
Electrodeposited Magnesium Nanoparticles Linking Particle Size to Activation Energy


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Supported Magnesium Nanoparticles
2.3. Magnesium Nanoparticles Characterisation
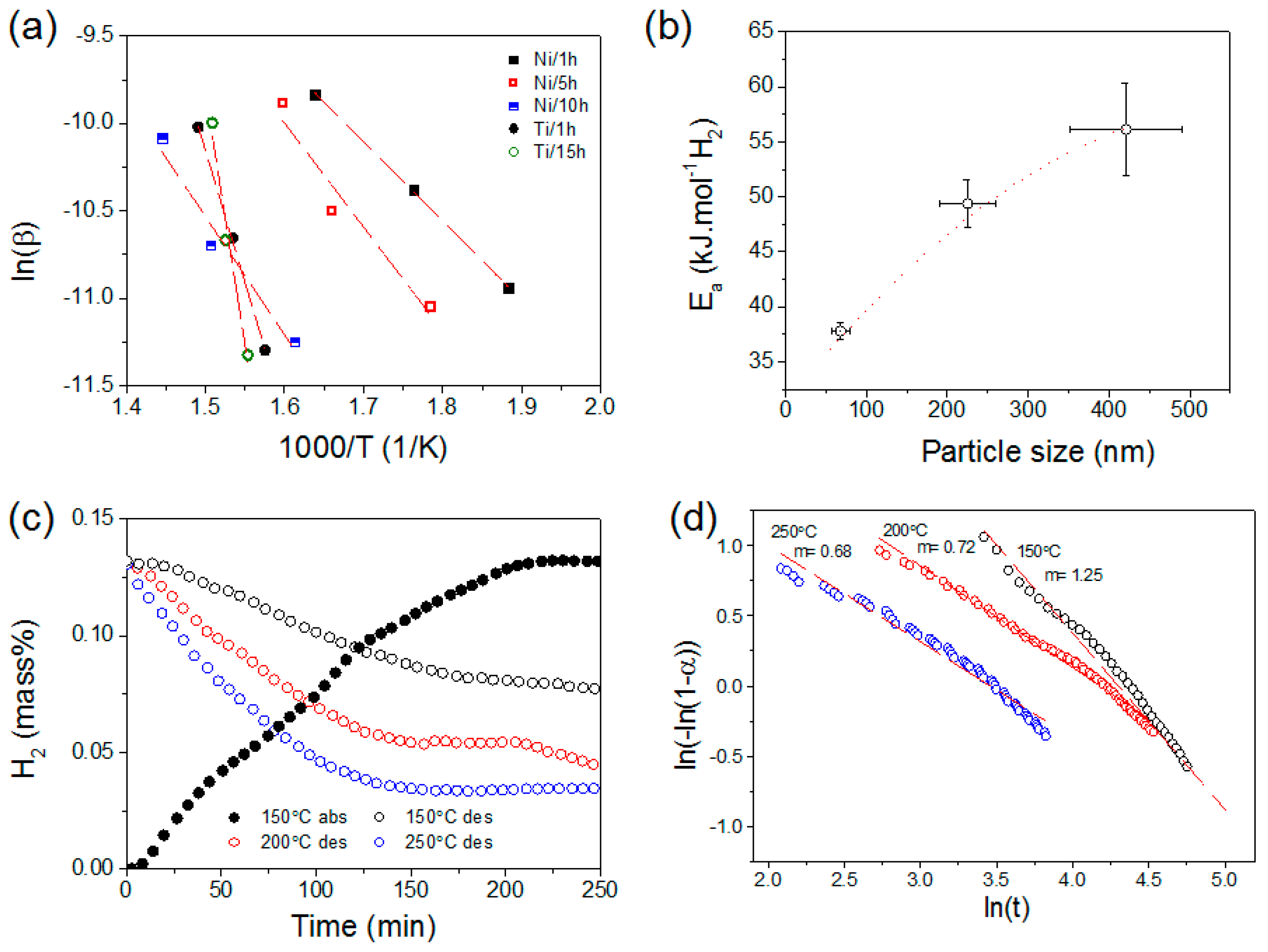
3. Results and Discussions
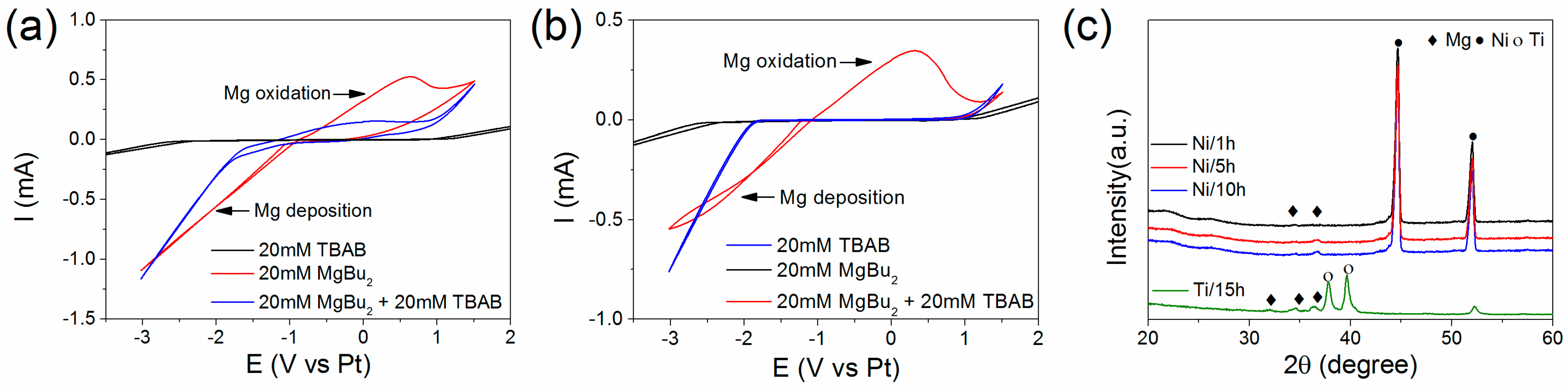
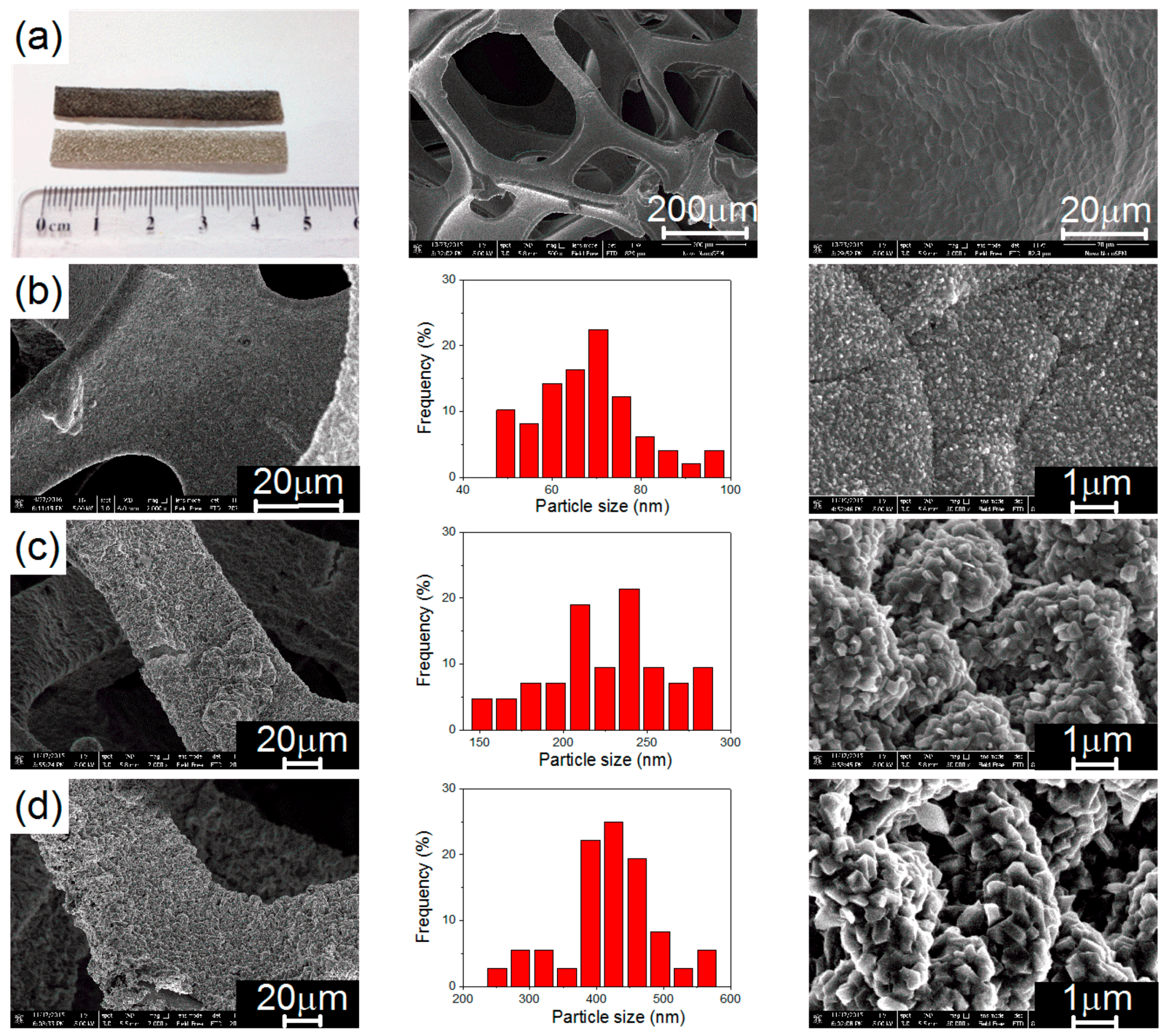
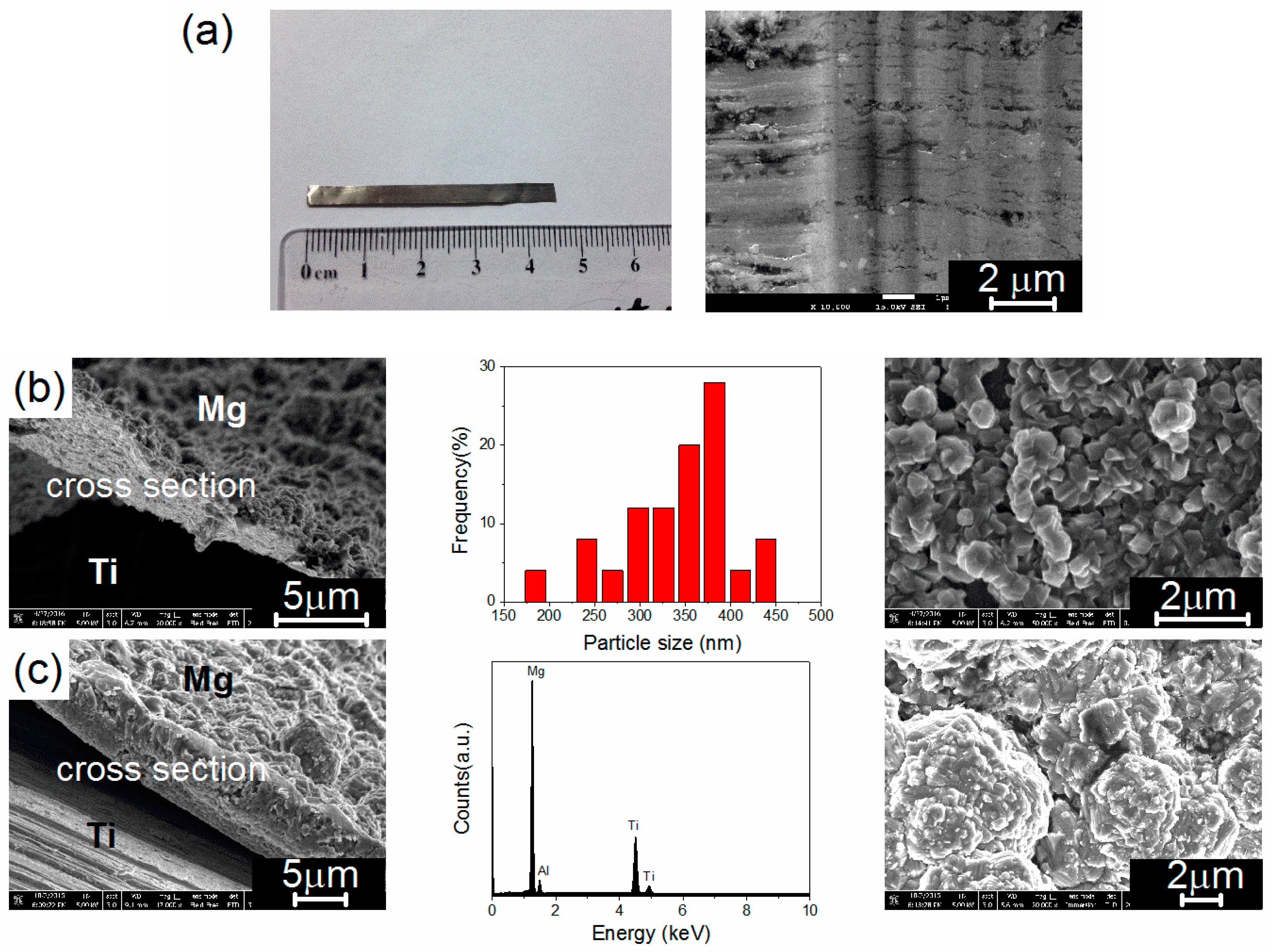
3.1. Electrochemical Deposition of Magnesium Nanoparticles and Structural Properties
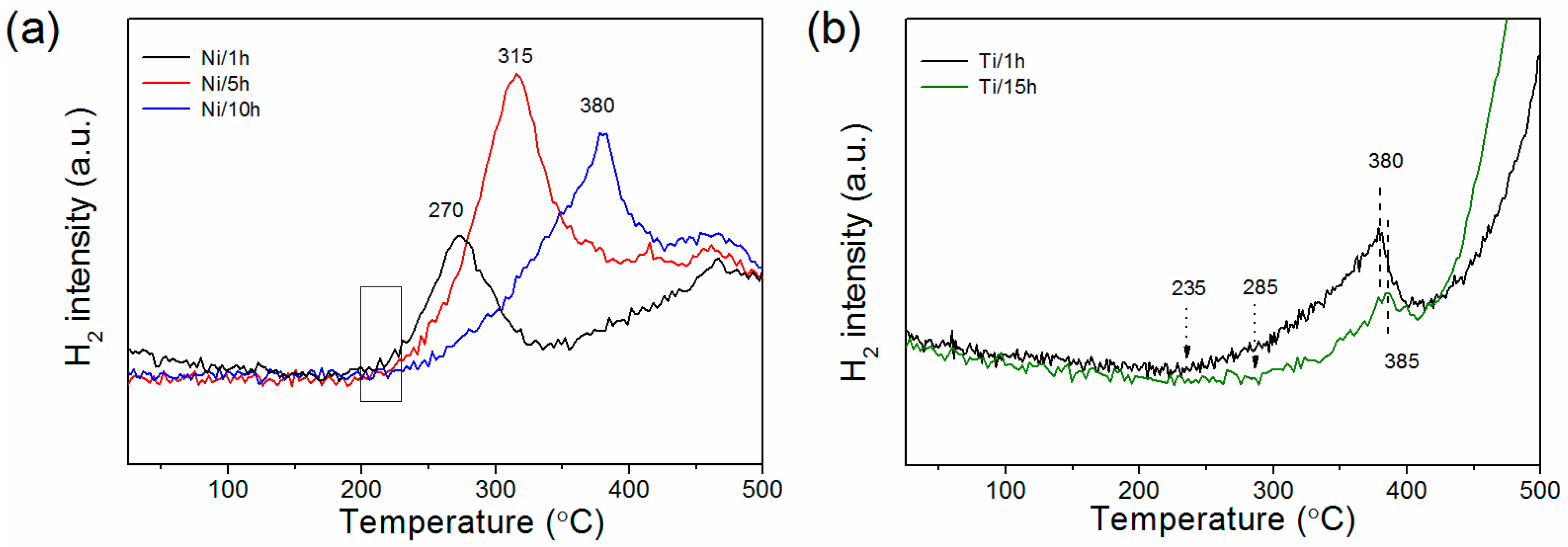
3.2. Hydrogen Sorption Properties
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aguey-Zinsou, K.-F.; Ares-Fernández, J.-R. Hydrogen in magnesium: New perspectives toward functional stores. Energy Environ. Sci. 2010, 3, 526–543. [Google Scholar] [CrossRef]
- Sakintuna, B.; Lamari-Darkrim, F.; Hirscher, M. Metal hydride materials for solid hydrogen storage: A review. Int. J. Hydrogen Energy 2007, 32, 1121–1140. [Google Scholar] [CrossRef]
- Bobet, J.L.; Kandavel, M.; Ramaprabhu, S. Effects of ball-milling conditions and additives on the hydrogen sorption properties of Mg + 5 wt % Cr2O3 mixtures. J. Mater. Res. 2011, 21, 1747–1752. [Google Scholar] [CrossRef]
- Bobet, J. Study of Mg-M (M=Co, Ni and Fe) mixture elaborated by reactive mechanical alloying—Hydrogen sorption properties. Int. J. Hydrogen Energy 2000, 25, 987–996. [Google Scholar] [CrossRef]
