1. Introduction
Gas hydrate formation has been an important subject in the petroleum industry, because when gas hydrate forms in oil and gas pipelines and processing equipment, it plugs them. This would cause flow assurance issues, and may create operational and safety contingencies that may lead to huge economic losses, potential environmental risk due to pollution, and safety hazards to operational personnel in the case of pipeline rupture [
1,
2,
3,
4].
More recently, there has been increased interest in the potential applications of gas-hydrate-related technology in the areas of gas separation [
5,
6,
7,
8,
9,
10,
11], gas transportation and storage [
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23], CO
2 capture and sequestration [
11,
13,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30], and as a potential energy source [
20,
31,
32,
33,
34]. In the area of gas transportation and storage, gas hydrate technology offers simplicity and safer conditions. Compared with building new pipelines and railway systems for transportation, which are expensive and labor-intensive, or storing the gas as compressed natural gas (CNG, requiring very high pressures; 20–25 MPa) and liquefied natural gas (LNG, requiring cryogenic temperatures below −161 °C), which are also capital-intensive and have a very high safety demand [
35], CO
2 capture and sequestration would contribute to a greener environment, while gas hydrates as an energy source will play a significant role in the transition to cleaner energy sources. To implement these gas-hydrate-related technologies at an industrial scale so that they can compete with existing technologies, emphasis must be placed on forming hydrates in a fast and efficient manner. This requires an improved understanding of the factors that influence the formation kinetics of gas hydrates. A brief review of some of these factors follows.
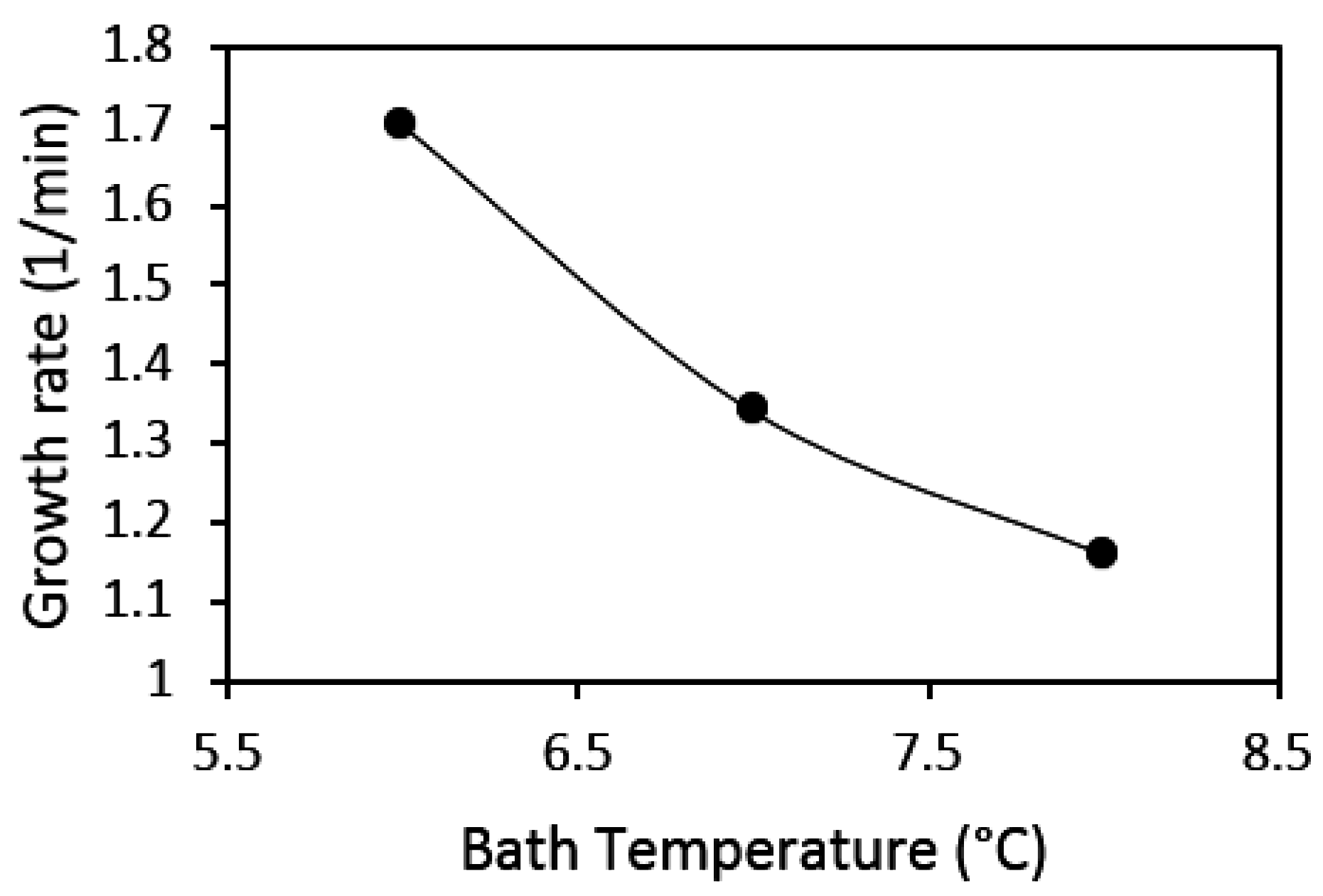
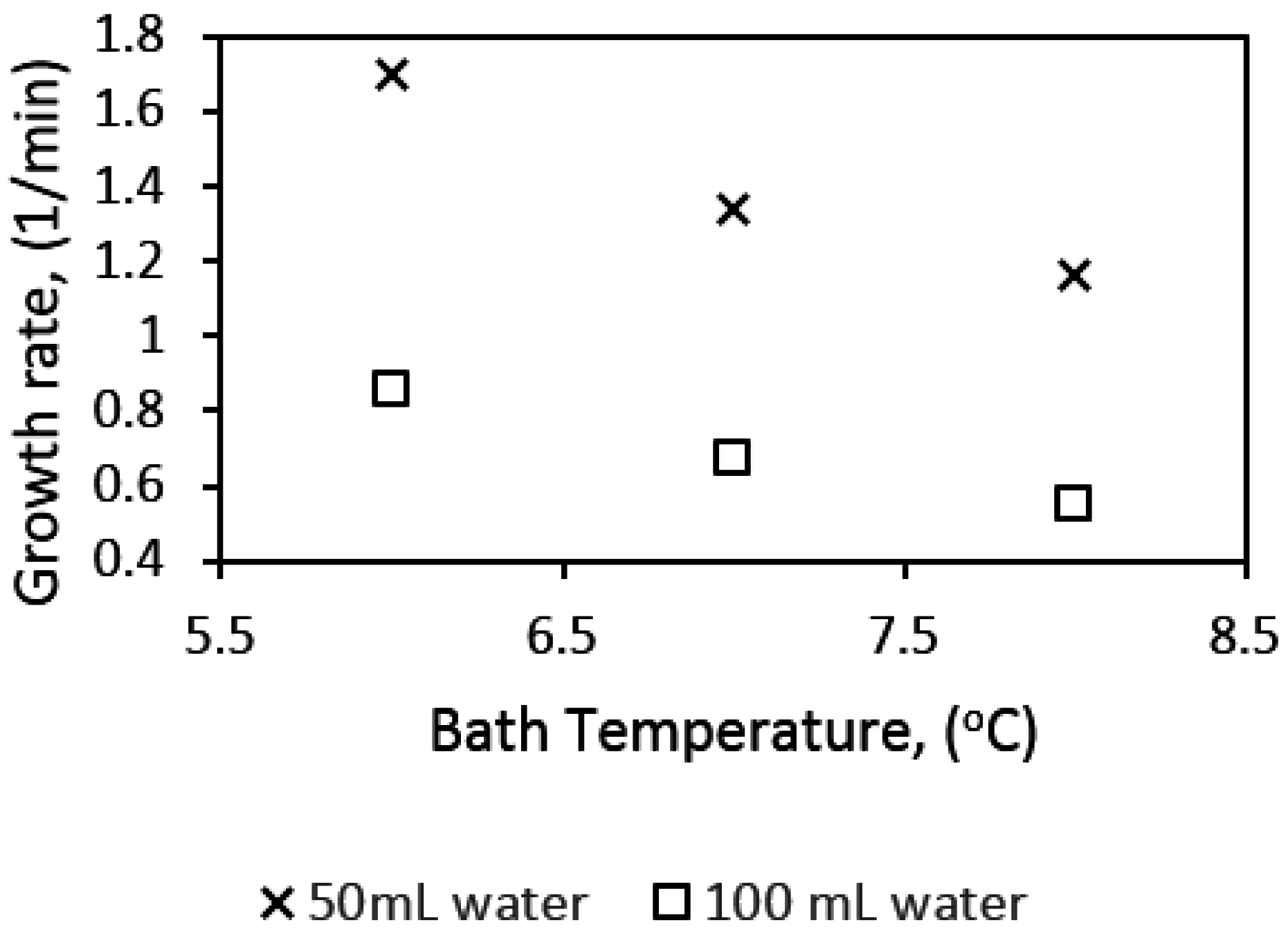
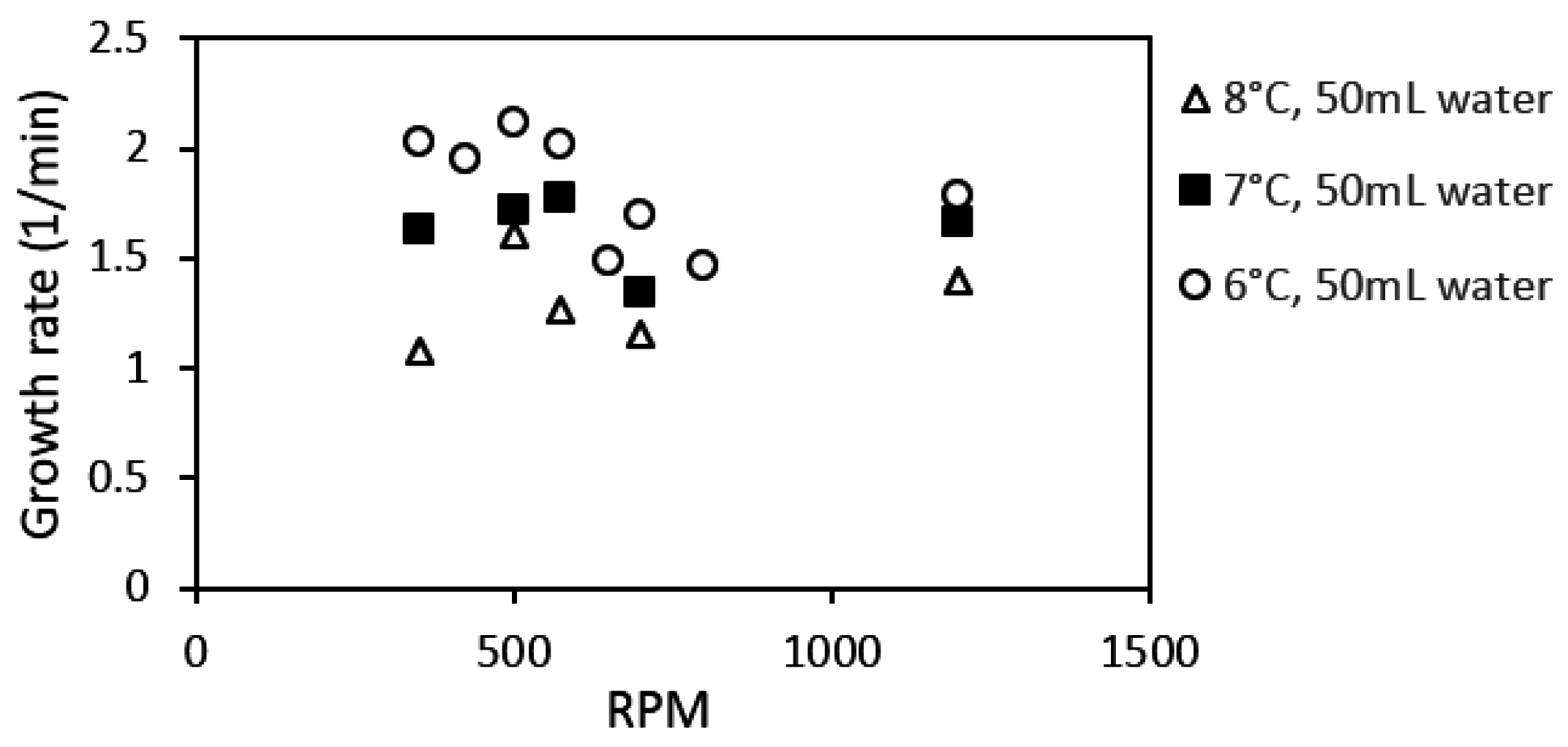
Factors that affect the kinetics of gas hydrate formation, such as temperature, pressure, driving force, agitation, and the concentration of guest and water phases, have been extensively studied [
4,
36,
37,
38,
39]. Some research findings have suggested that gas hydrate formation increases with decreasing system temperature, though an increase in pressure would promote hydrate growth rate; a unit increase in pressure has a less significant effect on gas hydrate growth rate at constant temperature [
4,
37,
38,
39,
40]. On the other hand, the combined effect of temperature and pressure on gas hydrate formation is coupled into a parameter called the “driving force” of gas hydrate formation. Models and correlations of gas hydrate nucleation and growth have presented the driving force as the fugacity, concentration, chemical potential, temperature differences at equilibrium, experimental conditions, and supersaturation of the system [
39,
41,
42,
43,
44]. Arjmandi et al. [
41], went on to prove that the temperature difference or subcooling (ΔT = T
eq − T
