Genetic Structure of Norway Spruce Ecotypes Studied by SSR Markers

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Site and Sampling
2.2. DNA Extraction and SSR Genotyping
2.3. Data Analysis
3. Results
3.1. Allele Frequencies and Statistic Parameters of Individual Loci
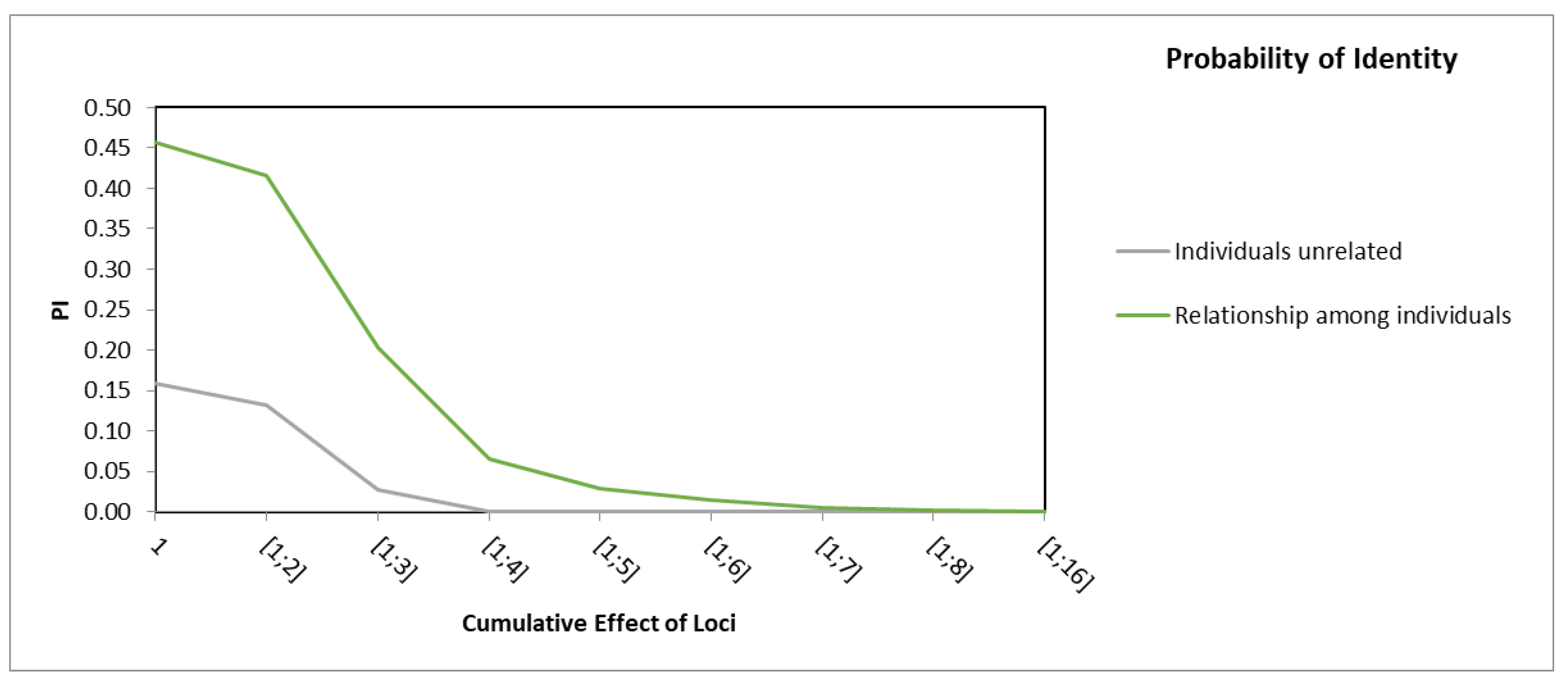
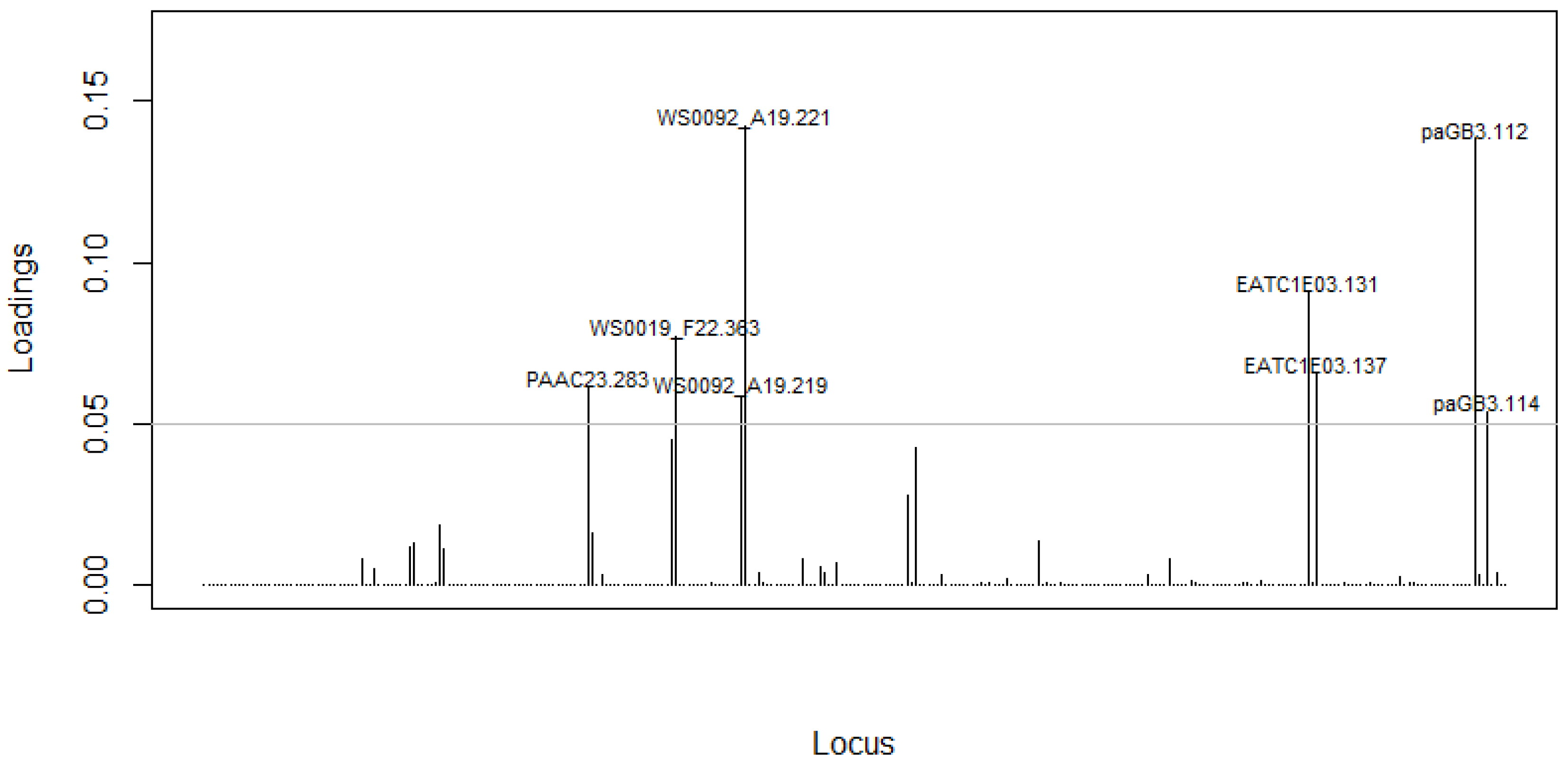
3.2. Discrimination Power of Assembled SSRs Markers
3.3. Parameters of Intra and Interpopulation Genetic Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koski, V.; Skrøppa, T.; Paule, L.; Wolf, H.; Turok, J. Technical Guidelines for Genetic Conservation of Norway Spruce (Picea abies (L.) Karst.); International Plant Genetic Resources Institute: Roma, Italy, 1997; ISBN 1581152833. [Google Scholar]
- Fajron, A. Picea abies. The IUCN Red List of Threatened Species 2017. Available online: https://www.iucnredlist.org/species/42318/71233492 (accessed on 20 February 2019).