- Song, M.; Kwon, S.; Hong, S.-H.; Park, H. Hydrogen storage properties of a Ni, Fe and Ti-added Mg-based alloy. Met. Mater. Int. 2012, 18, 279–286. [Google Scholar] [CrossRef]
- Liu, T.; Chen, C.; Wang, H.; Wu, Y. Enhanced hydrogen storage properties of Mg–Ti–V nanocomposite at moderate temperatures. J. Phys. Chem. C 2014, 118, 22419–22425. [Google Scholar] [CrossRef]
- Agueyzinsou, K.; Aresfernandez, J.; Klassen, T.; Bormann, R. Effect of Nb2O5 on MgH2 properties during mechanical milling. Int. J. Hydrogen Energy 2007, 32, 2400–2407. [Google Scholar] [CrossRef]
- Friedrichs, O.; Aguey-Zinsou, F.; Fernández, J.R.A.; Sánchez-López, J.C.; Justo, A.; Klassen, T.; Bormann, R.; Fernández, A. MgH2 with Nb2O5 as additive, for hydrogen storage: Chemical, structural and kinetic behavior with heating. Acta Mater. 2006, 54, 105–110. [Google Scholar] [CrossRef]
- Ares-Fernandez, J.R.; Aguey-Zinsou, K.F. Superior MgH2 kinetics with MgO addition: A tribological effect. Catalysts 2012, 2, 330–343. [Google Scholar] [CrossRef]
- Aguey-Zinsou, K.F.; Ares Fernandez, J.R.; Klassen, T.; Bormann, R. Using MgO to improve the (de)hydriding properties of magnesium. Mater. Res. Bull. 2006, 41, 1118–1126. [Google Scholar] [CrossRef]
- Pundt, A. Hydrogen in nano-sized metals. Adv. Eng. Mater. 2004, 6, 11–21. [Google Scholar] [CrossRef]
- De Jongh, P.E.; Wagemans, R.W.P.; Eggenhuisen, T.M.; Dauvillier, B.S.; Radstake, P.B.; Meeldijk, J.D.; Geus, J.W.; de Jong, K.P. The preparation of carbon-supported magnesium nanoparticles using melt infiltration. Chem. Mater. 2007, 19, 6052–6057. [Google Scholar] [CrossRef]
