1. Introduction
Each monomer comprising the DNA polymer is formed with a pentose, a phosphate group and one of four nitrogenous bases: adenine, guanine, cytosine and thymine. In 1953, scientists James Watson and Francis Crick [
1] discovered the double-helix spatial structure of the DNA molecule. This double chain is coiled around a single axis, and the strands are attached by hydrogen bridges between pairs of opposite bases. The direction of the polymer chain is determined by the pentose carbon atoms 5
′ and 3
′ (the beginning and ending positions of a DNA strand are denoted as 5′ and 3′, respectively; a DNA strand is read in the 5′ to 3′ direction, and the complementary strand runs in the opposite direction). The bases in each pair are complementary. The content of adenine (
A) is the same as the content of thymine (
T), and the cytosine (
C) content is the same as that of guanine (
G) (
Figure 1).
In 1975, Frederik Sanger [
2] proposed a DNA sequencing technique that involved detecting small dark bands in a thin gel using electrophoresis. Sanger proposed cutting the DNA molecule at specific points in the sequence with restriction enzymes [
3]. This method is slow and costly; with each digestion, the sample must be divided, and each new division must be cloned to obtain a sufficient amount of material. To reduce processing time, Sanger et al. [
4,
5] proposed splitting the DNA sequences at random points. The disadvantage of this method is that the order of the fragments is unknown, generating an NP-Complete (Non-deterministic polynomial time) [
6] computational problem. This method is known as the
shotgun technique.
Rodger Staden [
7] proposed a method to assemble the genome using a computer. As the DNA fragments are produced from many copies of the original genome, more than one fragment comes from the same region. The DNA fragments should be processed while looking for overlaps, coincidences in the extremes of the fragments. The total number of bases in the fragments divided by the total number of bases in the complete genome is called the
coverage. If the
coverage is high enough, it is possible to rebuild the genome, but it is difficult to obtain high-quality results because false overlaps can be generated due to sequencing errors, sample contamination with foreign DNA and chimeras (the cloning process is carried out using host bacteria, and sometimes the DNA of the sample is concatenated with that of the host bacteria, producing what is known as a
chimera). In our experiments, we noticed that some DNA sections might not be sampled and that complete genome reconstruction would therefore not be possible. What can be obtained is a set of
contigs covering most of the genome, and it is the job of a molecular biologist to assemble the
contigs using other techniques.
Later, James Weber and Eugene Myers [
4] proposed the generation of paired fragments in the
shotgun process. Traditional sequencing starts from the 5′ end of each piece of DNA, and only a limited number of bases can be sequenced. In NGS (Next-Generation Sequencing), the number of bases is always the same, yielding fixed-size fragments; however, the DNA pieces sequenced are generally longer. The goal of the paired fragments procedure is to obtain a sequence from the 3′ end as well as one from the 5′ end, obtaining two fragments from the same piece of DNA. This would help to establish an interval in the DNA sequence associated with the paired fragments. Unfortunately, sequencing from the 3′ end is difficult and produces a relatively large number of sequencing errors.
DNA sequencing technology has advanced, decreasing costs and process time. Today, there are databases with information about many genomes, including the human genome and those of many disease-causing microorganisms [
8]. Steven Salzberg [
9] developed a comparative study of DNA sequence assemblers; tests were carried out with fragment sets obtained using Illumina equipment (
Illumina:
http://www.illumina.com/, Illumina, Inc., San Diego, CA, USA). The fragments’ lengths were found by NGS technology to be in the range of 50 to 150 bases [
10]. The Salzberg study was developed in 2012; nevertheless, the paradigm remains the same today, and the Salzberg study is therefore still applicable. The primary metric in the evaluation was N50 (shortest sequence length close to the median of a set of
contigs). Salzberg employed four organisms in the test, including a
Staphylococcus aureus problem. These results are particularly interesting because the tests we present in this article have been obtained using a problem with the same bacterium. Salzberg did not include the
Edena assembler (
http://www.genomic.ch/edena.php, Genomic Research Institute, Geneva, Switzerland.), developed by David Hernández [
11] in his research. In our comparative study, this assembler is an important reference.
Initially,
greedy (Algorithmic technique that at each step tries to generate the optimal solution of that step of the problem without considering the rest of the problem) algorithms [
12] were used to find the order of the fragments; later, the
de Bruijn graph [
13] was introduced with different values for the
k-mer (section of k consecutive bases) [
13]. Most of the assemblers available today are based on
de Bruijn graphs. In our comparative study, we include the
Velvet assembler (
http://www.ebi.ac.uk/~zerbino/velvet/, EML-EBI, Cambridge, UK), developed by Zerbino [
14], which applies
de Bruijn graphs.