exp) can solely represent the driving force for pure component hydrate forming systems at given pressure conditions. Using the temperature difference as the driving force offers a simple option in modeling gas hydrate nucleation and growth. From previous works, and as we will come to see from this work, gas hydrate nucleation and growth rates increase with an increase in driving force/subcooling. However, in the Ph.D. work by Mork, it was suggested that increasing subcooling did not increase the gas hydrate growth rate [
45].
Another parameter crucial to gas hydrate formation is the degree of agitation of the reacting mixture or stirring rate. During stirring, more of the hydrate-forming guest molecules are brought in contact with the liquid bulk. This, along with an increase in the amount of dissolved gas, promotes gas hydrate formation. As various publications have shown, hydrate growth rate tends to increase with stirring [
39,
40,
46,
47,
48], though the effective gain in hydrate formation rate diminishes with each subsequent increase in stirring rate [
40].
Several works have also indicated that the reactor design, reactor type, and hydrate formation technique also play important roles in fast and efficient hydrate formation [
9,
14,
35,
49]. At industrial scale, energy efficiency and cost effectiveness are crucial measures of the feasibility of a process. Thus, an optimal combination of these factors in reactor design, for developing new gas-hydrate-related technologies, would hinge on gains in hydrate formation rate compared with the additional energy demands caused by increased pump rate, agitation rate, subcooling, or gas compression demands. As a way of reducing compression demands, Linga et al. [
50,
51] and Kumar et al. [
30] have used very low concentrations of additives like tetrahydrofuran (THF) and propane, which reduce the operating pressure for hydrate-based CO
2 capture applications. However, they noted that the use of these additives reduced the rate of hydrate formation. Other works have proposed to overcome the gas-water contact limitations without stirring, by contacting the gas phase with water dispersed in the pores of silica gel to form hydrate [
24,
28,
52,
53], or by the use of cyclopentane as an additive, which improves gas diffusion across the gas-water interface in an unstirred reactor [
9]. Linga et al. also proposed a reactor design with a second impeller close to the gas–liquid contact level in the reactor that enabled enhanced recirculation of the gas from the gas phase of the crystallizer, thereby improving the gas–liquid mass transfer after hydrates form [
54].
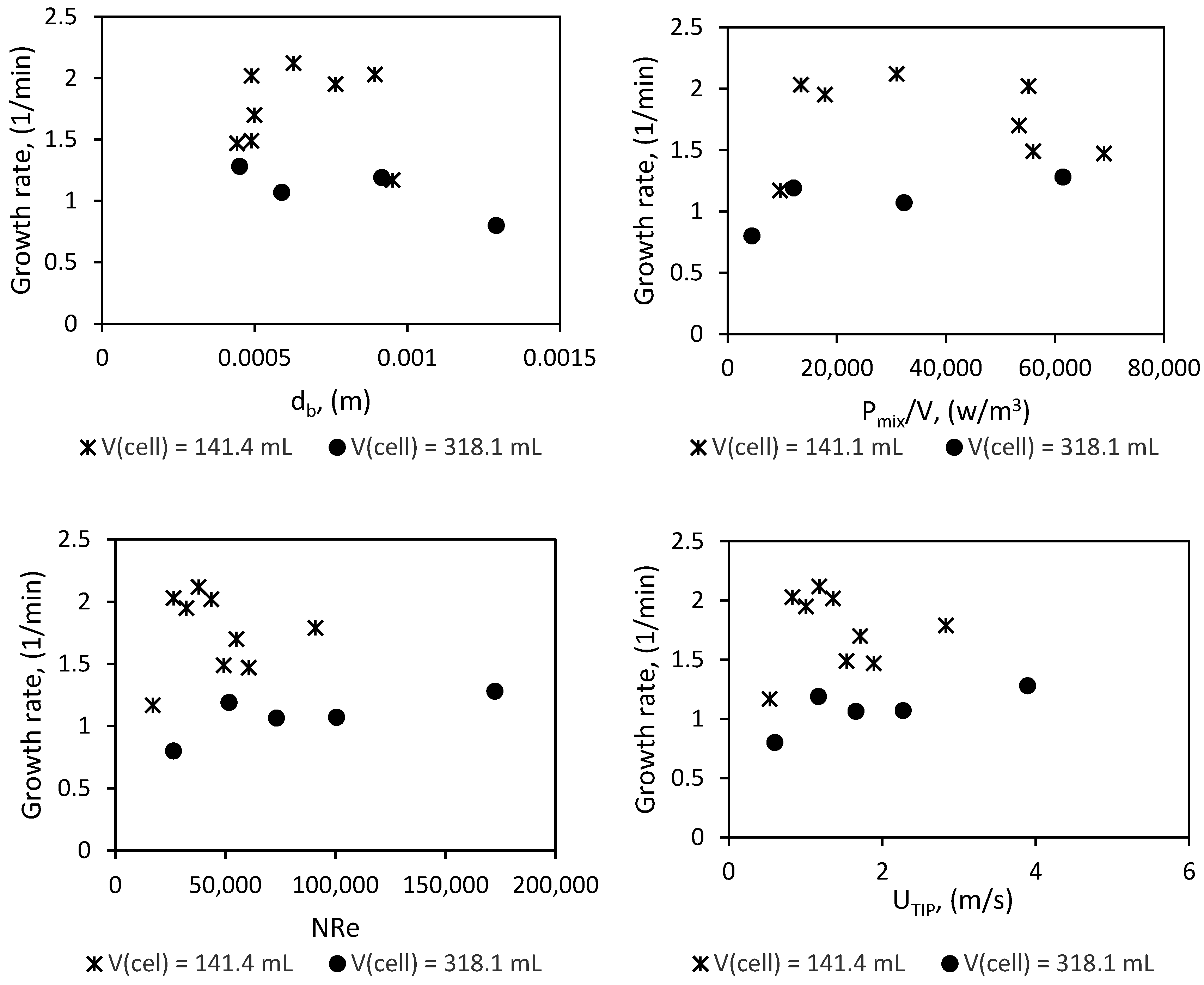
In this work, experiments were conducted in two different sized high-pressure semi-batch autoclave reactors with geometric similarity, at a pressure of 90 bars. We have carefully analyzed the effect of temperature, subcooling, water content, stirring rate, and reactor size on the growth kinetics of methane hydrate. Our findings show that system temperature, water content, and reactor size have a considerable impact on the growth rate of gas hydrates. The effect of stirring rate plateaus once the system attains heat and mass transfer equilibrium; furthermore, the effect of reactor size scale-up seems to be more a function of the specific thermal conductance of the system than the power input per unit volume of liquid into the system.