- Zlotea, C.; Oumellal, Y.; Hwang, S.J.; Ghimbeu, C.M.; de Jongh, P.E.; Latroche, M. Ultrasmall MgH2 nanoparticles embedded in an ordered microporous carbon exhibiting rapid hydrogen sorption kinetics. J. Phys. Chem. C 2015, 119, 18091–18098. [Google Scholar] [CrossRef]
- Zhao-Karger, Z.; Hu, J.; Roth, A.; Wang, D.; Kubel, C.; Lohstroh, W.; Fichtner, M. Altered thermodynamic and kinetic properties of MgH2 infiltrated in microporous scaffold. Chem. Commun. 2010, 46, 8353–8355. [Google Scholar] [CrossRef] [PubMed]
- Zlotea, C.; Latroche, M. Role of nanoconfinement on hydrogen sorption properties of metal nanoparticles hybrids. Colloids Surf. A Physicochem. Eng. Asp. 2013, 439, 117–130. [Google Scholar] [CrossRef]
- Smith, B.; Wepasnick, K.; Schrote, K.E.; Cho, H.H.; Ball, W.P.; Fairbrother, D.H. Influence of surface oxides on the colloidal stability of multi-walled carbon nanotubes: A structure-property relationship. Langmuir 2009, 25, 9767–9776. [Google Scholar] [CrossRef] [PubMed]
- Lai, Q.; Christian, M.; Aguey-Zinsou, K.-F. Nanoconfinement of borohydrides in CuS hollow nanospheres: A new strategy compared to carbon nanotubes. Int. J. Hydrogen Energy 2014, 39, 9339–9349. [Google Scholar] [CrossRef]
- Haas, I.; Gedanken, A. Synthesis of metallic magnesium nanoparticles by sonoelectrochemistry. Chem. Commun. 2008, 1795–1797. [Google Scholar] [CrossRef] [PubMed]
