Chinese Fir Breeding in the High-Throughput Sequencing Era: Insights from SNPs


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction, Genotyping-by-Sequencing and SNP Identification
2.3. Statistical Analyses
3. Results
3.1. Genome-Wide SNP Mining
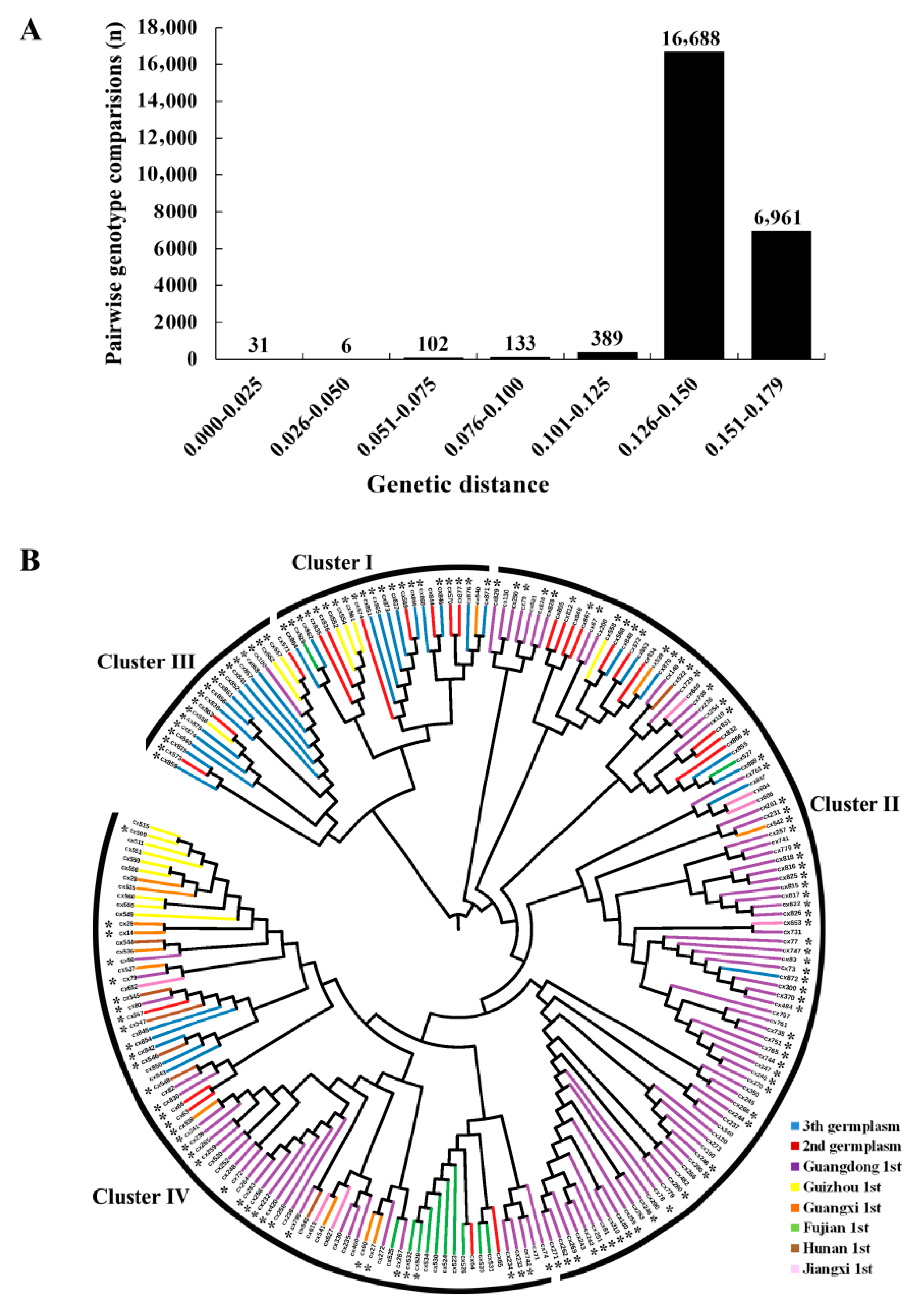
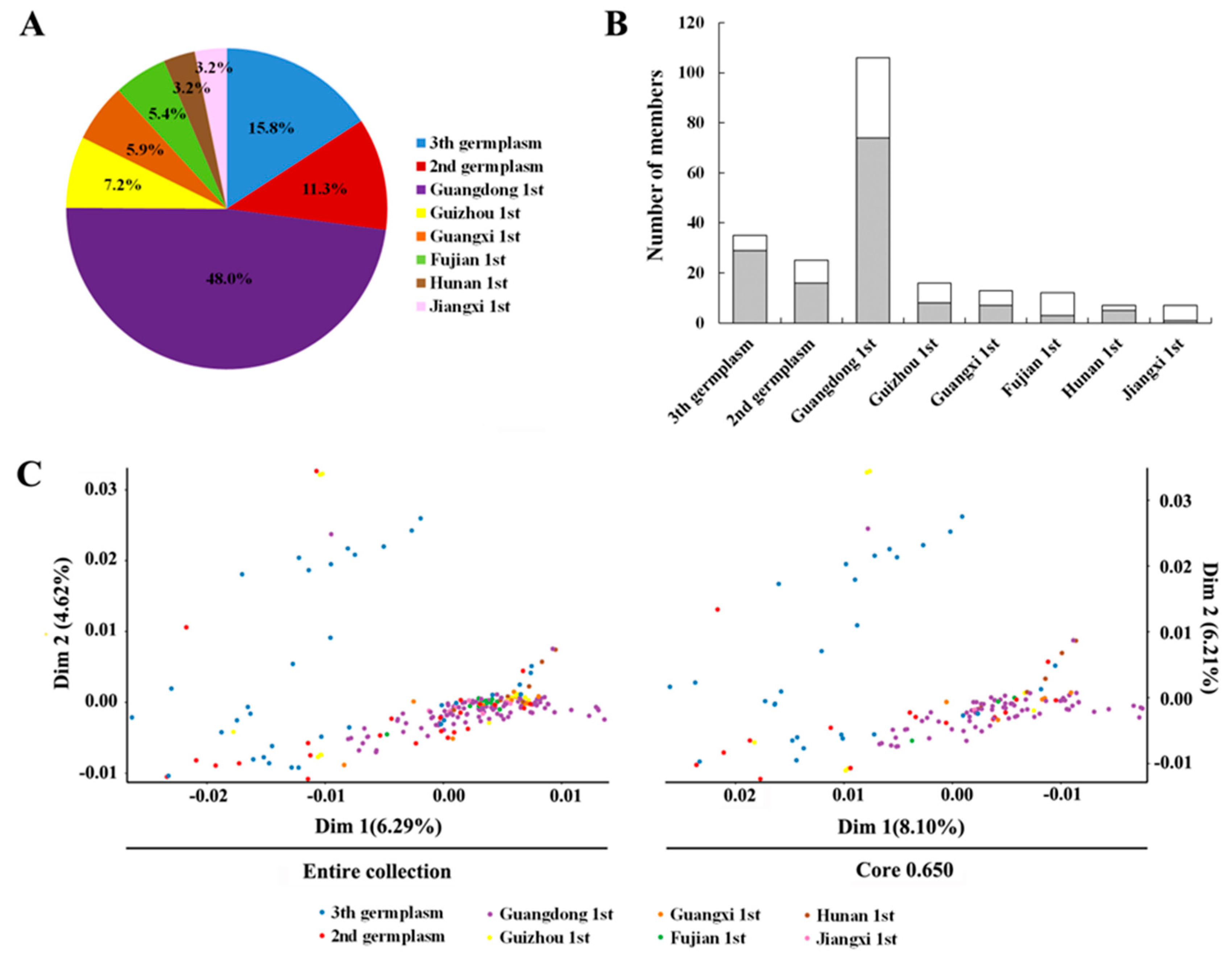
3.2. Genetic Diversity Assessment
3.3. Population Structure Inferring
3.4. Core Collection Development
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hodge, G.R.; Dvorak, W.S. Breeding southern US and Mexican pines for increased value in a changing world. New For. 2014, 45, 295–300. [Google Scholar] [CrossRef]
- Wu, H.X.; Hallingbäck, H.R.; Sánchez, L. Performance of seven tree breeding strategies under conditions of inbreeding depression. G3 Genes Genomes Genet. 2016, 6, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Isik, F. Genomic selection in forest tree breeding: The concept and an outlook to the future. New For. 2014, 45, 379–401. [Google Scholar] [CrossRef]
- Zapata-Valenzuela, J.; Whetten, R.W.; Neale, D.; McKeand, S.; Isik, F. Genomic estimated breeding values using genomic relationship matrices in a cloned population of loblolly pine. G3 Genes Genomes Genet. 2013, 3, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Thistlethwaite, F.R.; Ratcliffe, B.; Klápště, J.; Porth, I.; Chen, C.; Stoehr, M.U.; El-Kassaby, Y.A. Genomic prediction accuracies in space and time for height and wood density of Douglas-fir using exome capture as the genotyping platform. BMC Genom. 2017, 18, 930. [Google Scholar] [CrossRef] [PubMed]
- Grattapaglia, D.; Silva-Junior, O.B.; Resende, R.T.; Cappa, E.P.; Müller, B.S.F.; Tan, B.; Isik, F.; Ratcliffe, B.; El-Kassaby, Y.A. Quantitative genetics and genomics converge to accelerate forest tree breeding. Front. Plant Sci. 2018, 9, 1693. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.Q.; Duan, H.J.; Hu, D.H.; Li, Y.; Hao, Y.B. Genotypic variation of Cunninghamia lanceolata revealed by phenotypic traits and SRAP markers. Dendrobiology 2015, 74, 85–94. [Google Scholar] [CrossRef]
- Howe, G.T.; Yu, J.; Knaus, B.; Cronn, R.; Kolpak, S.; Dolan, P.; Lorenz, W.W.; Dean, J.F. A SNP resource for Douglas-fir: De novo transcriptome assembly and SNP detection and validation. BMC Genom. 2013, 14, 137. [Google Scholar] [CrossRef]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP markers and their impact on plant breeding. Int. J. Plant Genom. 2012, 2012, 728398. [Google Scholar] [CrossRef]