- Skrøppa, T. EUFORGEN Technical Guidelines for Genetic Conservation and Use for Norway Spruce (Picea abies); Bioversity International: Rome, Italy, 2003; p. 6. [Google Scholar]
- Potterf, M.; Nikolov, C.; Kočická, E.; Ferenčík, J.; Mezei, P.; Jakuš, R. Landscape-level spread of beetle infestations from windthrown- and beetle-killed trees in the non-intervention zone of the Tatra National Park, Slovakia (Central Europe). For. Ecol. Manag. 2019, 432, 489–500. [Google Scholar] [CrossRef]
- Priehausser, G. Die fichten-variationen und kominationen des bayr. waldes nach phänotypishen merkmalen mit bestimmungsschlüssel. Forstwiss. Cent. 1958, 77, 151–171. [Google Scholar] [CrossRef]
- Samek, V. Metodika výzkumu morfologické proměnlivosti smrku z hlediska fytogeografického. Zprávy Lesn. Výzkumu 1964, 10, 18–25. [Google Scholar]
- Schmidt-Vogt, H. Die Fichte. Band I: Taxonomie, Verbreitung, Morphologie, Ökologie, Waldgesellschaften; Verlag Paul Parey: Berlin, Germany, 1977; p. 647. [Google Scholar]
- Morgenstern, K.E. Geographic Variation in Forest Trees: Genetic Basis and Application of Knowledge in Silviculture; Chow, F.E., Ed.; UBC Press: Vancouver, BC, Canada; University of British Columbia: Vancouver, BC, Canada, 1996; ISBN 0-7748-0560-9. [Google Scholar]
- Hynek, V.; Buriánek, V.; Benedíková, M.; Frýdl, J.; Kaňák, J. Výběrové stromy a porosty uznané pro sběr osiva, základní kriteria. VÚLHM Jíloviště-Strnady 1997, 1, 51. [Google Scholar]
- Mráček, Z.; Pařez, J. Pěstování Smrku; Státní zemědělské nakladatelství: Praha, Czech Republic, 1986; p. 203. [Google Scholar]
- Androsiuk, P.; Shimono, A.; Westin, J.; Lindgren, D.; Fries, A.; Wang, X.-R. Genetic status of Norway spruce (Picea abies) breeding populations for northern Sweden. Silvae Genet. 2013, 62, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Fajron, A. Picea abies, Norway spruce. IUCN Red List Threat. Species 2015, 8235, 12–14. [Google Scholar]
- Sylvén, N. Studier öfver Granens Formrikedom, Särskildt dess Förgreningstyper och Deras Skogliga Värde; Statens skogsförsöksanstalt: Stockholm, Sweden, 1910. [Google Scholar]
- Kiellander, C.L. Picea, Abies, Pseudotsuga. In Manual of Plant Breeding VI—Breeding of Legumes and Fruits, Viniculture and Silviculture; Verlag Paul Parey: Berlin/Hamburg, Germany, 1962; pp. 854–873. [Google Scholar]
- Liesebach, M.; Rau, H.-M.; König, A.O. Fichtenherkunftsversuch von 1962 und IUFRO-Fichtenherkunftsversuch von 1972: Ergebnisse von mehr als 30-jähriger Beobachtung in Deutschland; Universitätsverlag Göttingen: Gottingen, Germany, 2010; Volume 5, ISBN 3941875760. [Google Scholar]
- Gruber, F. Phänotypen der fichte (Picea abies (L.) Karst.) I. verzweigungsphänotypen: Genotyp und modifikation. Allg. Forst-u. J.-Ztg 1989, 160, 157–165. [Google Scholar]
- Geburek, T.; Robitschek, K.; Milasowszky, N. A tree of many faces: Why are there different crown types in Norway spruce (Picea abies [L.] Karst.)? Flora Morphol. Distrib. Funct. Ecol. Plants 2008, 203, 126–133. [Google Scholar] [CrossRef]
- Lloyd, D.G. Variation strategies of plants in heterogeneous environments. Biol. J. Linn. Soc. 1984, 21, 357–385. [Google Scholar] [CrossRef]
- White, T.; Adams, W.; Neale, D. Forest Genetics; CABI: Wallingford, UK, 2007; ISBN 9780851990835. [Google Scholar]
- Maghuly, F.; Pinsker, W.; Praznik, W.; Fluch, S. Genetic diversity in managed subpopulations of Norway spruce [Picea abies (L.) Karst.]. For. Ecol. Manag. 2006, 222, 266–271. [Google Scholar] [CrossRef]