- Viyannalage, L.; Lee, V.; Dennis, R.V.; Kapoor, D.; Haines, C.D.; Banerjee, S. From Grignard’s reagents to well-defined Mg nanostructures: Distinctive electrochemical and solution reduction routes. Chem. Commun. 2012, 48, 5169–5171. [Google Scholar] [CrossRef] [PubMed]
- Aguey-Zinsou, K.-F.; Ares-Fernández, J.-R. Synthesis of colloidal magnesium: A near room temperature store for hydrogen. Chem. Mater. 2007, 20, 376–378. [Google Scholar] [CrossRef]
- Connor, J.H.; Reid, W.E.; Wood, G.B. Electrodeposition of metals from organic solutions v. electrodeposition of magnesium and magnesium alloys. J. Electrochem. Soc. 1957, 104, 38–41. [Google Scholar] [CrossRef]
- Gaddum, L.W.; French, H.E. The electrolysis of Grignard solution. J. Am. Chem. Soc. 1927, 49, 1295–1299. [Google Scholar] [CrossRef]
- French, H.E.; Drane, M. Electrolysis of Grignard solutions. J. Am. Chem. Soc. 1930, 52, 4904–4906. [Google Scholar] [CrossRef]
- Jolibois, P. On the formula of the organo-magnesian derivative and on the magnesium hydride. C. R. Hebd. Seances Acad. Sci. 1912, 155, 353–355. [Google Scholar]
- Aurbach, D.; Turgeman, R.; Chusid, O.; Gofer, Y. Spectroelectrochemical studies of magnesium deposition by in situ FTIR spectroscopy. Electrochem. Commun. 2001, 3, 252–261. [Google Scholar] [CrossRef]
- Setijadi, E.J.; Boyer, C.; Aguey-Zinsou, K.-F. MgH2 with different morphologies synthesized by thermal hydrogenolysis method for enhanced hydrogen sorption. Int. J. Hydrogen Energy 2013, 38, 5746–5757. [Google Scholar] [CrossRef]