2. Experimental Section
All experiments were conducted with distilled water and 99.9995 purity scientific grade methane gas (Methane 5.5).
2.1. Experimental Setup
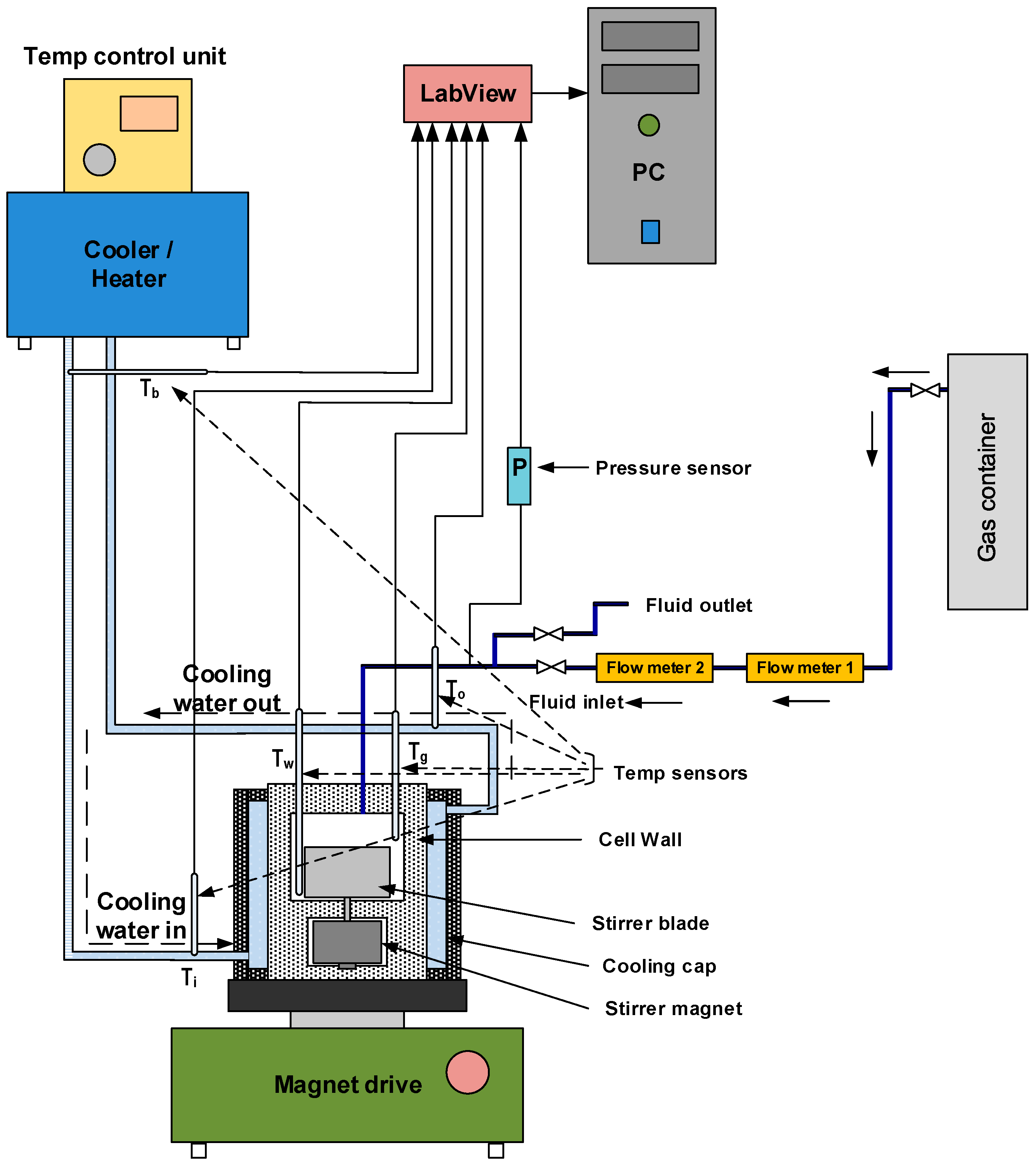
The growth kinetics of methane hydrate was studied in titanium autoclave reactors of two sizes, 142.4 mL and 318.1 mL. A schematic diagram of the experimental setup is shown in
Figure 1, and
Table 1 gives details of the geometry of the autoclave reactors and their component parts. The autoclave cell has an upper chamber where the reaction takes place, and a lower chamber where the stirrer magnet is seated. A single flat blade impeller is used. The impeller was screwed on to the stirrer magnet placed in the bottom chamber through a channel from the upper chamber. Gas was supplied by a gas bottle charged up to 200 bars, while the pressure in the cell was set to 90 bars during the experiments. The overpressure in the gas bottle ensured that the pressure in the cell remained within ±2 of 90 bars during hydrate growth. The gas was supplied to the cell through an opening in the top lid, via a mass flow meter. Depending on the stirring rate and the level of mixing of the gas and liquid in the cell, the gas supplied during the hydrate growth may make contact only with the gas phase, or with the gas-liquid mixture.
A Bronkhorst HIGH-TECH flow meter (BRONKHORST High-Tech B.V., Ruurlo, The Netherlands) is connected in the line between the gas container and the reactor cell for measuring gas flow rate into the cell during hydrate growth. Two 1/10 DIN Pt-100 temperature sensors (MRC Teamtrade AS, Dpt. Hyp Teck, Skotselv, Norway) (accuracy ± 0.03 °C) are installed through the top lid of the cell to enable temperature monitoring in the gas and bulk phase during experiments. Pressure monitoring is enabled using a Rosemount 3051TA absolute pressure transmitter (Emerson Electric Co., St. Louis, MO, USA) connected to the line along the inlet to the cell. To enable circulation of the cooling fluid, a coolant jacket envelops the cell body. Cooling and temperature control are enabled using a Julabo F34-HL refrigerating/heating circulator (JULABO GmbH, Seelbach, Germany). The cooling fluid used is water. The stirring rate is controlled using a magnetic stirrer drive (REO basic C) with an rpm sensor.
2.2. Experiment Runs
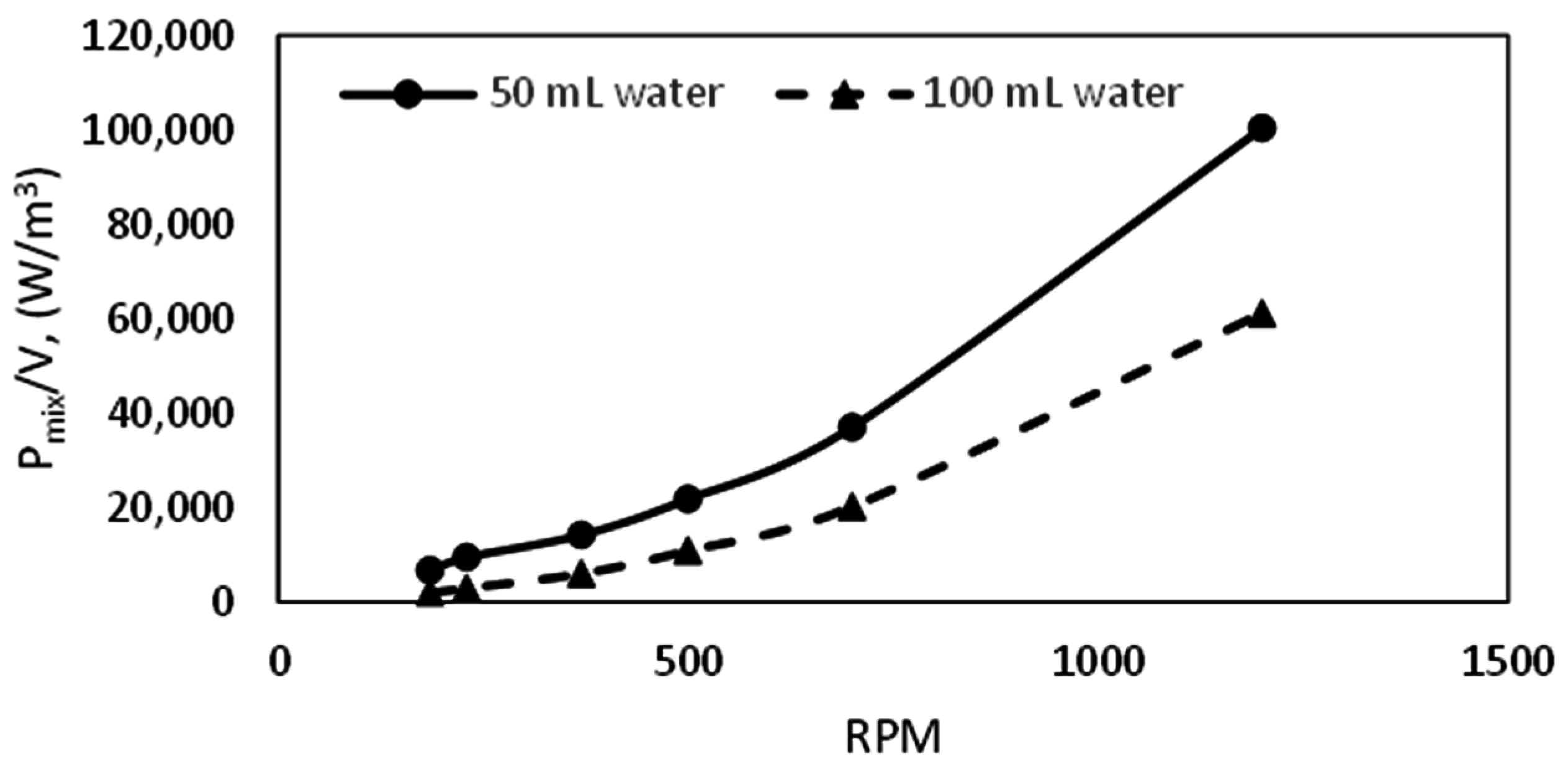
All experiments were run at a pressure of 90 bar. The cell content was cooled down at a constant cooling rate of 3 °C/h from an initial temperature of 13.5 °C to the required experimental temperature. System pressure was maintained constant at 90 bar within a deviance of ±2 bar, by adding “fresh” methane from the gas container through the flow meter. We ran experiments in the 141.4 mL cell at temperatures of 8, 7, and 6 °C, and at stirring rates of 350, 425, 500, 575, 650 700, 800, and 1200 rpm. Experiments in the 318.1 mL cell were at 8, 7, and 6 °C, and at stirring rates of 185, 370, 500, 700, and 1200 rpm. The experiments run in the 318.1 mL (large) cell allowed us to analyze scale-up effects on the growth kinetics. The water content used was 50 mL and 100 mL for the small cell, and 112.5 mL and 225 mL for the large cell for parallel experiments.