- Schubert, R.; Mueller-Starck, G.; Riegel, R. Development of EST-PCR markers and monitoring their intrapopulational genetic variation in Picea abies (L.) Karst. Theor. Appl. Genet. 2001, 103, 1223–1231. [Google Scholar] [CrossRef]
- Yazdani, R.; Scotti, I.; Jansson, G.; Plomion, C.; Mathur, G. Inheritance and diversity of simple sequence repeat (SSR) microsatellite markers in various families of Picea abies. Hereditas 2003, 138, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperisen, C.; Buchler, U.; Gugerli, F.; Matyas, G.; Geburek, T.; Vendramin, G.G. Tandem repeats in plant mitochondrial genomes: Application to the analysis of population differentiation in the conifer Norway spruce. Mol. Ecol. 2001, 10, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Vendramin, G.G.; Anzidei, M.; Madaghiele, A.; Sperisen, C.; Bucci, G. Chloroplast microsatellite analysis reveals the presence of population subdivision in Norway spruce (Picea abies K.). Genome 2000, 43, 68–78. [Google Scholar] [CrossRef]
- Acheré, V.; Faivre-Rampant, P.; Jeandroz, S.; Besnard, G.; Markussen, T.; Aragones, A.; Fladung, M.; Ritter, E.; Favre, J.-M. A full saturated linkage map of Picea abies including AFLP, SSR, ESTP, 5S rDNA and morphological markers. Theor. Appl. Genet. 2004, 108, 1602–1613. [Google Scholar] [CrossRef]
- Shi, Y.-Z.; Forneris, N.; Rajora, O.P. Highly informative single-copy nuclear microsatellite DNA markers developed using an AFLP-SSR approach in black spruce (Picea mariana) and red spruce (P. rubens). PLoS ONE 2014, 9, e103789. [Google Scholar] [CrossRef]
- Bozhko, M.; Riegel, R.; Schubert, R.; Müller-Starck, G. A cyclophilin gene marker confirming geographical differentiation of Norway spruce populations and indicating viability response on excess soil-born salinity. Mol. Ecol. 2003, 12, 3147–3155. [Google Scholar] [CrossRef]
- Tollefsrud, M.M.; Kissling, R.; Gugerli, F.; Johnsen, Ø.; Skrøppa, T.; Cheddadi, R.; van der Knaap, W.O.; Latalowa, M.; Terhürne-Berson, R.; Litt, T.; et al. Genetic consequences of glacial survival and postglacial colonization in Norway spruce: Combined analysis of mitochondrial DNA and fossil pollen. Mol. Ecol. 2008, 17, 4134–4150. [Google Scholar] [CrossRef]
- Paglia, G.P.; Olivieri, A.M.; Morgante, M. Towards second-generation STS (sequence-tagged sites) linkage maps in conifers: A genetic map of Norway spruce (Picea abies K.). Mol. Gen. Genet. MGG 1998, 258, 466–478. [Google Scholar] [CrossRef]
- Chałupka, W.; Mejnartowicz, L.; Lewandowski, A. Reconstitution of a lost forest tree population: A case study of Norway spruce (Picea abies [L.] Karst.). For. Ecol. Manag. 2008, 255, 2103–2108. [Google Scholar] [CrossRef]
- Magyari, E.K.; Major, Á.; Bálint, M.; Nédli, J.; Braun, M.; Rácz, I.; Parducci, L. Population dynamics and genetic changes of Picea abies in the South Carpathians revealed by pollen and ancient DNA analyses. BMC Evol. Biol. 2011, 11, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.S.; Ratcliffe, S.T.; French, B.W.; Liu, L.; Sappington, T.W. Utility of EST-derived SSRs as population genetics markers in a beetle. J. Hered. 2008, 99, 112–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caré, O.; Müller, M.; Vornam, B.; Höltken, A.; Kahlert, K.; Krutovsky, K.; Gailing, O.; Leinemann, L. High morphological differentiation in crown architecture contrasts with low population genetic structure of German Norway spruce stands. Forests 2018, 9, 752. [Google Scholar] [CrossRef] [Green Version]
