1. Introduction
Since 1953, when the engineers at IBM’s San Jose California laboratory invented the first “random access file” [
1], storage performance has continuously grown both in terms of capacity and speed data access. Inversely, the space/cost ratio has dropped drastically with time. Data recorded over the last 30 years show a very strong exponential correlation, that is, space per unit cost has doubled roughly every 14 months and, then, has increased by an order of magnitude every 48 months. The cost was estimated to follow the equation:
, with an extraordinarily high coefficient of correlation (
[
2].
Unfortunately, this positive trend has only mitigated the economical impact of the highly elevated data production rates of today’s biomedical instruments. Next Generation Sequencing (NGS) platforms, for example, are developing at a much greater rate than was seen for close technologies, yielding a 1000-fold drop in sequencing costs since 1990. Consequently, several new large data projects arose from one side (e.g., 1000 Genomes Project [
3], the ENCODE project [
4], Exome Sequencing Project (ESP) [
5]), which brought the downside of having to stock lots of data on the other side. Storage costs have in fact largely exceeded reagent costs, leading sometimes to the extreme decision of carrying out the experiment again rather than retain raw data for long time.
Next generation sequencers belong to the category of high-throughput instruments. They sequence short stretches of DNA/RNA molecules (also known as short-reads), previously extracted from biological samples, sheared and amplified according to manufacturer protocols. The sequencing procedure itself varies with the instrument. For example, the SOLiD platforms by Applied Biosystems (now Life Technology) perform five different steps, called
primer rounds. At any round, primers, which are strands of nucleic acids that serve as starting points for DNA synthesis, hybridize to the adapter sequences within the library templates and a fluorescence is emitted as a consequence of each ligation. After detection, dyes are cleaved off, giving way to the next differentially labeled probe that can hybridize to the target sequence. A quality value (QV) is associated to each dye, which typically ranges from 25 to 35, even if it can span a wider range of values (
i.e.,