Parsons et al. [
15] proposed an optimization with a genetic algorithm to maximize the sum of the fragment overlaps, but the obtained
contigs were relatively short and processing speed was low. Later, Mallén-Fullerton and Fernández-Anaya [
16] suggested a reduction of the fragments’ assembly problem to the traveling salesman problem (TSP) that has been studied extensively. Solution methods with relatively good efficiency exist for the TSP. Applying heuristics and algorithms, they obtained optimal solutions for several commonly used benchmarks, and for the first time, a real-world problem was solved by using optimization. Using graph theory and setting the appropriate objective functions, Mallén et al. [
17] developed a new assembly method from the perspective of a directed graph, looking for the reduction of the algorithm’s complexity.
In this publication, taking Mallén et al. [
17] as a starting point, we developed a new algorithm working with the
paired fragments (two records identified by “/1” and “/2”, respectively, conforming to an interval of the sequence of DNA) resulting from the sequencing results of the
Illumina equipment. In our initial tests, we found some
contigs that were not located in the published genome for the same organism, even though these
contigs were properly obtained. Empirically, we found that when these
contigs were split at certain points, all of the pieces could, in most cases, be found in the genome. Using the information from the paired fragments information, we could find the locations where a
contig should be split to increase the quality of the assembly.
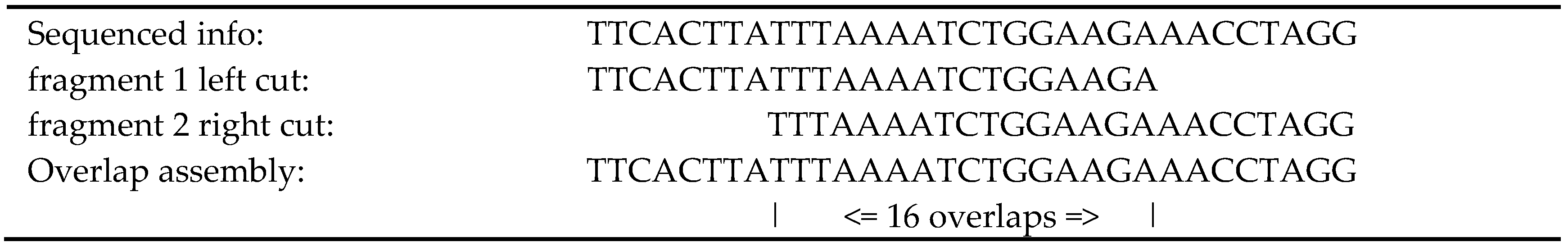
To obtain the overlaps between DNA fragments, we used a
Trie [
17]. Using the overlaps as edges, a directed graph has been obtained, and we could build a set of
contigs improving the N50 [
8] of the previous release [
17]. The interval of the paired fragments was a critical factor in our success. In our experiments, we worked with the sequencing data of real-life organisms obtained from
Illumina equipment, maximizing the lengths of the
contigs as the objective function.
In
Section 2 of this paper, we present the applied graph theory elements, data structures and algorithms. In
Section 3, we reveal the developed algorithms that solve the assembly problem.
Section 4 sets out the application of this new model to a real-life problem comparing our assembler with other assemblers (
Velvet and
Edena), and finally, in
Section 5, we present our conclusions and ideas for future work.