3. Theoretical Background
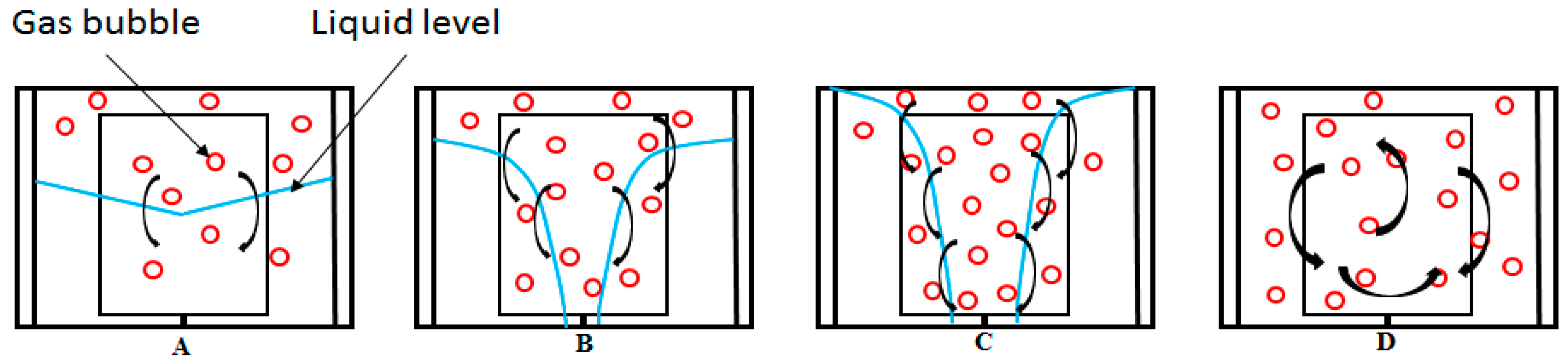
Gas hydrate formation involves coupled heat and mass transfer [
4]; therefore, in our effort to describe hydrate growth behavior, we see the importance of employing parameters that characterize the mass and heat transfer of our system. In stirred reactors, the gas disperses into the liquid in the form of bubbles. When gas hydrates form, the system will consist of gas bubbles and hydrate crystals, dispersed in a continuous water phase. The existence of three phases implies that there exists a gas–liquid interface between the gas and liquid phases, and a crystal–solution interface between the liquid phase and the hydrate crystal. Furthermore, the two-film theory suggests that there are film layers on the gas and liquid sides of the gas-liquid interface [
55]. Gas transport occurs through these film layers into the bulk liquid phase. Heat and mass transfer resistance are considered negligible on the gas side film layer, for sparingly soluble gases like methane [
56], thus gas transport into the liquid bulk is controlled by mass transfer of gas molecules across the liquid side film layer at the gas-liquid interface. Similarly, crystal growth theories suggest a two-step model for the hydrate crystal growth; first gas transport occurs from the liquid bulk across a hypothetical stagnant film to the crystal-solution interface, then gas molecules are included into the hydrate structure [
57,
58,
59].
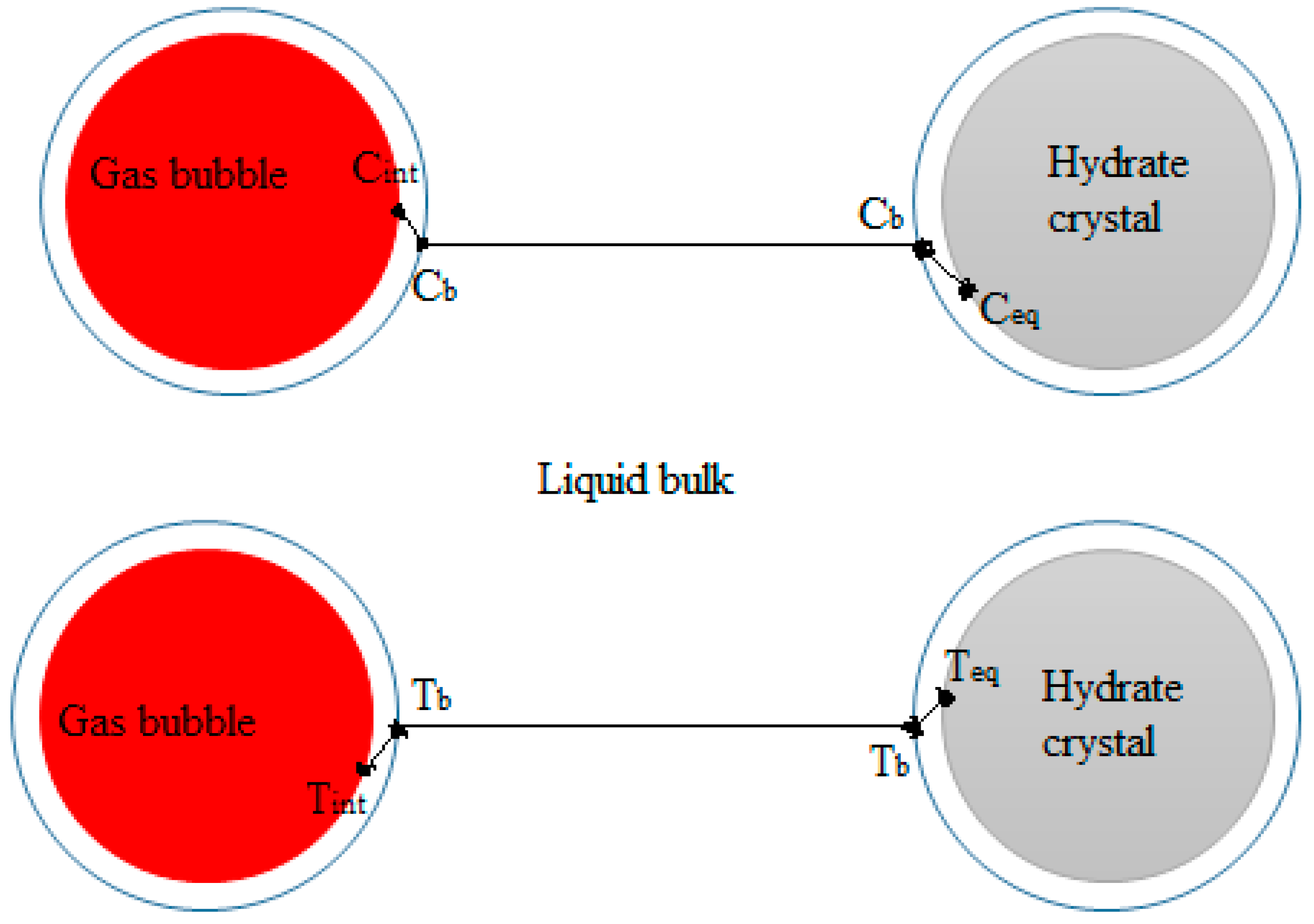
Figure 2 shows a schematic illustration of gas-liquid and crystal-solution interfaces for mass transfer of gas through the bulk phase to the crystal surface in a hydrate forming system. At the gas–liquid interface, the gas and liquid phases are at equilibrium at the system temperature and pressure. There is a drop in concentration of the gas from the gas–liquid interface across the liquid side film layer to the liquid bulk from C
int to C
b. Also, the temperature rises across the liquid side film layer due to gas dissolution from T
int to T
b. At the crystal-solution interface the gas is at the hydrate equilibrium conditions. The concentration of the gas drops to its value at the hydrate equilibrium conditions, C
eq, and the temperature increases to the hydrate equilibrium temperature, T
eq. The concentration and temperature gradients serve as driving forces for the transport of gas. Accordingly, the hydrate crystal growth rate has been modeled based on gas diffusion from the bulk phase to the crystal surface, e.g., by Englezos et al. [
38] and others [
60,
61]. Skovborg and Rasmussen [
62] have, on the other hand, modeled gas hydrate growth based on the mass transport of gas across the gas-liquid interface. The gas to liquid volumetric mass transfer coefficient (k
La) provides indispensable information in describing gas–liquid mass transfer processes; and is an intricate part of mass-transfer-based modeling attempts of gas hydrate growth [
62,
63].