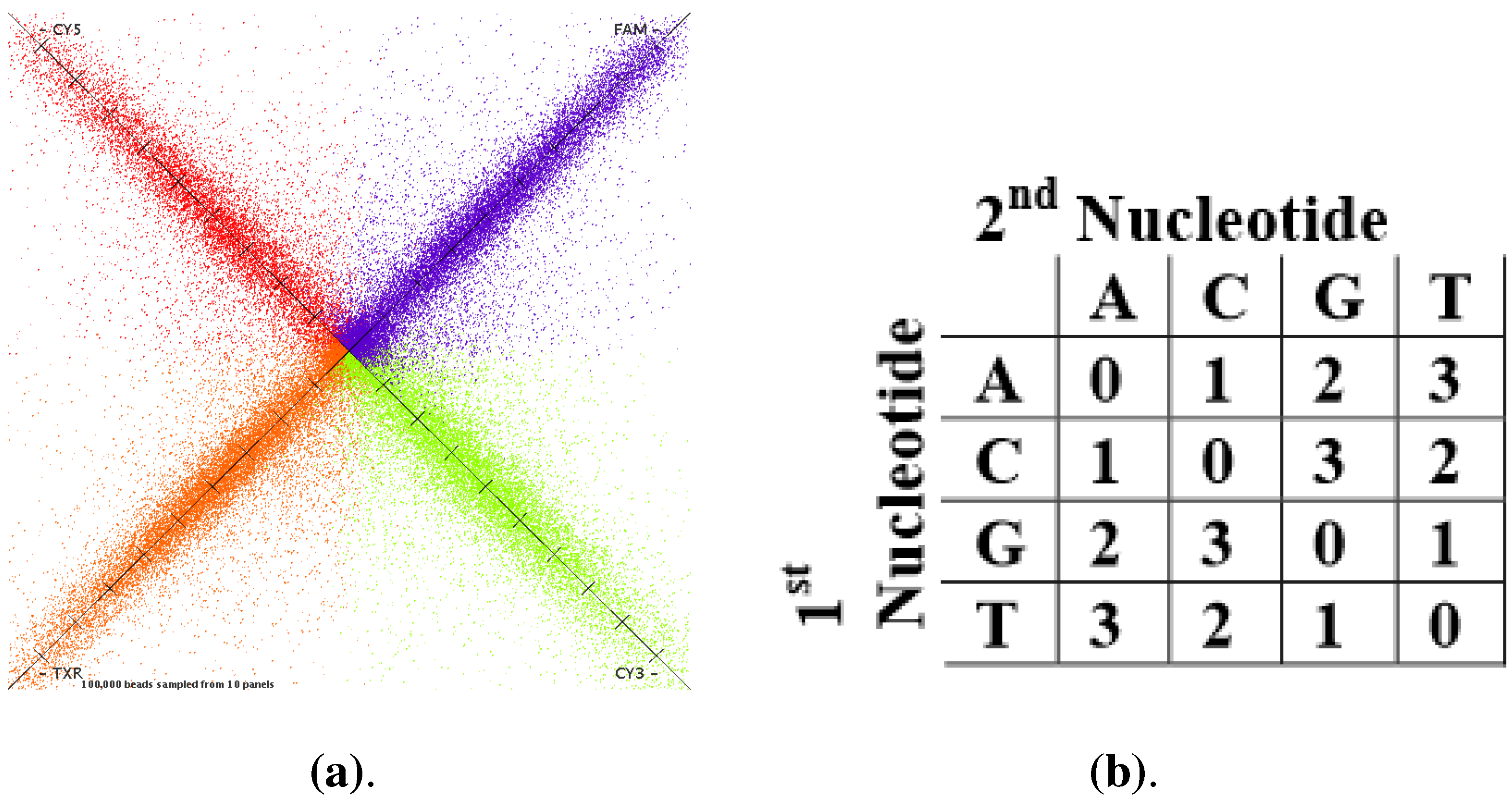
(missing color call) to 93, even though it rarely exceeds 60). When visualizing the dyes on a satay plot (
Figure 1a), those that are on the axis and far from the origin are the beads that have a bright signal in only one channel. In general, the brighter the dye, the greater the difference in signal between colors and the higher the QV.
Figure 1.
Elements of color-space sequencing. (a) Satay plot; (b) Color space matrix.
Figure 1.
Elements of color-space sequencing. (a) Satay plot; (b) Color space matrix.
Unlike other sequencers, which adhere to the standard FASTQ file, SOLiD outputs either a couple of Color Space FASTA (CSFASTA) and QV files, or an eXtensible SeQuence (XSQ) file. The former output encodes sequences of short-reads by four dye color codes (6-FAM = 0 (blue), CY3 = 1 (green), Texas Red = 2 (yellow), CY5 = 3 (red)). Each corresponds unambiguously to four combinations of adjacent nucleotides, according to
Figure 1b.
In fact, rather than reading one base per cycle, information on two bases is measured simultaneously, and in each cycle, one of four colors is called. In a separate file, the quality of color calls is reported. For the sake of completeness, we recall that each QV is calculated using a Phred-like score , where q is the quality value and p is the predicted probability that the color call is incorrect. Contrarily, the XSQ data format organizes in a hierarchical fashion and conforms to the HDF5 format. It losslessly compresses FASTA sequences and QVs in binary files. Average compression rates are around 60%.
Irrespective of their encoding, short-reads are mapped against a reference genome in order to sort them and reconstruct the target genome (
Figure 2). Aligned reads are piled up in textual (Sequence Alignment/Map, SAM) or binary (Binary Alignment/Map, BAM) archives [
6]. SAM is a tab-delimited text format consisting of a header section, which is optional, and an alignment section. Each alignment line has several mandatory fields for essential alignment information such as mapping position, and variable number of optional fields for aligner specific information. The SAM/BAM format is already supported by a number of read mappers and is becoming the de-facto standard alignment format.
Figure 2.
Short-Reads alignment.
Figure 2.
Short-Reads alignment.