3. Algorithms
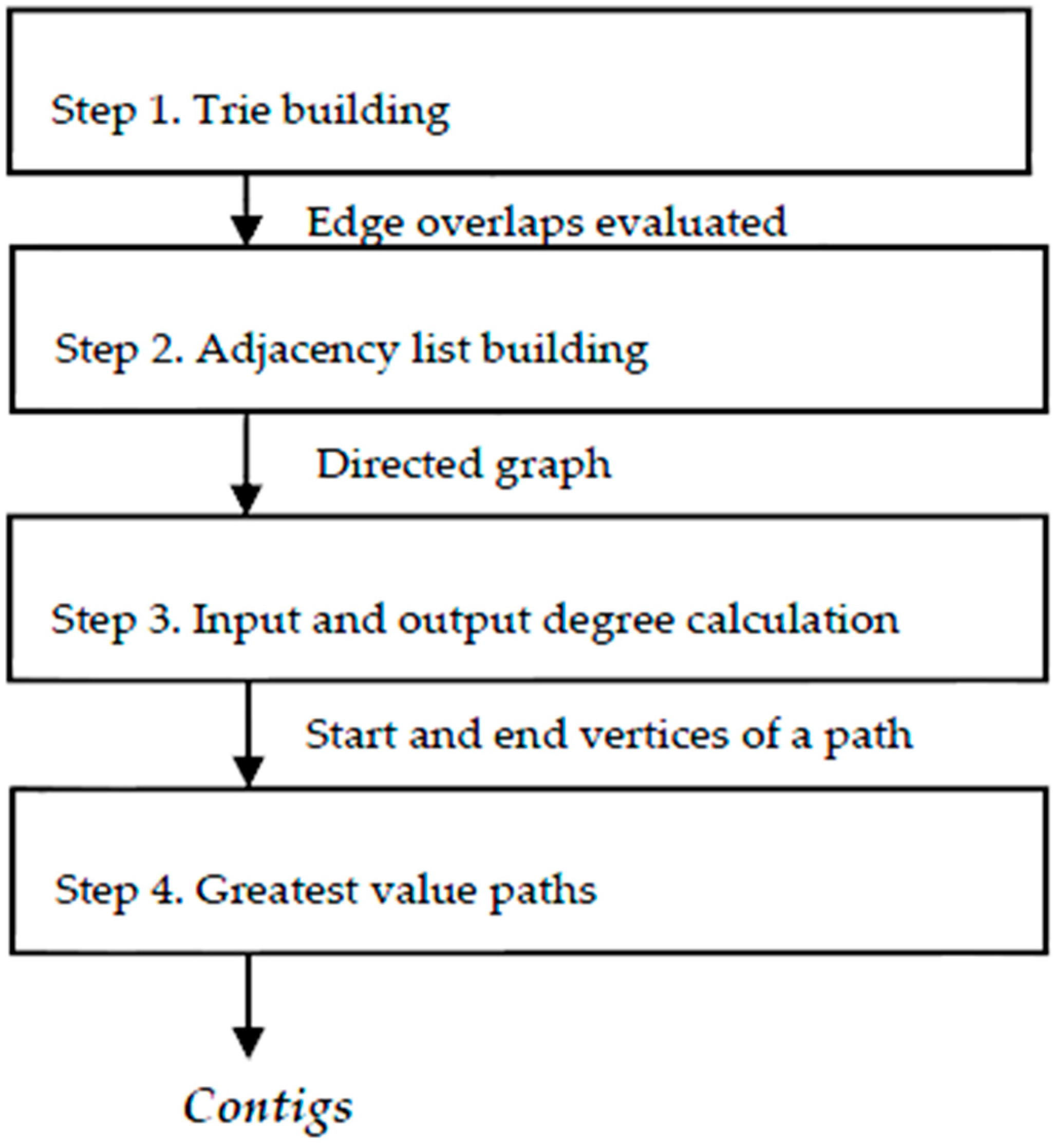
To resolve the assembly problem, we hereby propose the following sequence (
Figure 6):
Step 1. Build a
Trie (prefix tree) [
19] to identify the overlapped fragments, trying to get the greatest value from the overlapping while resolving duplicates, fragments without overlap and repetition bases with the same element.
Step 2. From the list of overlapping fragments that comprise a directed graph, an adjacency list is built [
21], with which we can calculate the input and output degrees of each vertex.
Step 3. Using the degree input and output information of the vertices, we could detect those that are a head of a path, with zero input degree, and build the path to those with zero output degree, accumulating the overlapping values of each edge. This accumulated value can change if, when on a new path, there is a value greater than the previous intersection of an intermediate vertex of the path.
Step 4. Finally, walking in reverse, beginning with the last vertex, rebuild the route marked with the greatest overlapping values until a vertex of zero input degree is reached. This path is a contig.