- Zheng, H.Q.; Duan, H.J.; Hu, D.H.; Wei, R.P.; Li, Y. Sequence-related amplified polymorphism primer screening on Chinese fir (Cunninghamia lanceolata (Lamb.) Hook). J. For. Res. 2015, 26, 101–106. [Google Scholar] [CrossRef]
- Taheri, S.; Lee Abdullah, T.; Yusop, M.R.; Hanafi, M.M.; Sahebi, M.; Azizi, P.; Shamshiri, R.R. Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules 2018, 23, 399. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Mitchell, S.E.; Elshire, R.J.; Buckler, E.S.; El-Kassaby, Y.A. Mining conifers’mega-genome using rapid and efficient multiplexed high-throughput genotyping-by-sequencing (GBS) SNP discovery platform. Tree Genet. Genomes 2013, 9, 1537–1544. [Google Scholar] [CrossRef]
- Karam, M.J.; Lefèvre, F.; Dagher-Kharrat, M.B.; Pinosio, S.; Vendramin, G.G. Genomic exploration and molecular marker development in a large and complex conifer genome using RADseq and mRNAseq. Mol. Ecol. Resour. 2015, 15, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Prunier, J.; Verta, J.P.; MacKay, J.J. Conifer genomics and adaptation: At the crossroads of genetic diversity and genome function. New Phytol. 2016, 209, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Utrilla, P.; Goswami, C.; Cottrell, J.E.; Pong-Wong, R.; Law, A.; A’Hara, S.W.; Lee, S.J.; Woolliams, J.A. QTL analysis and genomic selection using RADseq derived markers in Sitka spruce: The potential utility of within family data. Tree Genet. Genomes 2017, 13, 33. [Google Scholar] [CrossRef]
- Parchman, T.L.; Jahner, J.P.; Uckele, K.A.; Galland, L.M.; Eckert, A.J. RADseq approaches and applications for forest tree genetics. Tree Genet. Genomes 2018, 14, 39. [Google Scholar] [CrossRef]
- Ueno, S.; Uchiyama, K.; Moriguchi, Y.; Ujino-Ihara, T.; Matsumoto, A.; Wei, F.J.; Saito, M.; Higuchi, Y.; Futamura, N.; Kanamori, H.; et al. Scanning RNA-Seq and RAD-Seq approach to develop SNP markers closely linked to MALE STERILITY 1 (MS1) in Cryptomeria japonica D. Don. Breed. Sci. 2019, 69, 19–29. [Google Scholar] [CrossRef]