After alignment, SAM/BAM files are naturally subjected to deep scan, in search of point mutations (Single Nucleotide Polymorphisms, SNPs) or insertions/deletions (Indels) of short amino acid sequences. According to several factors, such as the ratio between alternate and reference alleles, depth of coverage and quality of call, a genotype is assigned to each locus. On average, a typical whole genome experiment identifies million SNPs, of which around are novel. This means about 1 out of every 1000 nucleotides yields a mutation, but only 1 out of every 6700 nucleotides yields a novel mutation. Variant callers usually format information of the detected variants in Variant Call Format (VCF), a text file format (most likely stored in uncompressed manner). It contains meta-information lines and data lines, which carry information about a locus in the genome.
Hence, NGS experiments require storage of three levels of data. Raw or unprocessed data often comprise images and standard textual representations of short biological sequences (also known as short-reads) and quality scores. Data occupancy highly depends on the sequencing platform, size of the target regions, wished average depth and breadth of coverage and sequencing modality. Roughly, raw files require about tens of GB to a few TB of free space to be stored. Size of SAM/BAM files depends on the experimental settings as well, but roughly ranges from tens to hundreds of GB. For example, assuming a 3 gigabase (GB) human genome length and an average depth of coverage of 30×, one would have 90 GB (roughly occupying 90 GB of disk space, if we map one character to one byte). Considering that a typical SAM file contains both mapped/unmapped short-reads and QVs, the total size would be around 180 GB (ignoring control lines and carriage returns). BAM files typically compress to of original size. A resulting 30× BAM should occupy about 100 GB of disk space. The size of a basic VCF file yet depends on the target genomic regions. Assuming each data line taking around 45 bytes, a whole human genome search would yield a file with a maximum size of 140 MB.
We focus here on data produced by SOLiD sequencers. We deal with software tools capable of compressing BAM files generated from XSQ files. We do not consider compression solutions for XSQ or VCF files in this study, since they do not actually require any special inflation that any general-purpose compressor software wound not be able to provide. In
Section 2, we describe our analysis strategy. We present our datasets, the software tools under evaluation and the criteria for measuring their compression performance. In
Section 3 and
Section 4, we comment results and conclude the paper, respectively.
2. Experimental Section
We present a simple analysis workflow, which deals with real sequencing data. In particular, we focus on the “exome”, which is the coding part of the genome. It represents an as important as small () fraction of the entire genome. After mapping of short-reads to a reference genome, we evaluate and compare the ability of four publicly available software tools to compress aligned reads, in terms of the extent of data lost. Levels of loss will be calculated on proportions of missed variants and reduced coverage.
2.1. Materials and Methods
DNA was extracted from ten different patients affected by mendelian neurogenetic disorders. Exonic regions were targeted using the Agilent SureSelect 50 Mb All Exon Kit. Paired-end libraries of reads were yielded for five samples, while single-fragment reads were produced for the other half, using the Exact Call Chemistry (ECC) system. Briefly, paired-end libraries consist of pairs of reads (e.g., 75/35 base pair long) that map to the extremities (5’ and 3’ ends) of the same DNA fragment. ECC provides a further primer cycle with the aim to correct eventual color mismatches.
Regardless of the sequencing technique, we have mapped short-reads to the hg19 reference genome, using Lifescope. The resulting alignments were sorted and checked for the presence of PCR duplicates by Picard [
7]. We used GATK [
8] to recalibrate QVs and correct misalignments. Statistics of coverage of the targeted regions (about
exonic regions, 51 million sites) were computed both using BEDTools [
9] and custom R scripts. We set SAMtools [
6] with standard parameters for exome analysis to call SNPs and short Indels. High quality variants (those with
,
) were annotated by using wANNOVAR [
10].
We have applied this analysis pipeline, as exhaustively documented in [
11], on BAM files before and after compression. We have calculated the site depth and breadth of coverage (DOC and BOC) before and after compression, with the aim to compare the compression tools. Generally, DOC is calculated for a given genomic site as the number of short-reads covering it. The deeper the coverage of a variant, the more accurate and truer will be its call and genotype. Contrarily, BOC measures the extent of target genomic regions covered by the short-reads. Furthermore, comparisons between sizes of the original and compressed BAM files as well as the compression/decompression times were provided.