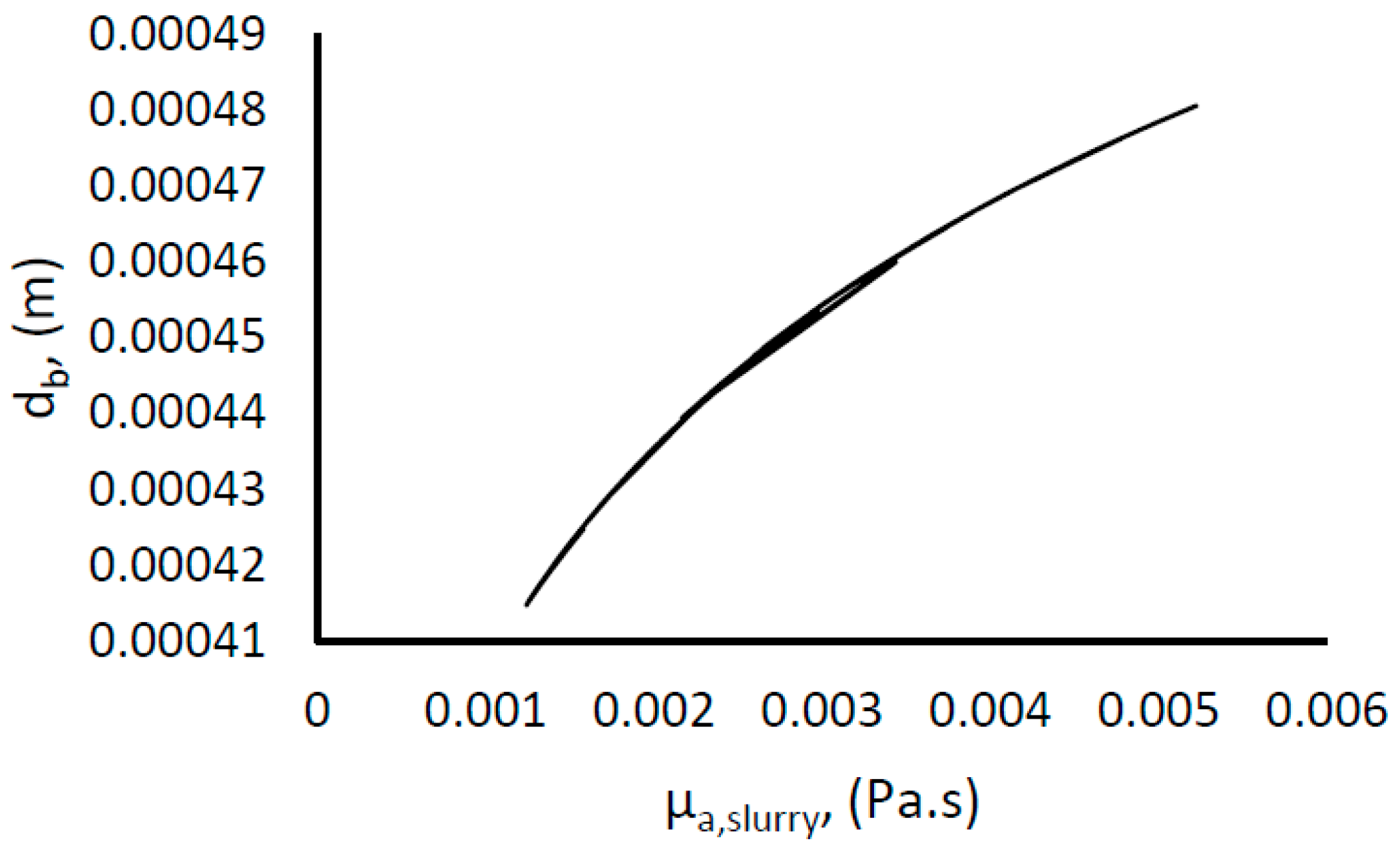
The mean bubble diameter and concentration of gas in the liquid phase (gas fraction), and the degree of dispersion of gas bubbles in the liquid will affect the volumetric gas liquid mass transfer coefficient. The degree of dispersion of the gas bubbles within the liquid phase is defined by the rate of turbulence energy dissipation, which is a function of the power input of the system. Though there are established techniques for measuring or estimating the gas fraction, and the mean bubble diameter, which are in turn used for the estimation of the gas–liquid contact area; we are unable to measure them directly in our lab facilities. However, there are some well-established correlations proposed by other researchers, which may be used to estimate these parameters [
64].
The volumetric mass transfer coefficient is a product of two parameters, the gas-liquid mass transfer coefficient (k
L), and the gas-liquid specific contact area (a). One can estimate the gas–liquid mass transfer coefficient if the gas-liquid phase diffusion coefficient and the density of the liquid phase are known. On the other hand, the gas–liquid specific contact area is a function of the gas fraction or gas hold-up, and the mean bubble diameter of the gas dispersed into the liquid phase. The simplest correlation for the gas–liquid specific contact area is generally given as:
where φ is the gas hold-up, and d
b is the mean bubble diameter.
Saravanan et al. [
65] measured the gas hold-up in a stirred tank by direct visual measurement of the increase in the height of the liquid column with aeration. The gas hold-up was calculated as φ = (H
G − H)/H
G. Where H
G is the liquid column height with aeration, and H is the liquid column height without aeration. Saravanan et al. also proposed a correlation for the gas hold-up as φ = a (P/V)
b (V
G)
c, where P is the power consumed by the impeller in aerated conditions (W), V is the volume of liquid in the vessel (m
3), V
G is the gas superficial velocity (m/s), and a, b, and c are constants that may be obtained from linear regression of experimental data. Their comparison of the experimental measurements to estimates from the correlations gave deviations within 15%. It is reasonable to expect errors in measurements using this technique due to fluctuations in the liquid surface from varying flow patterns and turbulence during stirring. A number of optical imaging techniques as well as computational fluid dynamics (CFD) simulations have also been used for estimating the gas hold-up, mean bubble diameter, and the gas and liquid phase velocities [
66,
67,
68,
69]. Yang et al. [
66] performed gas–liquid–solid dispersion studies using CFD simulations and compared with experimental measurements in a stirred reactor. They expressed the gas and liquid phase velocity in the axial and radial directions as functions of the velocity of the impeller tip (U
TIP) in good agreement with the experimental data. The average liquid phase velocity in the radial direction was about 0.25 U
TIP, in the axial direction it was about 0.07 U
TIP, while that of the gas phase velocity in the radial direction was about 0.2 U
TIP. Chung et al. also reported average gas phase velocities in the range of 0.03–0.2 U
TIP in the radial direction, and 0.02–0.11 U
TIP in the axial direction [
68]. The velocities were also higher within the impeller region, reaching a maximum value around the tip of the impeller, and decreases towards the axis of rotation of the impeller and the reactor wall [
66,
67,
68,
69]. The gas hold-up in the impeller region was as high as 0.1–0.2, but decreased as we moved from the impeller in the radial direction to almost zero near the reactor wall. The mean bubble size in Yang et al.’s simulations was based on the model by Zhang et al. [
70], which estimates the bubble diameter with consideration of the influence of the turbulence dissipation rate in the liquid phase:
where σ
lg is the surface tension, ρ
l is the density of the liquid phase, and ε
l is the liquid phase turbulence eddy dissipation.
Both the simulation results and those of Chung et al. showed a very small size distribution of the bubbles in the impeller region (0.0008–0.0024 m) compared with the bulk region (0.003–0.005).
Several semi-empirical correlations have also been used to estimate gas hold-up, mean bubble size, and the gas–liquid specific interfacial area. However, Garcia-Ochoa and Gomez [
64] proposed a theoretical approach for predicting these parameters, using equations proposed by different scientists to calculate the gas hold-up, mean bubble size, and the gas–liquid specific interfacial area. The following equations were used by Garcia-Ochoa and Gomez [
64].
3.1. Liquid Phase Mass Transfer Coefficient (kL)
For non-Newtonian fluids that obey the power law viscosity model,
where D
L is the liquid phase diffusion coefficient, k is a consistency index, and n is a flow index.
For non-Newtonian fluids that obey the Casson viscosity model,
where μ
c is the Casson viscosity and α
r is the apparent yield stress to shear stress ratio.
3.2. Gas Hold-Up
A modified equation for the gas hold-up based on the derivation by Kudrewizki [
71] using isotropic turbulence theory as follows:
If the liquid phase is viscous, a modification is made to the equation to account for viscous forces:
where μ
L is liquid viscosity and μ
G is gas viscosity.
3.3. Mean Bubble Diameter
The mean bubble diameter is given by [
72]:
3.4. Volumetric Mass Transfer Coefficient, kLa
The volumetric mass transfer coefficient (kLa) was calculated by multiplying kL and a. Garcia-Ochoa and Gomez’s simulation results of kLa by this approach were found to fit the experimental data from different reactor sizes (2 L–500 L), and impeller blade configurations. Simulations for non-Newtonian fluids required the use of non-Newtonian viscosity models, with the Casson viscosity model giving a better fit with the experimental data.