- Aurbach, D.; Gizbar, H.; Schechter, A.; Chusid, O.; Gottlieb, H.E.; Gofer, Y.; Goldberg, I. Electrolyte solutions for rechargeable magnesium batteries based on organomagnesium chloroaluminate complexes. J. Electrochem. Soc. 2002, 149, A115–A121. [Google Scholar] [CrossRef]
- Oniciu, L.; Muresan, L. Some fundamental-aspects of leveling and brightening in metal electrodeposition. J. Appl. Electrochem. 1991, 21, 565–574. [Google Scholar] [CrossRef]
- Hanada, N.; Ichikawa, T.; Fujii, H. Catalytic effect of nanoparticle 3d-transition metals on hydrogen storage properties in magnesium hydride MgH2 prepared by mechanical milling. J. Phys. Chem. B 2005, 109, 7188–7194. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zou, J.; Zeng, X.; Wu, X.; Li, D.; Ding, W. Hydrogen storage properties of a Mg–Ni nanocomposite coprecipitated from solution. J. Phys. Chem. C 2014, 118, 18401–18411. [Google Scholar] [CrossRef]
- Minachev, K.M.; Dmitriev, R.V.; Rustamov, M.I.; Kasimov, C.K.; Abad-zade, K. Thermal desorption of hydrogen from supported nickel catalysts. Bull. Acad. Sci. USSR Divis. Chem. Sci. 1981, 30, 1170–1176. [Google Scholar] [CrossRef]
- Christmann, K.; Schober, O.; Ertl, G.; Neumann, M. Adsorption of hydrogen on nickel single crystal surfaces. J. Phys. Chem. 1974, 60, 4528–4540. [Google Scholar] [CrossRef]
- Wright, S.; Skelly, J.F.; Hodgson, A. Energy disposal during desorption of D2 from the surface and subsurface region of Ni(111). Faraday Discuss. 2000, 117, 133–146. [Google Scholar] [CrossRef]