2.2. Tools
Several compression packages exist that deal with raw sequence data or directly with SAM/BAM files. From the latter class, we considered CRAMtools 1.0 [
12], Quip 1.1.4 [
13], NGC [
14] and the
ReduceReads module of the GATK suite [
8]. The first three differ from ReduceReads as the latter does not allow to restore the uncompressed form of the BAM files. The compressed BAM is indeed a valid BAM file, with reduced size, thereby preserving “essential” information.
CRAM is a framework technology that builds on early proof-of-principle for reference-based compression. The basis of this method is the efficient storage of information that is identical or near-identical to input “reference” sequences. The key feature is that new sequences identical to the reference have minimal impact on storage regardless of their length or depth of sequencing coverage. Most reads in a resequencing run match the sequence perfectly or near-perfectly, thereby requiring the efficient storage of mapping properties and deviations from the reference in an efficient manner. Much of the efficiency of this method relies on appropriate use of Golomb codes [
15] that are now a standard compression technique for storing integer values.
CRAMtools: java -jar cramtools-1.0.jar cram
--input-bam-file sample.bam
--reference-fasta-file hg19.fa
--output-cram-file sample.cram
Quip is a lossless compression algorithm for NGS data in the FASTQ and SAM/BAM formats. It uses an arithmetic coding, a form of entropy coding, which refines the Huffman coding. Arithmetic coding is a particularly elegant means of compression in that it allows a complete separation between statistical modeling and encoding. In Quip, the same arithmetic coder is used to encode quality scores, read identifiers, nucleotide sequences and alignment information, but with very different statistical models for each, which gives it a tremendous advantage over general-purpose compression algorithms that lump everything into a single context. Furthermore, the algorithm is adaptive, as it tunes parameters as data are compressed.
Quip: quip --input=bam
-output=quip
-r hg19.fa
sample.bam
NGC enables lossless and lossy compression strategies. It builds on the same idea of CRAM, thereby traversing the bases in an alignment of reads in a per-column way rather than handling each read individually. This leads, ultimately, to a reduction of coding and, in consequence, to a more efficient data compression. QVs are categorized and compressed in a lossy way, yet preserving 99%–100% of all called variants on average.
NGC: java -jar ngc-core-0.0.1-standalone.jar compress
-i sample.bam
-o sample.ngc
-r hg19.fa
ReduceReads is a one-way lossy compressor that aims at keeping only essential information for variant calling. It is tunable in terms of compression levels. The default values have been shown to reduce a typical WES BAM file 100×. The higher the coverage, the bigger the savings in file size and performance of the downstream tools. Generally, it distinguishes between variable and consensus regions around variants and holds the variable windows around the disagreement between the reads with sufficient information for subsequent analysis. A disagreement can be triggered for any generic analysis goal with different thresholds (e.g., heterozygous sites, insertions, deletions). Furthermore, the original surviving reads are downsampled to a “more than reasonable” coverage for analysis.
ReduceReads: java -jar GenomeAnalysisTK.jar -R hg19.fa
-T ReduceReads
-I sample.bam
-o sample.reduced.bam
Generally, a compression-decompression round was carried out for each of the first three tools and for each sample. Quip has proven to be unable to handle five of the ten BAM files, i.e., those produced with the SOLiD ECC technique and yielded by Lifescope. To perform comparisons of their performance, we used a computer cluster consisting of two nodes, each equipped with 48 AMD Opteron 6172, 2.1 GHz processors, with a 256 GB shared RAM. Each tool was run with standard parameters (as suggested in the reference manuals).
3. Results and Discussion
Generally, WES detects 20,000–30,000 SNPs, even if only 40% of these cause amino acid changes [
16]; an even smaller portion, in the order of a few hundreds, are private missense mutations, and still fewer are pathogenic [
17,
18]. Briefly, we obtained about 14,000–16,000 variants (SNPs and short Indels) from the ten original exomic samples. As expected, 50%–55% of such variants were synonymous (
i.e., silent) SNPs, while 40%–42% were nonsynonymous (
i.e., amino acid changing mutations), 2%–5% were short exonic Indels and 0.1%–0.5% were causative either of a premature stop of the translation of the protein or, contrarily, of a loss of the stop coding signal.