- Hamrick, J.L.; Godt, M.J.W. Allozyme diversity in cultivated crops. Crop Sci. 1997, 37, 26. [Google Scholar] [CrossRef]
- Jaramillo-Correa, J.P.; Verdu, M.; Gonzalez-Martinez, S.C. The contribution of recombination to heterozygosity differs among plant evolutionary lineages and life-forms. BMC Evol. Biol. 2010, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Shimono, A.; Wang, X.-R.; Torimaru, T.; Lindgren, D.; Karlsson, B. Spatial variation in local pollen flow and mating success in a Picea abies clone archive and their implications for a novel “breeding without breeding” strategy. Tree Genet. Genomes 2011, 7, 499–509. [Google Scholar] [CrossRef]
- Buschiazzo, E.; Ritland, C.; Bohlmann, J.; Ritland, K. Slow but not low: Genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol. Biol. 2012, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Müller-Starck, G. Survey of genetic variation as inferred from enzyme gene markers. In Genetic Variation in European Populations of Forest Trees; JD Sauerländers Verlag: Frankfurt am Main, Germany, 1995; pp. 20–37. [Google Scholar]
- Scotti, I.; Paglia, G.; Magni, F.; Morgante, M. Population genetics of Norway spruce (Picea abies Karst.) at regional scale: Sensitivity of different microsatellite motif classes in detecting differentiation. Ann. For. Sci. 2006, 63, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Tollefsrud, M.M.; Sønstebø, J.H.; Brochmann, C.; Johnsen, Ø.; Skrøppa, T.; Vendramin, G.G. Combined analysis of nuclear and mitochondrial markers provide new insight into the genetic structure of North European Picea abies. Heredity 2009, 102, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meloni, M.; Perini, D.; Binelli, G. The distribution of genetic variation in Norway spruce (Picea abies Karst.) populations in the western Alps. J. Biogeogr. 2007, 34, 929–938. [Google Scholar] [CrossRef]
- Westergren, M.; Bozic, G.; Kraigher, H. Genetic diversity of core vs. peripheral Norway spruce native populations at a local scale in Slovenia. iFor. Biogeosci. For. 2018, 11, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Lagercrantz, U.; Ryman, N. Genetic Structure of Norway Spruce (Picea abies): Concordance of Morphological and Allozymic Variation. Evolution 1990, 44, 38. [Google Scholar]
- Cornes, R.C.; van der Schrier, G.; van den Besselaar, E.J.M.; Jones, P.D. An ensemble version of the E-OBS temperature and precipitation data sets. J. Geophys. Res. Atmos. 2018, 123, 9391–9409. [Google Scholar] [CrossRef] [Green Version]
- Rungis, D.; Bérubé, Y.; Zhang, J.; Ralph, S.; Ritland, C.E.; Ellis, B.E.; Douglas, C.; Bohlmann, J.; Ritland, K. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor. Appl. Genet. 2004, 109, 1283–1294. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Olivieri, A.M.; Morgante, M. Identification and characterization of microsatellites in Norway spruce (Picea abies K.). Genome 1997, 40, 411–419. [Google Scholar] [CrossRef]
- Scotti, I.; Magni, F.; Fink, R.; Powell, W.; Binelli, G.; Hedley, P.E. Microsatellite repeats are not randomly distributed within Norway spruce (Picea abies K.) expressed sequences. Genome 2000, 43, 41–46. [Google Scholar] [CrossRef]
- Scotti, I.; Paglia, G.; Magni, F.; Morgante, M. Efficient development of dinucleotide microsatellite markers in Norway spruce (Picea abies Karst.) through dot-blot selection. Theor. Appl. Genet. 2002, 104, 1035–1041. [Google Scholar] [CrossRef]