3.1. Trie
The
Trie data structure was developed by Fredking [
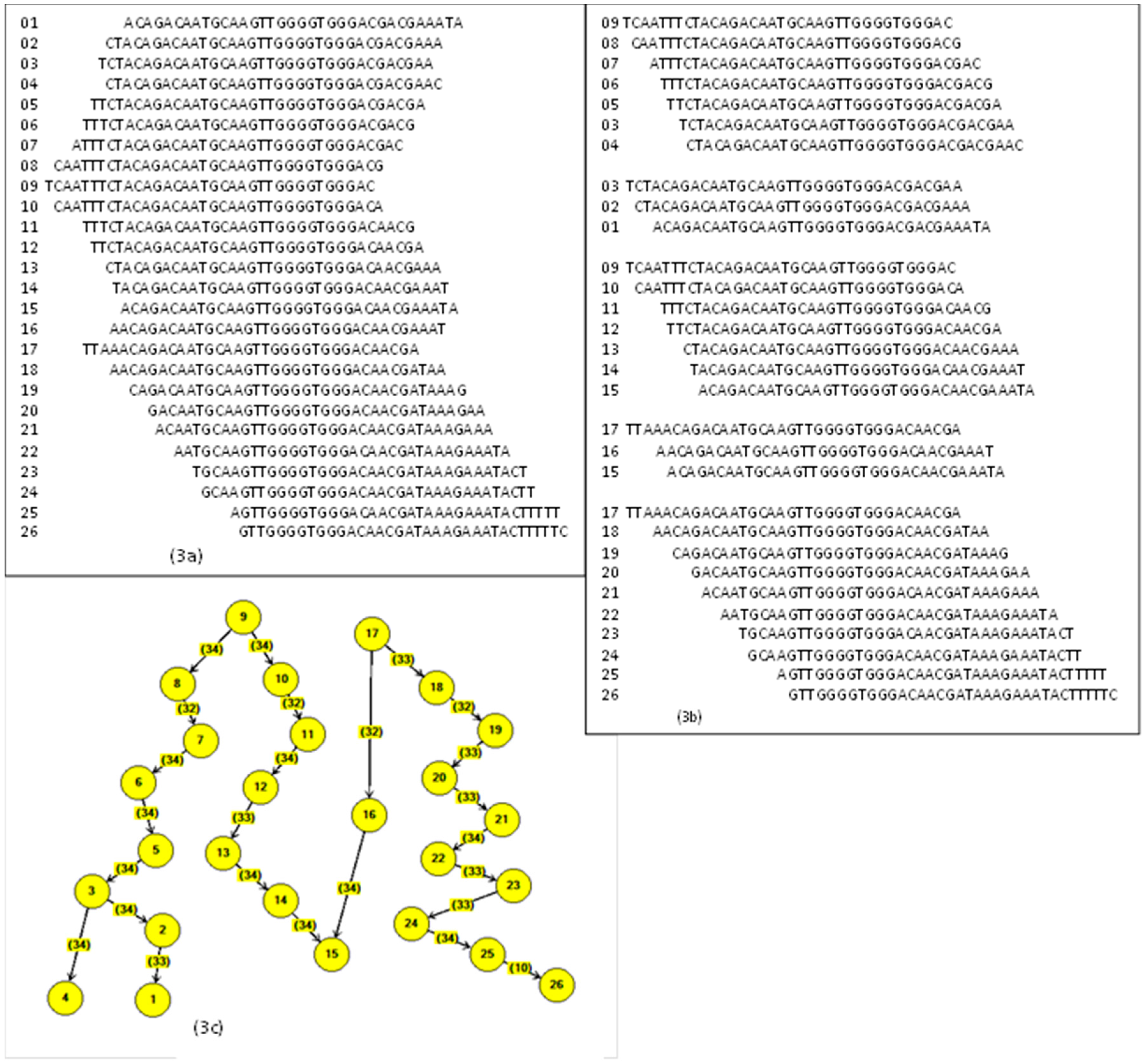
22]. It is a tree structure that forms prefix texts that are converted into a search index for the new texts. The new texts fit with the prefix and complement the branch of the tree with a suffix. In our problem, the prefixes and suffixes are letters from the alphabet: A, C, G, T.
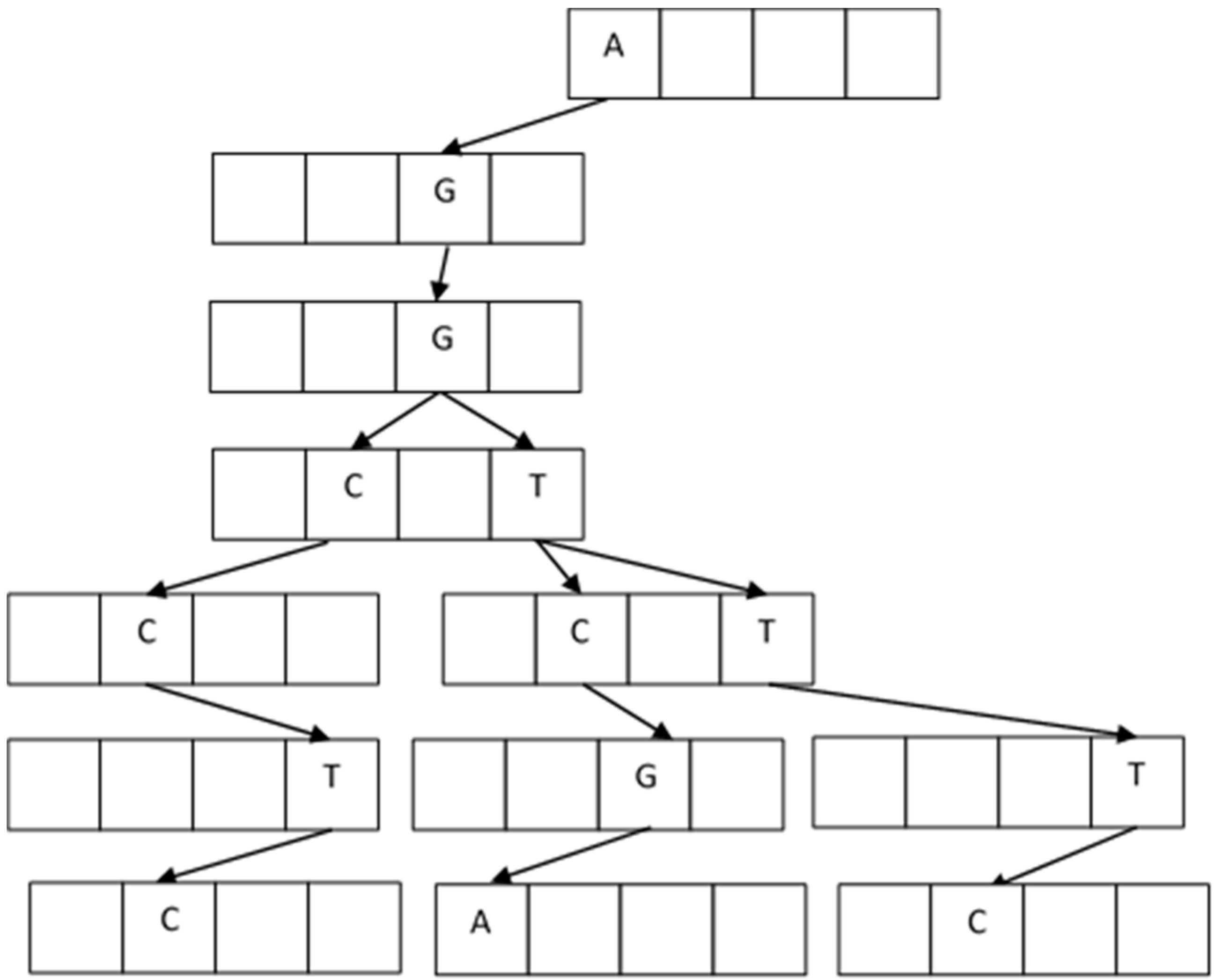
In the building process, the first node with four locations is empty and ready to receive the four elements A, C, G, T. The first fragment arrives and accommodates depending on the letter value. For example, the first string is AGGTCGA, and it goes on creating new blank nodes. The next one arrives with AGGTTTC, and it accommodates from AGGT; the next fragment is AGGCCTC, and now it accommodates from AGG.
Figure 7 shows the resulting tree.
The duplicate fragments are easily handled because they do not provide new values to the branches and are eliminated. The construction of the
Trie is, in the worst case,
O(ln) [
19] when there are no coincidences between the fragments (
l is the fragment length and
n is the number of fragments). In our data problem, the number of coincidences is large. Algorithm 1 describes the process of building the
Trie.
| Algorithm 1. Trie construction. |
- 1.
For each fragment - 1.1
For each letter x from the fragment - 1.1.1
If box node(x) is free: - 1.1.1.1
Take up the box and create new empty node
- 1.1.2
Else - 1.1.2.1
Go to next node
|