- Su, Y.; Hu, D.H.; Zheng, H.Q. Detection of SNPs based on DNA specific-locus amplified fragment sequencing in Chinese fir (Cunninghamia lanceolata (Lamb.) Hook). Dendrobiology 2016, 76, 73–79. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Han, X.J.; Sang, J.; He, X.L.; Liu, M.Y.; Qiao, G.R.; Zhuo, R.Y.; He, G.P.; Hu, J.J. Transcriptome analysis of immature xylem in the Chinese fir at different developmental phases. PeerJ 2016, 4, e2097. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.H.; Su, Y.; Wu, S.J.; Wu, J.Z.; Wang, R.H.; Yan, S.; Wei, R.P.; Zheng, H.Q. Association of SRAP markers with juvenile wood basic density and growth traits in Cunninghamia lanceolata (Lamb.) Hook. Dendrobiology 2018, 79, 111–118. [Google Scholar] [CrossRef]
- Zheng, H.Q.; Hu, D.H.; Wang, R.H.; Wei, R.P.; Yan, S. Assessing 62 Chinese fir (Cunninghamia lanceolata) breeding parents in a 12-year grafted clone test. Forests 2015, 6, 3799–3808. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Beukelaer, H.; Smýkal, P.; Davenport, G.F.; Fack, V. Core Hunter II: Fast core subset selection based on multiple genetic diversity measures using Mixed Replica search. BMC Bioinform. 2012, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birol, I.; Raymond, A.; Jackman, S.D.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.; Keeling, C.I.; Brand, D.; Vandervalk, B.P.; et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics 2013, 29, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Stevens, K.A.; Crepeau, M.W.; Puiu, D.; Wegrzyn, J.L.; Yorke, J.A.; Langley, C.H.; Neale, D.B.; Salzberg, S.L. An improved assembly of the loblolly pine mega-genome using long-read single-molecule sequencing. GigaScience 2017, 6, 1–4. [Google Scholar] [PubMed]
- Stevens, K.A.; Wegrzyn, J.L.; Zimin, A.; Puiu, D.; Crepeau, M.; Cardeno, C.; Paul, R.; Gonzalez-Ibeas, D.; Koriabine, M.; Holtz-Morris, A.E.; et al. Sequence of the sugar pine megagenome. Genetics 2016, 204, 1613–1626. [Google Scholar] [CrossRef] [PubMed]
- Neale, D.B.; McGuire, P.E.; Wheeler, N.C.; Stevens, K.A.; Crepeau, M.W.; Cardeno, C.; Zimin, A.V.; Puiu, D.; Pertea, G.M.; Sezen, U.U.; et al. The Douglas-fir genome sequence reveals specialization of the photosynthetic apparatus in Pinaceae. 2017. G3 Genes Genomes Genet. 2017, 7, 3157–3167. [Google Scholar]
- Kuzmin, D.A.; Feranchuk, S.I.; Sharov, V.V.; Cybin, A.N.; Makolov, S.V.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Stepwise large genome assembly approach: A case of Siberian larch (Larix sibirica Ledeb). BMC Bioinform. 2019, 20 (Suppl. 1), 37. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Puiu, D.; Crepeau, M.W.; Stevens, K.; Salzberg, S.L.; Langley, C.H.; Neale, D.B. Genomic architecture of complex traits in loblolly pine. New Phytol. 2019, 221, 1789–1801. [Google Scholar] [CrossRef]