- Li, T.; Ma, G.H.; Sun, H.J. Ammonia-alkali solution coordination-precipitation route for preparation of beta-Ni(OH)2 nano-particles. Rare Met. Mater. Eng. 2014, 43, 563–566. [Google Scholar] [CrossRef]
- Fry, C.M.P.; Grant, D.M.; Walker, G.S. Catalysis and evolution on cycling of nano-structured magnesium multilayer thin films. Int. J. Hydrogen Energy 2014, 39, 1173–1184. [Google Scholar] [CrossRef]
- Shahi, R.R.; Tiwari, A.P.; Shaz, M.A.; Srivastava, O.N. Studies on de/rehydrogenation characteristics of nanocrystalline MgH2 co-catalyzed with Ti, Fe and Ni. Int. J. Hydrogen Energy 2013, 38, 2778–2784. [Google Scholar] [CrossRef]
- Li, P.; Wan, Q.; Li, Z.; Zhai, F.; Li, Y.; Cui, L.; Qu, X.; Volinsky, A.A. MgH2 dehydrogenation properties improved by MnFe2O4 nanoparticles. J. Power Sources 2013, 239, 201–206. [Google Scholar] [CrossRef]
- Juahir, N.; Mustafa, N.S.; Sinin, A.M.; Ismail, M. Improved hydrogen storage properties of MgH2 by addition of Co2NiO nanoparticles. RSC Adv. 2015, 5, 60983–60989. [Google Scholar] [CrossRef]
- Perejón, A.; Sánchez-Jiménez, P.E.; Criado, J.M.; Pérez-Maqueda, L.A. Magnesium hydride for energy storage applications: The kinetics of dehydrogenation under different working conditions. J. Alloys Compd. 2016, 681, 571–579. [Google Scholar] [CrossRef]
- Yao, X.; Zhu, Z.H.; Cheng, H.M.; Lu, G.Q. Hydrogen diffusion and effect of grain size on hydrogenation kinetics in magnesium hydrides. J. Mater. Res. 2008, 23, 336–340. [Google Scholar] [CrossRef]