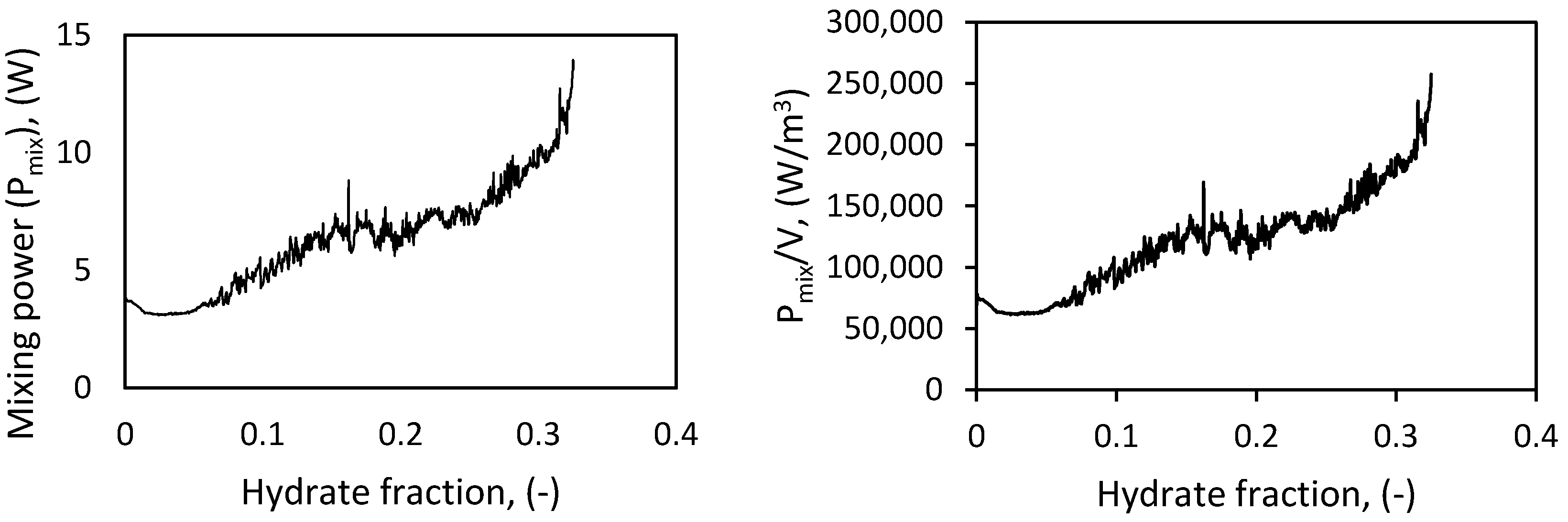
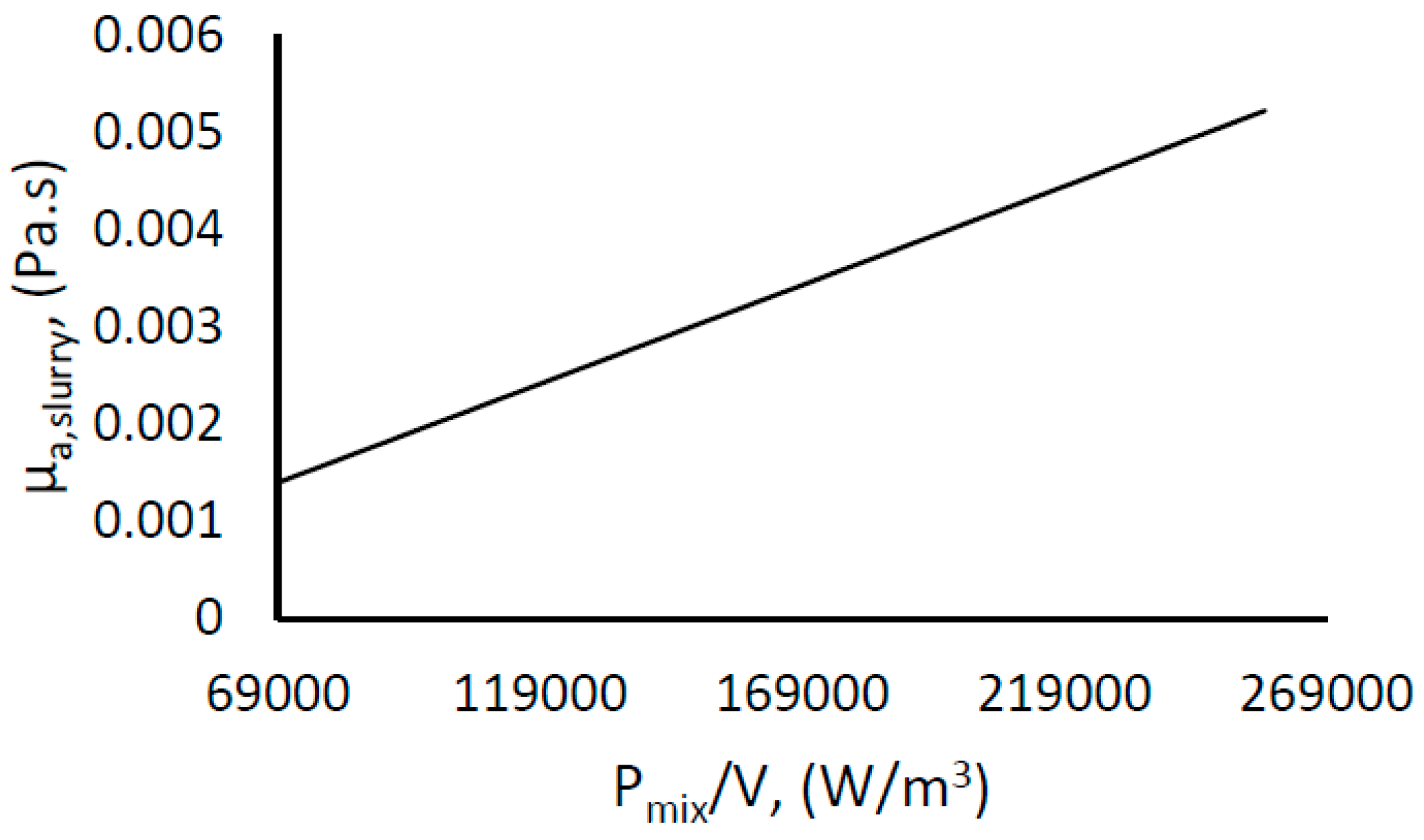
3.5. Rheological Properties of the Hydrate-Water Slurry
In gas hydrate systems, as gas hydrates form, the viscosity and density of the hydrate–water slurry will change with hydrate concentration. While the slurry viscosity increases, the slurry density will decrease with increasing hydrate concentration [
73]. In Andersson and Gudmundsson’s work on the rheological properties of gas hydrate–water slurry [
74], they could describe the viscosity of gas hydrate–water slurry with the Bingham viscosity model for up to 12% hydrate concentration. However, at higher concentrations, the power-law viscosity model better described the gas hydrate–water slurry viscosity. The following relation relates the power input per unit volume of fluid in the cell (P/V) to the shear stress (τ) and the shear rate (γ) [
75]:
For water, which is a Newtonian fluid, the viscosity is equal to the ratio of the shear stress to the shear rate:
Combining Equations (8) and (9) gives the shear rate as