- Wakeley, J. The excess of transitions among nucleotide substitutions: New methods of estimating transition bias underscore its significance. Trends Ecol. Evol. 1996, 11, 158–162. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1996, 351, 1291–1298. [Google Scholar]
- Frankel, O.H.; Brown, A.H.D. Plant genetic resources today: A critical appraisal. In Crop Genetic Resources: Conservation and Evaluation; Holden, J.H.W., Williams, J.T., Eds.; Georges Allen & Unwin Ltd.: London, UK, 1984; pp. 249–257. [Google Scholar]
- Duan, H.J.; Cao, S.; Zheng, H.Q.; Hu, D.H.; Lin, J.; Cui, B.B.; Lin, H.Z.; Hu, R.Y.; Wu, B.; Sun, Y.H.; et al. Genetic characterization of Chinese fir from six provinces in southern China and construction of a core collection. Sci. Rep. 2017, 7, 13814. [Google Scholar] [CrossRef]
- Liu, F.M.; Zhang, N.N.; Liu, X.J.; Yang, Z.J.; Jia, H.Y.; Xu, D.P. Genetic diversity and population structure analysis of Dalbergia odorifera germplasm and development of a core collection using microsatellite markers. Genes 2019, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- Guardo, M.D.; Scollo, F.; Ninot, A.; Rovira, M.; Hermoso, J.F.; Distefano, G.; Malfa, S.L.; Batlle, I. Genetic structure analysis and selection of a core collection for carob tree germplasm conservation and management. Tree Genet. Genomes 2019, 15, 41. [Google Scholar] [CrossRef]
- Le Cunff, L.; Fournier-Level, A.; Laucou, V.; Vezzulli, S.; Lacombe, T.; Adam-Blondon, A.F.; Boursiquot, J.M.; This, P. Construction of nested genetic core collections to optimize the exploitation of natural diversity in Vitis vinifera L. subsp. sativa. BMC Plant Biol. 2008, 8, 31. [Google Scholar] [CrossRef]
- Haouane, H.; El Bakkali, A.; Moukhli, A.; Tollon, C.; Santoni, S.; Oukabli, A.; El Modafar, C.; Khadari, B. Genetic structure and core collection of the World Olive Germplasm Bank of Marrakech: Towards the optimised management and use of Mediterranean olive genetic resources. Genetica 2011, 139, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Lassois, L.; Denancé, C.; Ravon, E.; Guyader, A.; Guisnel, R.; Hibrand-Saint-Oyant, L.; Poncet, C.; Lasserre-Zuber, P.; Feugey, L.; Durel, C.E. Genetic diversity, population structure, parentage analysis, and construction of core collections in the French apple germplasm based on SSR markers. Plant Mol. Biol. Rep. 2016, 34, 827–844. [Google Scholar] [CrossRef]