As anticipated, we compared the compression performance of the aforementioned tools, by comparing the number and properties (
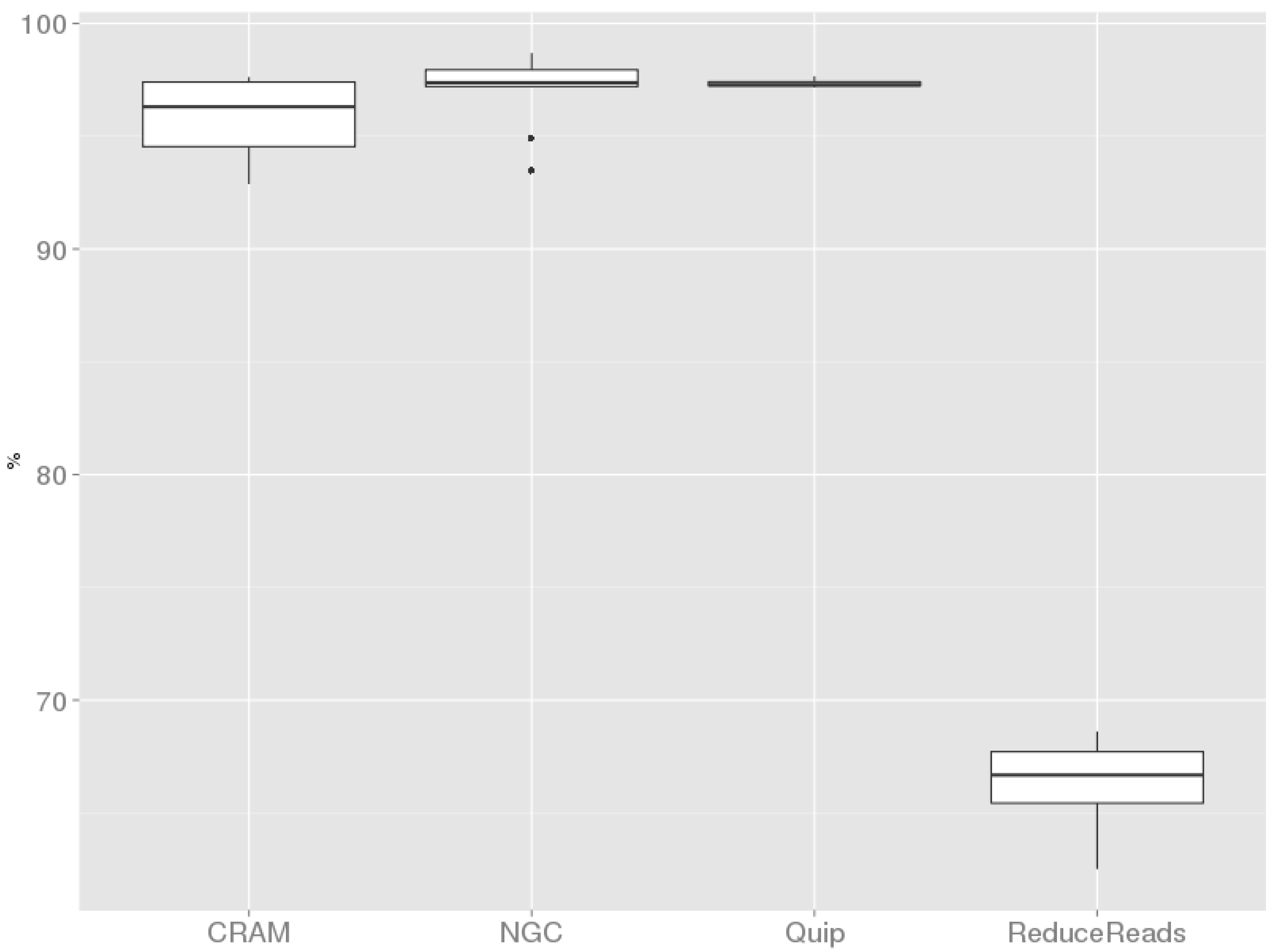
i.e., genomic position, mutated allele and genotype) of the called variants before and after compression. We correctly retrieved more than 95% of the original variants after decompression of the BAM files compressed by CRAMtools, NGC and Quip. Contrarily, ReduceReads caused an evident data loss: Only 66.4%, on average, of the originally called variants surviving the compression step.