The subsequent apparent viscosity of the hydrate–water slurry when hydrate forms will be
which, when combined with Equations (8) and (10), gives
The density of the hydrate slurry is estimated as a volume-weighted average of the hydrate and water phases as
4. Data from a Typical Experimental Run
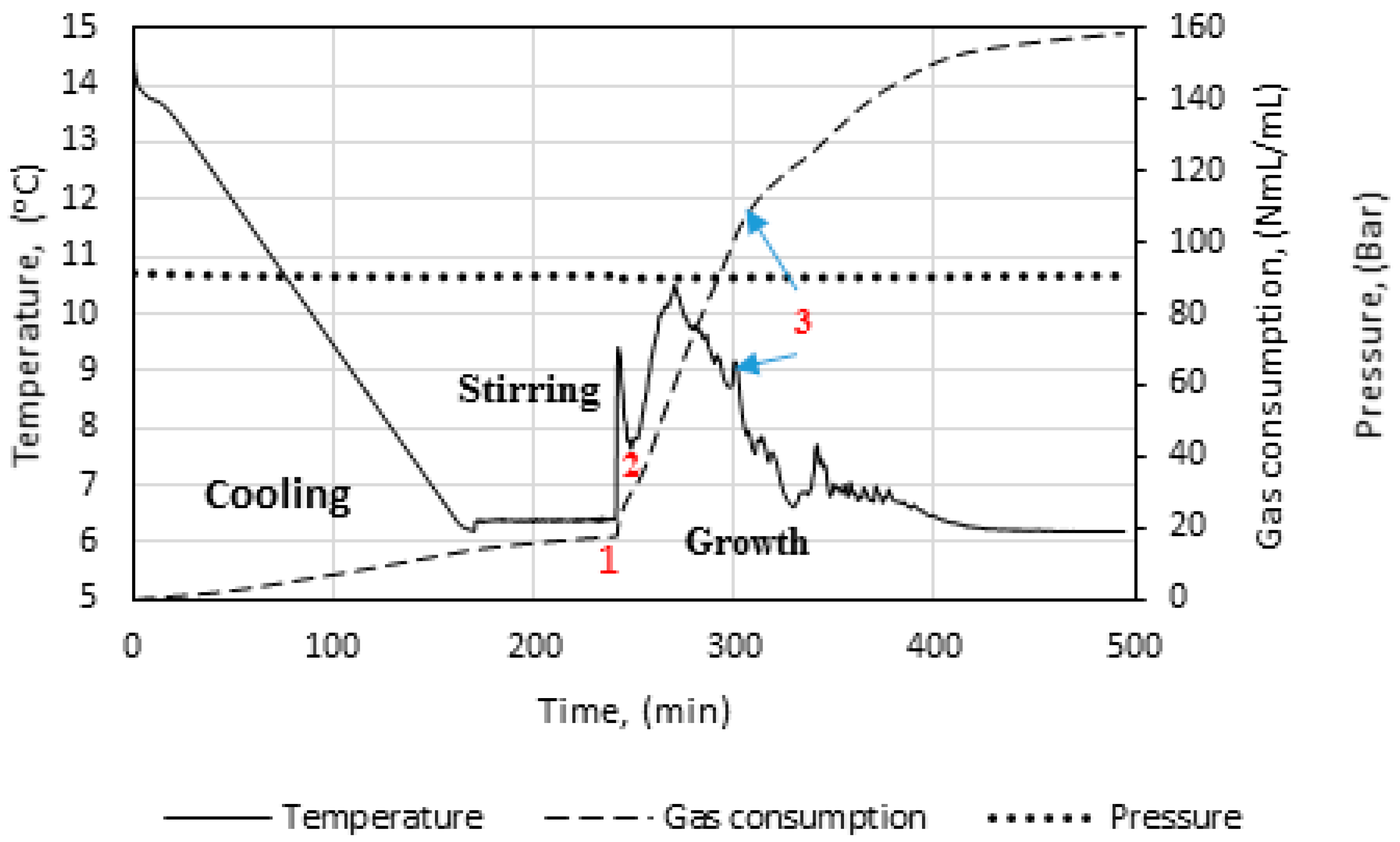
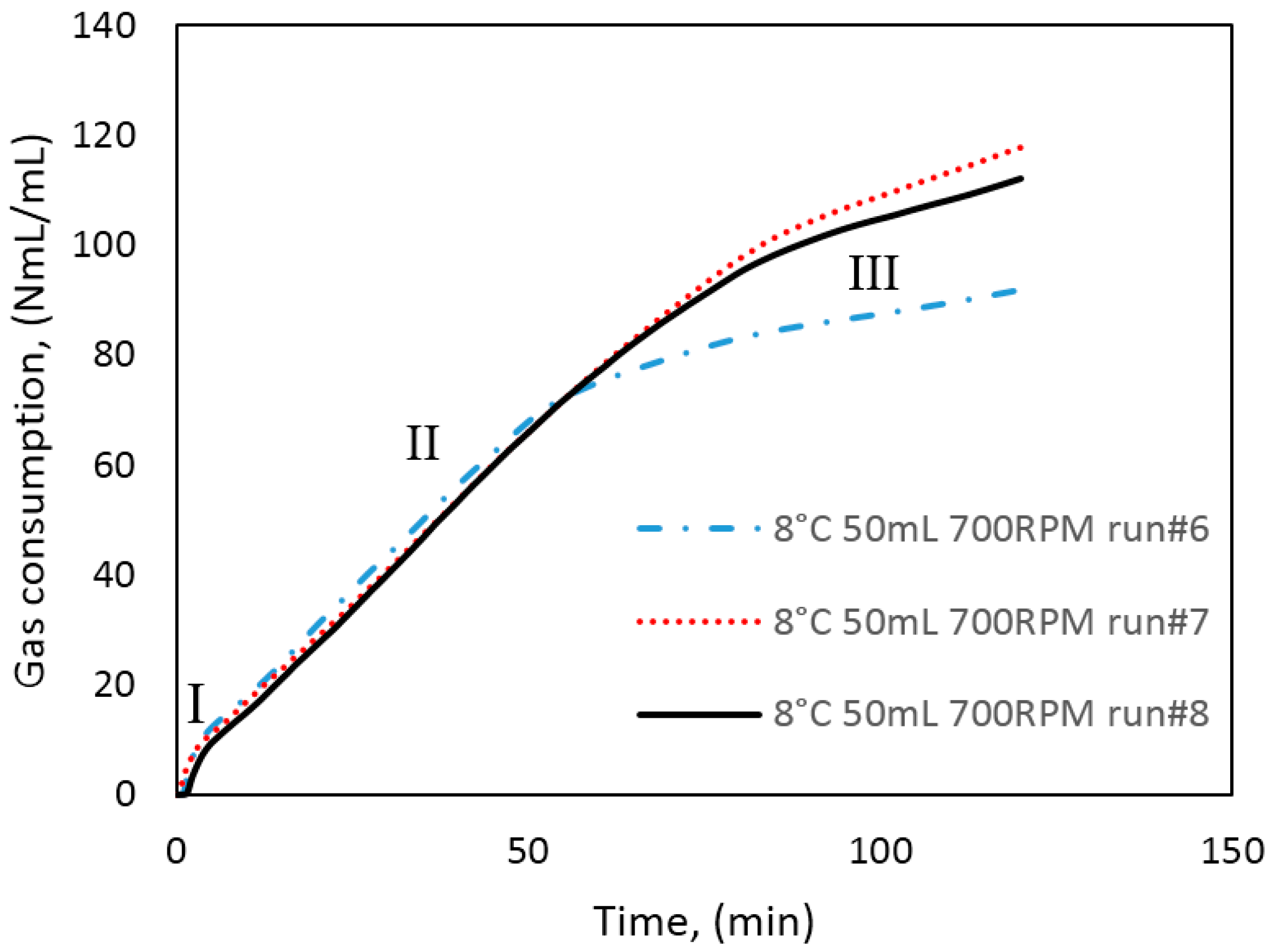
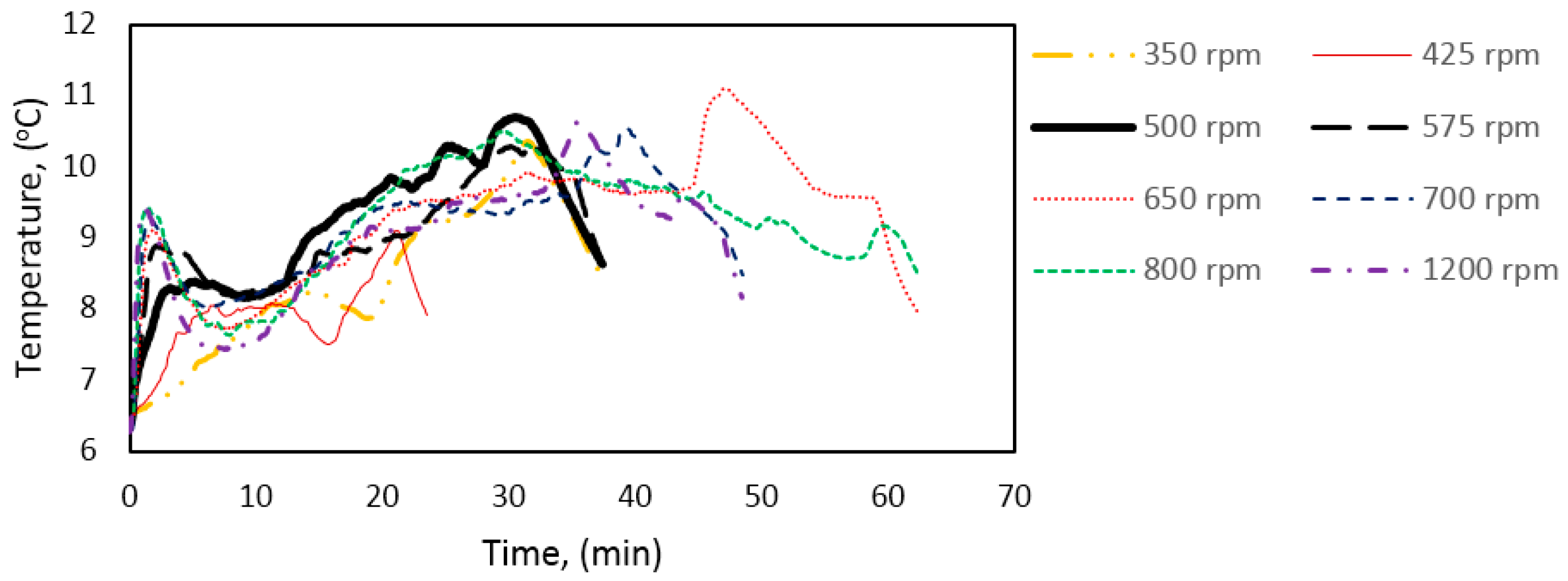
Figure 3 illustrates the temperature, pressure, and gas consumption during a typical experimental run. While cooling the cell content down to the experimental temperature, the stirring was turned off. We see that some gas is consumed during the cooling stage. This is due to gas compensation, mainly for the drop in pressure, and partly to the increase in gas solubility as a function of decreasing temperature. The amount of gas consumed during the initial stirring prior to the start of growth is low, and the temperature remains constant, indicating that the gas saturation process has a low influence on the measured gas consumption during initial hydrate formation.
