hagen, M.; Noakes, T.; Kennedy, S. Echidna—the new high-resolution powder diffractometer being built at OPAL. Phys. B: Condens. Matter 2006, 385, 1010–1012. [Google Scholar] [CrossRef]
- Chevreau, H.; Duyker, S.G.; Peterson, V.K. Using neutron powder diffraction and first-principles calculations to understand the working mechanisms of porous coordination polymer sorbents. Acta Crystallogr. Sect. B 2015, 71, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chevreau, H.; Booth, N.; Duyker, S.G.; Ogilvie, S.H.; Imperia, P.; Peterson, V.K. Powder sample-positioning system for neutron scattering allowing gas delivery in top-loading cryofurnaces. J. Appl. Crystallogr. 2016, 49, 705–711. [Google Scholar] [CrossRef]
- Larson, A.C.; Von Dreele, R.B. General Structural Analysis System (GSAS); LAUR 86-748; Los Alamos National Laboratory Report: Los Alamos, NM, USA, 2000.
- Toby, B.H. EXPGUI, a graphical user interface for GSAS. J. Appl. Crystallogr. 2001, 34, 210–213. [Google Scholar] [CrossRef]
- Momma, K.; Izuma, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Site | Atom | Wyckoff Site | Site Symmetry | Fractional Coordinates | Fractional Occupancy | Uiso (10−2 Å2) | ||
|---|---|---|---|---|---|---|---|---|
| x (a) | y (b) | z (c) | ||||||
| ACO2 | C1a | 8d | 1 | 0.818(4) | 0.819(8) | 0.359(5) | 0.52(2) | 21.3(2) |
| O1a | 8d | 1 | 0.831(7) | 0.852(7) | 0.4347(4) | 0.52(2) | 21.3(2) | |
| O1b | 8d | 1 | 0.853(7) | 0.808(8) | 0.2850(4) | 0.52(2) | 21.3(2) | |
| BCO2 | C2a | 4a | .2. | 0.693(1) | 0 | 0.25 | 0.199(17) | 17.5(3) |
| O2a | 8d | 1 | 0.673(8) | 0.007(1) | 0.1670(2) | 0.199(17) | 17.5(3) | |
| CCO2 | C3a | 4a | .2. | 0 | 0.945(1) | 0.5 | 0.111(11) | 4.6(2) |
| O3a | 8d | 1 | 0.915(8) | 0.950(8) | 0.553(1) | 0.111(11) | 4.6(2) | |
| Guest:Y | a (Å) | c (Å) | Fractional Occupancy | Rwp (%) | |||
|---|---|---|---|---|---|---|---|
| Site A | Site B | Site C | Total | ||||
| None 1 | 10.2998(1) | 13.8635(2) | - | - | - | - | 3.46 |
| 1 CD4 1 | 10.2970(2) | 13.8581(3) | 0.39(4) | 0.274(8) | - | 0.67(5) | 4.85 |
| 2 CD4 | 10.2949(3) | 13.8432(4) | 0.740(5) | 1.006(12) | - | 1.75(6) | 5.81 |
| 1 O2 1 | 10.2974(3) | 13.8600(5) | 0.722(16) | 0.220(14) | 0.198(10) | 1.14(4) | 5.58 |
| 2 O2 | 10.29363(15) | 13.8522(2) | 1.32(2) | 0.337(14) | 0.440(12) | 2.09(5) | 3.97 |
| 1 CO2 | 10.2976(2) | 13.8639(3) | 0.52(2) | 0.199(17) | 0.111(11) | 0.86(5) | 4.85 |
| 2 CO2 | 10.2709(7) | 13.8579(6) | 0.966(12) | 0.37(2) | 0.260(11) | 1.59(4) | 7.04 |
| Guest:Y | O1–Y–O1 (°) | Change from Dehydrated (%) | O3–Y–O3 (°) | Change from Dehydrated (%) |
|---|---|---|---|---|
| Hydrated | 167.9(3) | - | 137.6(3) | - |
| Dehydrated | 139.3(5) | - | 120.8(5) | - |
| 1 CO2 | 143.1(11) | 3.8 | 115.9(8) | −4.9 |
| 2 CO2 | 143(3) | 3.7 | 115.7(19) | −5.1 |
| 1 CD4 | 141.9(10) | 2.6 | 114.3(8) | −6.5 |
| 2 CD4 | 147.2(11) | 7.9 | 114.6(9) | −6.2 |
| 1 O2 | 141.9(7) | 2.6 | 117.3(6) | −3.5 |
| 2 O2 | 147.5(8) | 8.2 | 121.4(6) | 0.6 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auckett, J.E.; Ogilvie, S.H.; Duyker, S.G.; Southon, P.D.; Kepert, C.J.; Peterson, V.K. Flexible Yttrium Coordination Geometry Inhibits “Bare-Metal” Guest Interactions in the Metal-Organic Framework Y(btc). Energies 2016, 9, 836. https://doi.org/10.3390/en9100836
Auckett JE, Ogilvie SH, Duyker SG, Southon PD, Kepert CJ, Peterson VK. Flexible Yttrium Coordination Geometry Inhibits “Bare-Metal” Guest Interactions in the Metal-Organic Framework Y(btc). Energies. 2016; 9(10):836. https://doi.org/10.3390/en9100836
Chicago/Turabian StyleAuckett, Josie E., Stephen H. Ogilvie, Samuel G. Duyker, Peter D. Southon, Cameron J. Kepert, and Vanessa K. Peterson. 2016. "Flexible Yttrium Coordination Geometry Inhibits “Bare-Metal” Guest Interactions in the Metal-Organic Framework Y(btc)" Energies 9, no. 10: 836. https://doi.org/10.3390/en9100836
APA StyleAuckett, J. E., Ogilvie, S. H., Duyker, S. G., Southon, P. D., Kepert, C. J., & Peterson, V. K. (2016). Flexible Yttrium Coordination Geometry Inhibits “Bare-Metal” Guest Interactions in the Metal-Organic Framework Y(btc). Energies, 9(10), 836. https://doi.org/10.3390/en9100836