- Langhammer, C.; Zhdanov, V.P.; Zorić, I.; Kasemo, B. Size-dependent kinetics of hydriding and dehydriding of Pd nanoparticles. Phys. Rev. Lett. 2010, 104, 135502. [Google Scholar] [CrossRef] [PubMed]
- Baldé, C.P.; Hereijgers, B.P.C.; Bitter, J.H.; de Jong, K.P. Sodium alanate nanoparticles—Linking size to hydrogen storage properties. J. Am. Chem. Soc. 2008, 130, 6761–6765. [Google Scholar] [CrossRef] [PubMed]
- Hoang, K.; Janotti, A.; van de Walle, C.G. The particle-size dependence of the activation energy for decomposition of lithium amide. Angew. Chem. Int. Ed. 2011, 50, 10170–10173. [Google Scholar] [CrossRef] [PubMed]
- Mintz, M.H.; Zeiri, Y. Hydriding kinetics of powders. J. Alloys Compd. 1995, 216, 159–175. [Google Scholar] [CrossRef]
- Hancock, J.D.; Sharp, J.H. Method of comparing solid-state kinetic data and its application to decomposition of kaolinite, brucite, and BaCO3. J. Am. Ceram. Soc. 1972, 55, 74–77. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Deposition Time (h) | Particle Size (nm) | Crystallite Size (nm) | H2 Desorption Temperature (°C) | Ea (kJ·mol−1 H2) |
|---|---|---|---|---|---|
| Ni | 1 | 68 ± 11 | - | 295 | 37.8 ± 0.7 |
| 5 | 225 ± 35 | 12 ± 2 | 330 | 49.4 ± 2.1 | |
| 10 | 421 ± 70 | 14 ± 2 | 390 | 56.1 ± 4.2 | |
| Ti | 1 | 341 ± 60 | - | 380 | 125.4 ± 2.6 |
| 15 | - | 14 ± 2 | 385 | 240.1 ± 12.7 |
| Mechanism | Functional Time Dependence G (α) | m |