At point 1, hydrate growth commences, as seen from the rapid increase in temperature and gas consumption. The period between point 1 and 2 is referred to as growth stage I in this work. During growth stage I there is a minimal impact from mass transfer and heat transfer restrictions. The gas consumption rate at this point will be strongly dependent on the hydrodynamics of the system, which is affected by the hydrate content and the stirring rate. We see that stage I is a rather brief period that makes way for growth stage II between points 2 and 3. The start of growth stage II is marked by a drop in the initial temperature. The initial gas consumption rate is also characterized by a similar trend of spike and drop. The average rate of gas consumption during growth stage II is sustained until point 3, where the stirring stops in the cell due to plugging by the agglomerated hydrate mass.
The saturation level of methane in water is lower in a system with hydrate present than in a system without hydrates [
4]. At incipient hydrate formation, the methane saturation adjusts to a lower value [
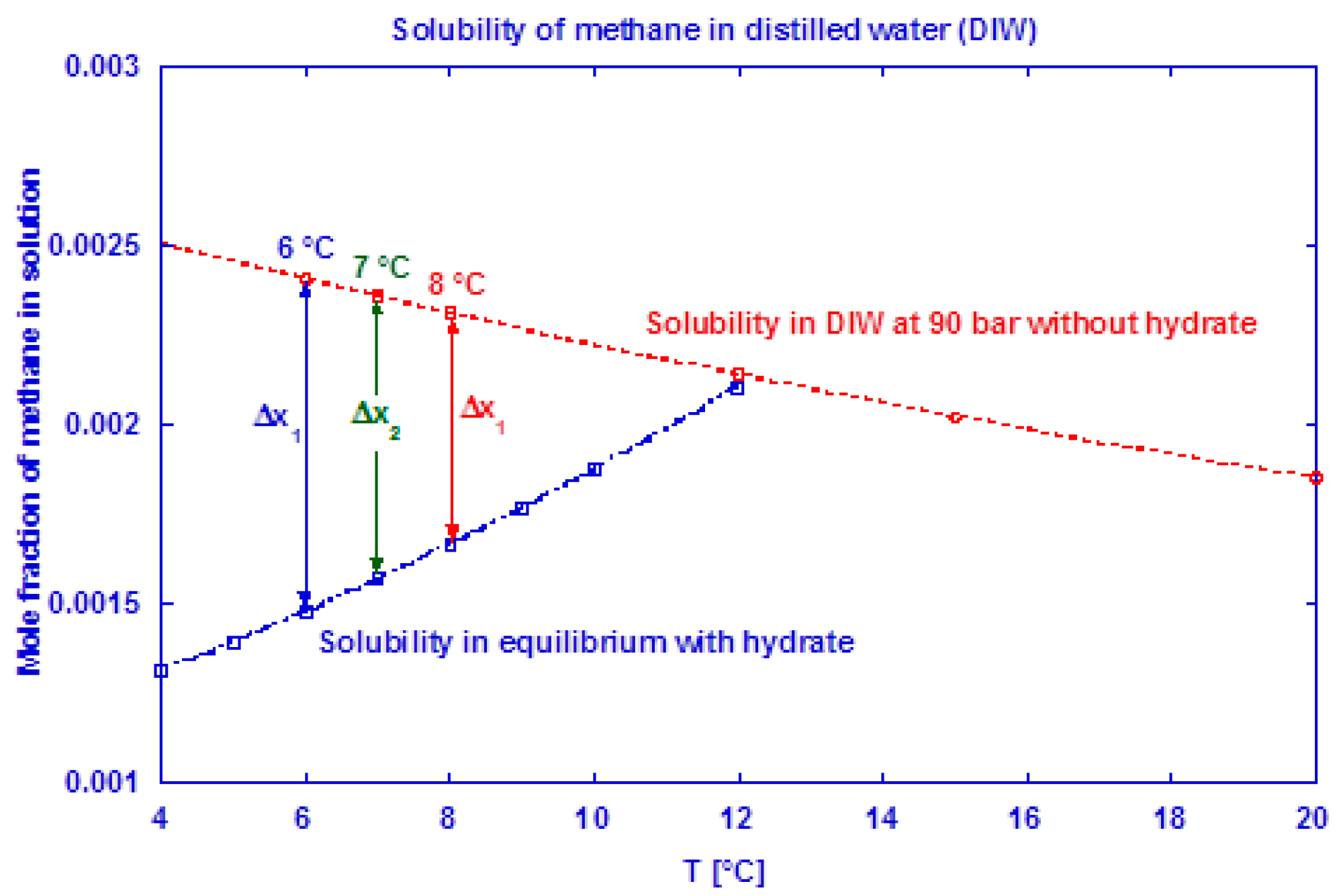
76] and the “released” excess gas contributes to the growth. The saturation level of methane in water at 90 bar is shown in
Figure 4 for both a hydrate-free system and a system in equilibrium with methane hydrate. At incipient hydrate formation, excess gas is available due to a reduction in gas solubility (Δx
n at given T
n). This affects the measured gas consumption rate during the initial growth stage I, which most probably becomes underestimated. We have used the method of Duan et al. [
77] to predict the mole fraction of methane dissolved in pure water and CSMGem program [
4] to estimate the amount of methane dissolved in water in a system containing hydrate.
Table 2 shows the amount of excess gas released from the solution at incipient hydrate formation.
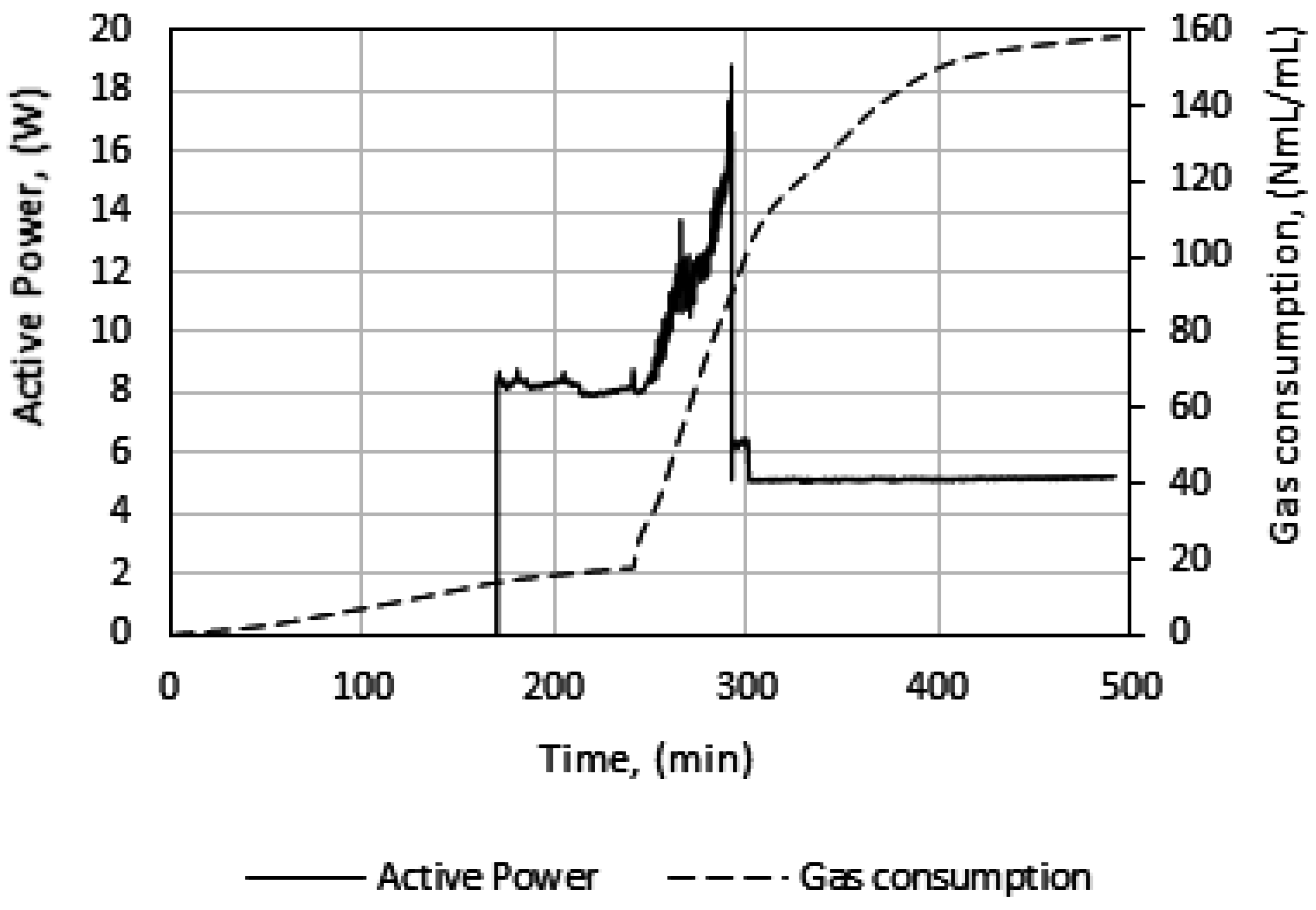
In
Figure 5, we see power readings during an experimental run. As expected, the power is zero when the power supply to the stirrer motor is turned off during cooling, but upon start of stirring the active power attains a value, about 8 watts in this case, and remains roughly constant. At hydrate growth, the power builds up due to the increased resistance to flow from increasing hydrate content. Eventually the power drops sharply, indicating a complete impediment to stirring at point 3, due to plugging in the cell by the hydrate mass. Therefore, we do not analyze the growth data beyond point 3. We thus consider growth stage II to be representative of the actual instantaneous growth kinetics of the process, taking into consideration the heat transfer limitations caused by a temperature increase in the cell. It is also during growth stage II that the bulk of the hydrate forms, so further parametric analysis and discussion of the data focus on growth stage II.





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}