Figure 3 shows these performances. The performance of Quip is biased since the calculation was made on five out of ten samples. Interestingly, we observe that the sizes of the classes of exonic variants (synonymous SNPs, nonsynonymous SNPs, short Indels,
etc.) were proportionally retained throughout the compression experiments.
Figure 3.
Proportion of variants that survived the compression step. Point dimensions indicate tool-specific sample size (n): for Cramtools, NGC and ReduceReads; for Quip.
Figure 3.
Proportion of variants that survived the compression step. Point dimensions indicate tool-specific sample size (n): for Cramtools, NGC and ReduceReads; for Quip.
We have also analyzed the mean distributions of DOC and BOC: Coverage data were preserved by all compression tools except for ReduceReads (as expected), which downsamples, by design, the DOC to a reasonable level for analysis.
Table 1 shows the coverage reduction levels achieved by ReduceReads only.
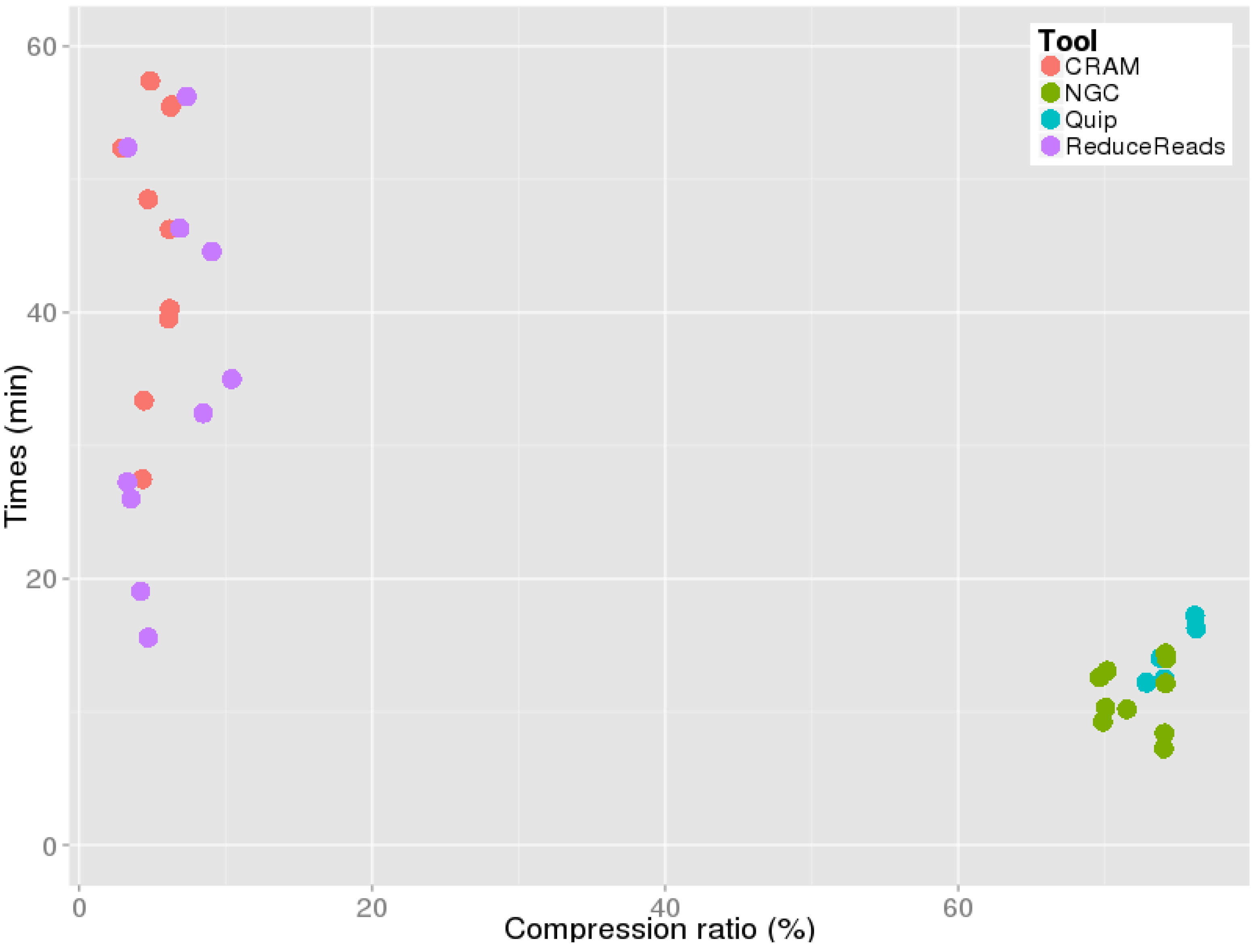
Finally, we have observed the wall clock times and disk occupancy saving provided by each tool. CRAMtools and ReducedReads took from 20 to 60 min to compress the ten original BAM files, whose sizes ranged from
to
GB. The ratios between uncompressed and compressed files were proportional. NGC and Quip exhibit a more conservative compression policy, with expected reduced running times (see
Figure 4). Furthermore, NGC and Quip deliver compressed files with comparable sizes, irrespective of their original sizes.
Table 1.
DOC and BOC of original and compressed BAMs by ReduceReads.
Table 1.
DOC and BOC of original and compressed BAMs by ReduceReads.
| Mapped | BOC | Mean DOC | Mean DOC | DOC deflation |
|---|
| reads | (%) | (Original) | (ReduceReads) | rate (%) |
|---|
| 36,371,970 | 93.14 | 32.28 | 5.56 | 83 |
| 52,081,471 | 94.37 | 46.36 | 5.08 | 89 |
| 50,599,995 | 93.7 | 45.02 | 6.67 | 85 |
| 40,586,880 | 93.34 | 35.88 | 4.22 | 88 |
| 35,004,528 | 93.58 | 30.96 | 4.29 | 86 |
| 49,141,745 | 93.37 | 61.41 | 7.18 | 88 |
| 49,142,549 | 93.09 | 61.3 | 7.13 | 88 |
| 42,024,818 | 92.97 | 52.32 | 6.55 | 87 |
| 23,754,890 | 92.04 | 29.56 | 7.2 | 76 |
| 29,203,073 | 92.5 | 36.37 | 5.66 | 84 |
Figure 4.
Compression performance.
Figure 4.
Compression performance.