Algorithm 2 shows the search for the overlapped fragments. In the worst case, it is O(l) for a single fragment, where l is the length of the fragment, because, at that point, it has completely walked through the branch of the tree.
| Algorithm 2. Searching a fragment in the Trie. |
- 1.
For each fragment (l) - 1.1
For each fragment (l ← l-1 until overlap limit value) - 1.1.1
For each fragment-letter vs. Trie-node-letter - 1.1.1.a
If equal: continue to next fragment-letter and node - 1.1.1.b
If empty box: finish cycle 1.1
- 1.2
If end of fragment: - 1.2.1
Drain the branch - 1.2.2
Identify both fragments and create an edge (from, to, overlap)
|
3.2. Adjacency List
This data structure is built by taking the list of edges. The list is a conventional data structure for each fragment.
3.3. Contig Assembly
In this step, the longest path will be sought, starting with all the vertices with zero input degree, these being the potential starting points of a path that could eventually become a contig. The walkthrough is carried out until a vertex with zero output degree is found, while accumulating the overlapping values. Once a path is finished, the walkthrough shifts to the next vertex with zero input degree and the process is repeated. If a node that has been used in another path is detected, the process will evaluate the greatest value and will leave the greater value as the result. Once the complete graph has been processed, each starting node is a contig. Algorithm 3 shows the process used to obtain the greatest weight of a path.
| Algorithm 3. Contigs assembly. |
- 1.