- Urrestarazu, J.; Kägi, C.; Bühlmann, A.; Gassmann, J.; Santesteban, L.G.; Frey, J.E.; Kellerhals, M.; Miranda, C. Integration of expert knowledge in the definition of Swiss pear core collection. Sci. Rep. 2019, 9, 8934. [Google Scholar] [CrossRef]
- Miyamoto, N.; Ono, M.; Watanabe, A. Construction of a core collection and evaluation of genetic resources for Cryptomeria japonica (Japanese cedar). J. For. Res. 2015, 20, 186–196. [Google Scholar] [CrossRef]
- Wang, J.; Guan, Y.; Wang, Y.; Zhu, L.; Wang, Q.; Hu, Q.; Hu, J. A strategy for finding the optimal scale of plant core collection based on Monte Carlo simulation. Sci. World J. 2014, 2014, 503473. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Value |
|---|---|
| Number of reads | 261,513,887 |
| Average Q30 percentage | 88.86% |
| Average GC percentage | 38.12% |
| Average depth | 7.32 |
| Total number of SLAFs | 748,509 |
| Number of polymorphic SLAFs | 263,099 |
| Number of no polymorphic SLAFs | 485,410 |
| Total single nucleotide polymorphisms (SNPs) | 1,396,279 |
| The high-qualified SNPs (Integrity>0.8, minor allele frequency >0.05) | 108,753 |
| Transition | |
| A/G | 519,627 (37.97%) |
| C/T | 518,869 (37.92%) |
| Transversion | |
| A/C | 99,065 (7.24%) |
| A/T | 64,848 (4.74%) |
| C/G | 66,144 (4.83%) |
| G/T | 99,886 (7.30%) |
| Sub-Origin Set | 3rd Germplasm | 2nd Germplasm | Guangdong 1st | Guizhou 1st | Guangxi 1st | Fujian 1st | Hunan 1st | Jiangxi 1st |
|---|---|---|---|---|---|---|---|---|
| 3rd germplasm | - | |||||||
| 2nd germplasm | 0.0285 | - | ||||||
| Guangdong 1st | 0.0355 | 0.0305 | - | |||||
| Guizhou 1st | 0.0539 | 0.0510 | 0.0461 | - | ||||
| Guangxi 1st | 0.0621 | 0.0532 | 0.0396 | 0.0716 | - | |||
| Fujian 1st | 0.0633 | 0.0573 | 0.0420 | 0.0734 | 0.0688 | - | ||
| Hunan 1st | 0.0791 | 0.0770 | 0.0639 | 0.0958 | 0.0865 | 0.0946 | - | |
| Jiangxi 1st | 0.0769 | 0.0717 | 0.0536 | 0.0913 | 0.0846 | 0.0861 | 0.0990 | - |
| Source of Variation | df | Sum of Square Difference | Mean of Square Difference | Components of Covariance (%) |
|---|---|---|---|---|
| Among sub-origin sets | 7 | 0.0640 | 0.0091 | 5.85 |