4. Conclusions
Given the dramatic expansion of NGS technology in biomedical research and the consequent production of huge amount of data, data compression has become of great importance. Many algorithms have been published in order to shrink NGS data sizes, trying to compress raw image data (i.e., fluorescence measurements during the sequencing cycles) or DNA sequence data, at level of the genomic sequence or of the reference-aligned fragments. In this work we have tested some of the most recent and used compression tools for SAM/BAM files.
The original contribution of this work is the test of such tools on data coming from SOLiD sequencing platforms and their official analysis toolkit: Lifescope. This aim was not ever pursued since SOLiD sequencers were not so popular. Contrarily, today this trend is going inversely. As described in
Section 1, SOLiD differs from other similar tools, not only for its original function, but for the data format of its output files. Thus, we tried to assess whether and how original information were preserved from these files, by measuring the starting and residual called variants and target coverage, before and after compression.
The CRAMtools, Quip and NGC Package basically showed to preserve almost all original information, meaning that quite any changes made during compression could be undone after decompression. Contrastingly, ReduceReads proved to irreversibly lose more than of original variants after compression. Generally, CRAMtools and ReduceReads achieved very high compression performance, with as high storage saving.
Our results provide useful indications about compression performance of SOLiD data. We were unable to retrieve all the original variants from the compressed datasets with any of these tools. However, CRAMtools exhibited the best performance among all tested softwares. NGC resulted to be the easiest to use, even though the compression rates were not so impressive. Quip performed quite well, even though it bumped against the fragment-based BAM files generated by Lifescope. On the other hand, the ReduceReads module of GATK exhibited extraordinary compression rates at the expense of a significant number of lost variants.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