For all elements with Din = 0 - 1.1
Dout minus 1 - 1.2
For each adjacency element until Dout = 0 - 1.2.1
Accumulate weight
- 1.3
If accumulated weight > previous weight - 1.3.1
Label maximum path
|
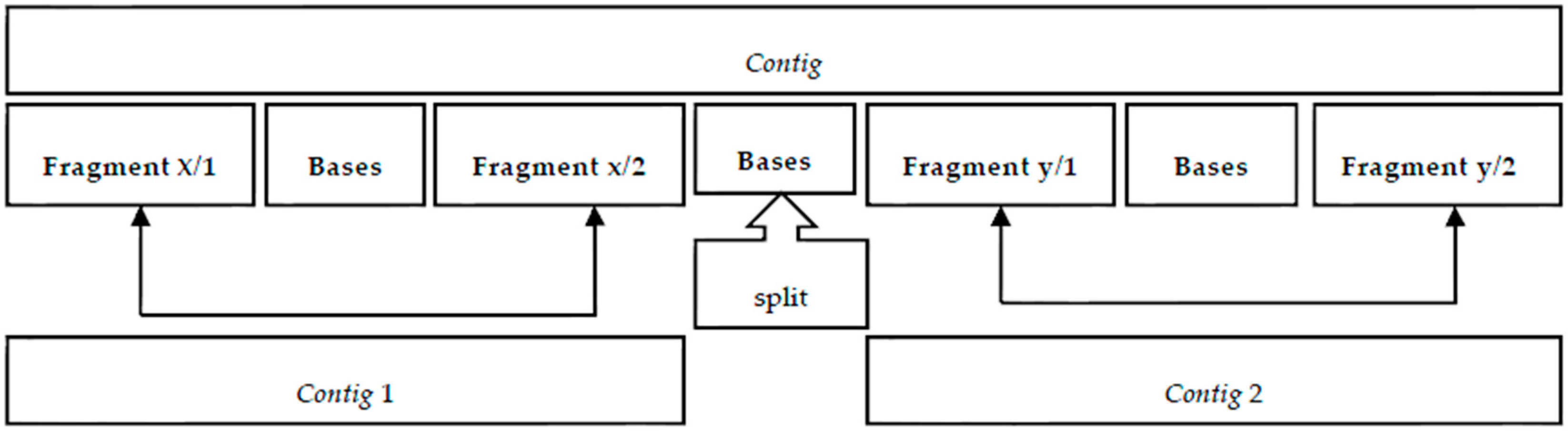
While assembling the branch, a content counter is generated for the column; at the end of the assembly process, the consensus is reviewed with the counter, and it is possible to remove the sections not in compliance with the predefined value.
In the FASTA file containing the original fragments [
18], the paired values are identified as “/1” and “/2”; these identifiers will be used to confirm the
contig by searching the interval inside the
contig. Algorithm 4 describes how the paired fragments confirm intervals.
| Algorithm 4. Confirm intervals. |
- 1.
Interval-counter ← 0 (counts the intervals into a contig). - 2.
For each fragment in the contig - 2.1
If type “/1”, search type “/2” in the contig - 2.1.1
If Found type “/2” in the contig - 2.1.1.1
Yes: interval counter = interval counter+1
- 3.