| Within genotypes | 213 | 0.8083 | 0.0038 | 94.15 |
| Total | 220 | 0.8723 | 0.0040 | 100.00 |
| Core Collection | Size (n) | MR | MRmin | CE | CEmin | SH | HE | NE | PN | CV | The Most Feasible K Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Core 0.200 | 44 | 0.3819 | 0.2818 | 0.3945 | 0.2962 | 11.9985 | 0.2515 | 1.3855 | 0.0013 | 0.9987 | 1 |
| Core 0.300 | 66 | 0.3806 | 0.1186 | 0.3931 | 0.1257 | 11.9992 | 0.2513 | 1.3839 | 0.0002 | 0.9999 | 1 |
| Core 0.400 | 88 | 0.3796 | 0.1138 | 0.3921 | 0.1203 | 12.0002 | 0.2517 | 1.3845 | 0.0000 | 1.0000 | 2 |
| Core 0.500 | 110 | 0.3790 | 0.1138 | 0.3915 | 0.1203 | 12.0001 | 0.2513 | 1.3833 | 0.0000 | 1.0000 | 2 |
| Core 0.600 | 132 | 0.3784 | 0.0995 | 0.3909 | 0.1058 | 12.0002 | 0.2513 | 1.3829 | 0.0000 | 1.0000 | 3 |
| Core 0.625 | 138 | 0.3789 | 0.0995 | 0.3914 | 0.1058 | 12.0010 | 0.2518 | 1.3839 | 0.0000 | 1.0000 | 3 |
| Core 0.650 | 143 | 0.3788 | 0.0995 | 0.3913 | 0.1058 | 12.0009 | 0.2518 | 1.3837 | 0.0000 | 1.0000 | 4 |
| Core 0.675 | 149 | 0.3787 | 0.0995 | 0.3912 | 0.1058 | 12.0008 | 0.2516 | 1.3833 | 0.0000 | 1.0000 | 4 |
| Core 0.700 | 154 | 0.3780 | 0.1083 | 0.3904 | 0.1149 | 11.9999 | 0.2510 | 1.3820 | 0.0000 | 1.0000 | 4 |
| Core 0.800 | 176 | 0.3774 | 0.0995 | 0.3898 | 0.1058 | 11.9995 | 0.2505 | 1.3811 | 0.0000 | 1.0000 | 4 |
| Core 0.900 | 198 | 0.3771 | 0.0995 | 0.3894 | 0.1058 | 11.9989 | 0.2500 | 1.3801 | 0.0000 | 1.0000 | 4 |
| Entire collection | 221 | 0.3760 | 0.0995 | 0.3883 | 0.1058 | 11.9982 | 0.2495 | 1.3791 | 0.0000 | 1.0000 | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, H.; Hu, D.; Wei, R.; Yan, S.; Wang, R. Chinese Fir Breeding in the High-Throughput Sequencing Era: Insights from SNPs. Forests 2019, 10, 681. https://doi.org/10.3390/f10080681
Zheng H, Hu D, Wei R, Yan S, Wang R. Chinese Fir Breeding in the High-Throughput Sequencing Era: Insights from SNPs. Forests. 2019; 10(8):681. https://doi.org/10.3390/f10080681
Chicago/Turabian StyleZheng, Huiquan, Dehuo Hu, Ruping Wei, Shu Yan, and Runhui Wang. 2019. "Chinese Fir Breeding in the High-Throughput Sequencing Era: Insights from SNPs" Forests 10, no. 8: 681. https://doi.org/10.3390/f10080681
APA StyleZheng, H., Hu, D., Wei, R., Yan, S., & Wang, R. (2019). Chinese Fir Breeding in the High-Throughput Sequencing Era: Insights from SNPs. Forests, 10(8), 681. https://doi.org/10.3390/f10080681