- Scotti, I.; Magni, F.; Paglia, G.; Morgante, M. Trinucleotide microsatellites in Norway spruce (Picea abies): Their features and the development of molecular markers. Theor. Appl. Genet. 2002, 106, 40–50. [Google Scholar] [CrossRef]
- Besnard, G.; Acheré, V.; Faivre Rampant, P.; Favre, J.M.; Jeandroz, S. A set of cross-species amplifying microsatellite markers developed from DNA sequence databanks in Picea (Pinaceae). Mol. Ecol. Notes 2003, 3, 380–383. [Google Scholar] [CrossRef]
- Achere, V.; Favre, J.M.; Besnard, G.; Jeandroz, S. Genomic organization of molecular differentiation in Norway spruce (Picea abies). Mol. Ecol. 2005, 14, 3191–3201. [Google Scholar] [CrossRef] [PubMed]
- Fluch, S.; Burg, A.; Kopecky, D.; Homolka, A.; Spiess, N.; Vendramin, G.G. Characterization of variable EST SSR markers for Norway spruce (Picea abies L.). BMC Res. Notes 2011, 4, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, T.C.; Slate, J.; Krukk, L.E.B.; Pemberton, J.M. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 1998, 7, 639–655. [Google Scholar] [CrossRef] [Green Version]
- Slate, J.; Marshall, T.; Pemberton, J. A retrospective assessment of the accuracy of the paternity inference program cervus. Mol. Ecol. 2000, 9, 801–808. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx Tutorial Part 1—Introduction to Population Genetic Analysis; Australian National University: Canberra, Australia, 2009; pp. 1–51. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-Checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Pritcharda, J.K.; Wena, X.; Falush, D. Documentation for STRUCTURE Software, Version 2.3; Thermo Fisher Scientific: Waltham, MA, USA, 2010. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; VonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Barker, M.; Rayens, W. Partial least squares for discrimination. J. Chemom. 2003, 17, 166–173. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbylaitė, R.; Pliūra, A.; Lygis, V.; Suchockas, V.; Jankauskienė, J.; Labokas, J. Genetic diversity and its spatial distribution in self-regenerating Norway spruce and scots pine stands. Forests 2017, 8, 470. [Google Scholar] [CrossRef] [Green Version]
- Unger, G.M.; Konrad, H.; Geburek, T. Does spatial genetic structure increase with altitude? An answer from Picea abies in Tyrol, Austria. Plant Syst. Evol. 2011, 292, 133–141. [Google Scholar] [CrossRef]
- Máchová, P.; Trčková, O.; Cvrčková, H. Use of nuclear microsatellite loci for evaluating genetic diversity of selected populations of Picea abies (L.) karsten in the Czech Republic. Forests 2018, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Stojnić, S.; V. Avramidou, E.; Fussi, B.; Westergren, M.; Orlović, S.; Matović, B.; Trudić, B.; Kraigher, H.; A. Aravanopoulos, F.; Konnert, M. Assessment of genetic diversity and population genetic structure of Norway spruce (Picea abies (L.) Karsten) at its southern lineage in Europe. Implications for conservation of forest genetic resources. Forests 2019, 10, 258. [Google Scholar]
- Cvjetkovic, B.; Konnert, M.; Fussi, B.; Mataruga, M.; Sijacic-Nikolic, M.; Danicic, V.; Lucic, A. Norway spruce (Picea abies Karst.) variability in progeny tests in Bosnia and Herzegovina. Genetika 2017, 49, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Nowakowska, J.A.; Zachara, T.; Konecka, A. Genetic variability of Scots pine (Pinus sylvestris L.) and Norway spruce (Picea abies L. Karst.) natural regeneration compared with their maternal stands. For. Res. Pap. 2014, 75, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Porth, I.; El-Kassaby, Y. Assessment of the genetic diversity in forest tree populations using molecular markers. Diversity 2014, 6, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. In Population Genetics of Forest Trees; Springer: Dordrecht, The Netherlands, 1992; pp. 95–124. [Google Scholar]