For each fragment in the contig - 3.1
If interval counter = 0 - 3.1.1
If fragment in in-between, cut of the contig - 3.1.2
If fragment in extremes, drop the section
|
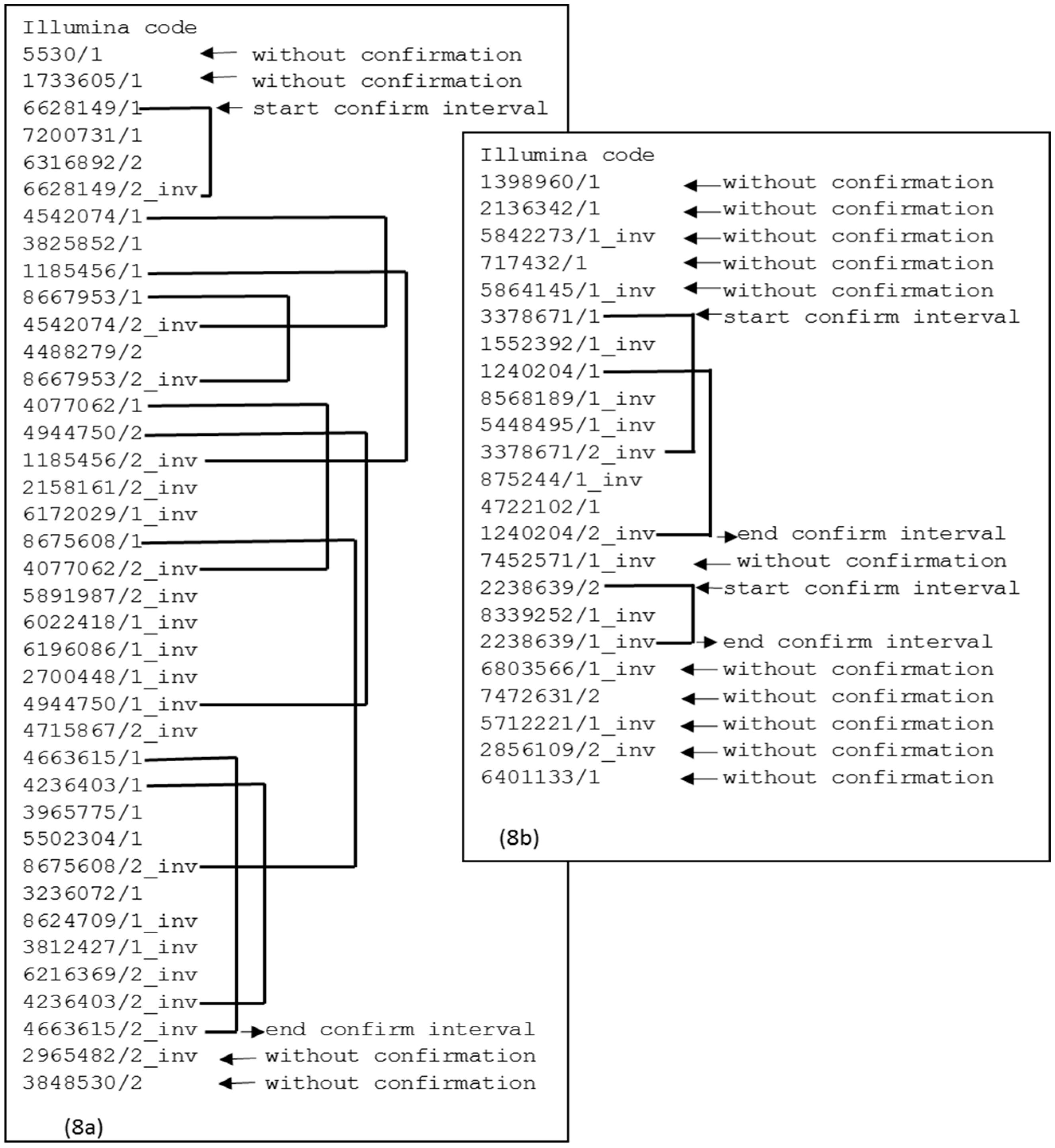
Figure 8a shows a case of a
contig that demonstrates continuity between the first confirmation interval and the subsequent interval. If there is no continuity between the confirmation intervals, then there is a split point, as shown in
Figure 8b. This split point will produce two
contigs. The elements at the extremes are also removed because there is no confirmation interval.
4. Experiments
The application of the algorithms was carried out with the
Staphylococcus aureus problem, taken from GAGE (Genome Assembly Gold-Standard Evaluations, 2011, [
9]). The information consisted of 647,062 fixed-length fragments of 101 bases. The bacterium had a genomic chromosome with 2,903,081 bases, a first plasmid with 27,428 bases and a second plasmid with 3170 bases.
The
Illumina equipment generates three types of DNA sequences: single record, two records paired in a lineal sample and two records paired in a circular sample. In the paper of Mallén et al. [
17], the work was applied to a single record; in this new approach, we have worked with lineal paired records. These paired records contained the information necessary to confirm intervals in the assembled DNA.
The reverse-complementary fragments were generated (inverted sequence exchanging AxT and CxG in reverse order) for both types of records, “/1” and “/2”. The correspondence of the pairs is:
Type “/1” paired with the reverse complement of “/2”,
Type “/2” paired with the reverse complement of “/1”.
The generation of these reverse-complementary pairs generated duplicated contigs; they were eliminated from the result.
The
Velvet release we used was 1.2.10, and we searched for the execution with the best results of N50 [
9], varying the value of the
k-mer [
13]. Ultimately, the best value obtained was 31. The
Eden release we used was 3.13, and, in the same way, we took the best result with different overlap values to obtain the best N50 [
9]. The best value was 30 overlapping bases.