|---|---|---|
| Surface control | ||
| S1 | α | 1.24 |
| Random nucleation and growth (Avrami equations) | ||
| A1 | [−ln(1 − α)]1/4 | 4.00 |
| A2 | [−ln(1 − α)]1/3 | 3.00 |
| A3 | [−ln(1 − α)]2/5 | 2.50 |
| A4 | [−ln(1 − α)]1/2 | 2.00 |
| A5 | [−ln(1 − α)]2/3 | 1.50 |
| Shrinking core with constant velocity: controlled by interface reaction | ||
| IP2—contracting surface | 1 − (1 − α)1/2 | 1.11 |
| IP3—contracting volume | 1 − (1 − α)1/3 | 1.07 |
| Shrinking core with decelerating velocity: controlled by diffusion | ||
| D1—1-D diffusion | α2 | 0.62 |
| D2—2-D diffusion | (1 − α)ln(1 − α) + α | 0.57 |
| D3—Jander, 3-D diffusion | (1 − (1 − α)1/3)2 | 0.54 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, C.; Aguey-Zinsou, K.-F. Electrodeposited Magnesium Nanoparticles Linking Particle Size to Activation Energy. Energies 2016, 9, 1073. https://doi.org/10.3390/en9121073
Shen C, Aguey-Zinsou K-F. Electrodeposited Magnesium Nanoparticles Linking Particle Size to Activation Energy. Energies. 2016; 9(12):1073. https://doi.org/10.3390/en9121073
Chicago/Turabian StyleShen, Chaoqi, and Kondo-Francois Aguey-Zinsou. 2016. "Electrodeposited Magnesium Nanoparticles Linking Particle Size to Activation Energy" Energies 9, no. 12: 1073. https://doi.org/10.3390/en9121073
APA StyleShen, C., & Aguey-Zinsou, K.-F. (2016). Electrodeposited Magnesium Nanoparticles Linking Particle Size to Activation Energy. Energies, 9(12), 1073. https://doi.org/10.3390/en9121073