Illumina equipment delivers two fragment data files. The first contains the record type “/1” and the second contains the record type “/2”. In both Edena and Velvet, it is possible to define a parameter for how to use these files. During the tests, this was considered.
With our programs (in the C/C++ programming language), we also had different parameter values with respect to overlapping. The best result of N50 [
9] was found with 50 bases. In the results, we produced two versions, A and B. In our programs, we merge both data files because, during the initial tests, we found these records in the same file. We deleted the records with “N” values for the bases; this value means that the base is undetermined. In addition, we removed all characters except for the A, C, G, and T values. During the
Trie process, we removed duplicate fragments, and, finally, we eliminated repetitions of the same base value with more than 15 occurrences. This decision was based on an analysis of the data as a special case and would not be applicable to other organisms.
The A version of our program was designed for paired records; the B version also included both records, but the confirmation interval was applied to the
contigs. In this version, we also eliminated the
contigs with a length shorter than 1.5 times the fragment size.
Edena also carries out this process, but this decision cannot be modified by the user; it is a consistency requirement. This action resulted in a modest improvement in N50 [
9].
Table 1 shows the results of the executions in the “Generated” section. To determine how close we were to obtaining total genome reconstruction, we did a search of every
contig in the original genome with our results, with those of
Edena and with those of
Velvet.
The results presented in
Table 1 have been obtained from QUAST (v 4.4. CAB: Center for Algorithmic Biotechnology. St. Petersburg, Russia) [
23]. The search was also done with Mummer [
24]. The last column of
Table 1 shows the result from Mummer, representing the
contigs that have been found completely in the original genome. This result was applied to
Edena,
Velvet and our programs.
We performed all program executions two consecutive times to guarantee an execution time independent of computer dependencies such as those from the cache memory, the hard disk or the processor. With the Linux command “time” in terminal mode, we obtained the real time and the user time. We took the real time in all the proofs and the lower of the two times from the first and the second executions.
All of the runs were made on the same computer, employing 64 bit Linux with Ubuntu 14.4 Long Term Support (London, UK), 16 gigabytes of RAM, an Intel (Santa Clara, CA, USA) i5-4460 processor @ 3.2 GHz × 4 and a hard disk with an EXT4 partition type. The programming language of our programs (versions A and B) was C/C++, with the standard compilation C++ ISO (-std = C++11), and without any optimization.
Table 2 shows the execution times for each of the cases tested.
5. Conclusions
The advantages of our algorithms are described below.
Before applying the confirmation intervals, our N50 [
9] was superior to
Edena but not to
Velvet. It is clear that
Velvet uses
de Bruijn graphs [
13], and it contains a rebuilding
contigs step. The resultant N50 of
Velvet is better than those of
Edena and our programs (see
Table 3).
After applying the confirmation intervals, our N50 [
9] for the paired fragments was smaller, but the quality of the
contigs was greater than those of the other programs, as seen in the percentage of Mummer-located
contigs (see
Table 4).
The values found by Mummer (see
Table 1) do not help
Velvet (67.3%), and our program yielded the best value (93.4%). If Mummer locates the
contig completely in the genome, it appears as a 100% value; if Mummer cannot locate the
contig, it adjusts the difference, splitting or tolerating certain errors and trying to relocate the pieces, reporting these as a percent value less than 100%. These values are not included in
Table 1.
Regarding the execution times (see
Table 2), in the first step,
Edena took 6 min to calculate the overlap, and it took the longest time for the samples. In our version, an equivalent process took approximately 1 min, so we concluded that our process is much faster than
Edena, though that is not the case when compared to
Velvet. Our program requested approximately 4 gigabytes of RAM to manage the
Trie [
22]. Meanwhile, as the
Trie grows, it becomes asymptotic; the construction of the
Trie is rapid, with times on the order of five seconds.
A potential future study could search the “/2” type records outside the
contig with a “/1” type record without its matching pair. These might generate a
contig with a greater confirmation interval and, consequently, a better N50 value [
9].
To obtain an improvement in the execution time, it would be advisable to eliminate the transitive edges of the graph. With this action, the data volume would be reduced significantly.
The identification of the sequences of the contigs could represent valuable information for molecular biologists, and these could probably be obtained by looking for paired fragments that span multiple contigs. The records with undetermined values (“N”) could probably also contribute some information about the contig ordering.
Finally, the scaffolding process (contigs assembly) could generate important results for biologists, including a likely new step of detecting overlapping within contigs and giving rise to some adjustments in the quality assurance of the assembly.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}