- Piotti, A.; Garbarino, M.; Avanzi, C.; Berretti, R.; Motta, R.; Piovani, P.; Leonardi, S. Influence of spatiotemporal dynamics on the fine-scale spatial genetic structure of differently managed Picea abies stands. Forests 2018, 9, 622. [Google Scholar] [CrossRef] [Green Version]
- Rajora, O.P.; Rahman, M.H.; Dayanandan, S.; Mosseler, A. Isolation, characterization, inheritance and linkage of microsatellite DNA markers in white spruce (Picea glauca) and their usefulness in other spruce species. Mol. Gen. Genet. MGG 2001, 264, 871–882. [Google Scholar] [PubMed]
- Bérubé, Y.; Zhuang, J.; Rungis, D.; Ralph, S.; Bohlmann, J.; Ritland, K. Characterization of EST-SSRs in loblolly pine and spruce. Tree Genet. Genomes 2007, 3, 251–259. [Google Scholar] [CrossRef]
- Hodgetts, R.B.; Aleksiuk, M.A.; Brown, A.; Clarke, C.; Macdonald, E.; Nadeem, S.; Khasa, D.; Macdonald, E. Development of microsatellite markers for white spruce (Picea glauca) and related species. Theor. Appl. Genet. 2001, 102, 1252–1258. [Google Scholar] [CrossRef]
- Piotti, A.; Leonardi, S.; Piovani, P.; Scalfi, M.; Menozzi, P. Spruce colonization at treeline: Where do those seeds come from? Heredity 2009, 103, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Ryman, N.; Palm, S.; André, C.; Carvalho, G.R.; Dahlgren, T.G.; Jorde, P.E.; Laikre, L.; Larsson, L.C.; Palmé, A.; Ruzzante, D.E. Power for detecting genetic divergence: Differences between statistical methods and marker loci. Mol. Ecol. 2006, 15, 2031–2045. [Google Scholar] [CrossRef]
- Vendramin, G.G.; Lelli, L.; Rossi, P.; Morgante, M. A set of primers for the amplification of 20 chloroplast microsatellites in Pinaceae. Mol. Ecol. 1996, 5, 595–598. [Google Scholar] [CrossRef]
- Echt, C.S.; Vendramin, G.G.; Nelson, C.D.; Marquardt, P. Microsatellite DNA as shared genetic markers among conifer species. Can. J. For. Res. 1999, 29, 365–371. [Google Scholar] [CrossRef]
- Varshney, R.K.; Sigmund, R.; Börner, A.; Korzun, V.; Stein, N.; Sorrells, M.E.; Langridge, P.; Graner, A. Interspecific transferability and comparative mapping of barley EST-SSR markers in wheat, rye and rice. Plant Sci. 2005, 168, 195–202. [Google Scholar] [CrossRef]
- Chagné, D.; Chaumeil, P.; Ramboer, A.; Collada, C.; Guevara, A.; Cervera, M.T.; Vendramin, G.G.; Garcia, V.; Frigerio, J.-M.; Echt, C.; et al. Cross-species transferability and mapping of genomic and cDNA SSRs in pines. Theor. Appl. Genet. 2004, 109, 1204–1214. [Google Scholar] [CrossRef]
- Bandopadhyay, R.; Sharma, S.; Rustgi, S.; Singh, R.; Kumar, A.; Balyan, H.S.; Gupta, P.K. DNA polymorphism among 18 species of Triticum–Aegilops complex using wheat EST–SSRs. Plant Sci. 2004, 166, 349–356. [Google Scholar] [CrossRef]
- Slatkin, M.; Barton, N.H. A comparison of three indirect methods for estimating average levels of gene flow. Evolution 1989, 43, 1349–1368. [Google Scholar] [CrossRef] [PubMed]
- Schuster, W.S.; Alles, D.L.; Mitton, J.B. Gene flow in limber pine: Evidence from pollination phenology and genetic differentiation along an elevational transect. Am. J. Bot. 1989, 76, 1395. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
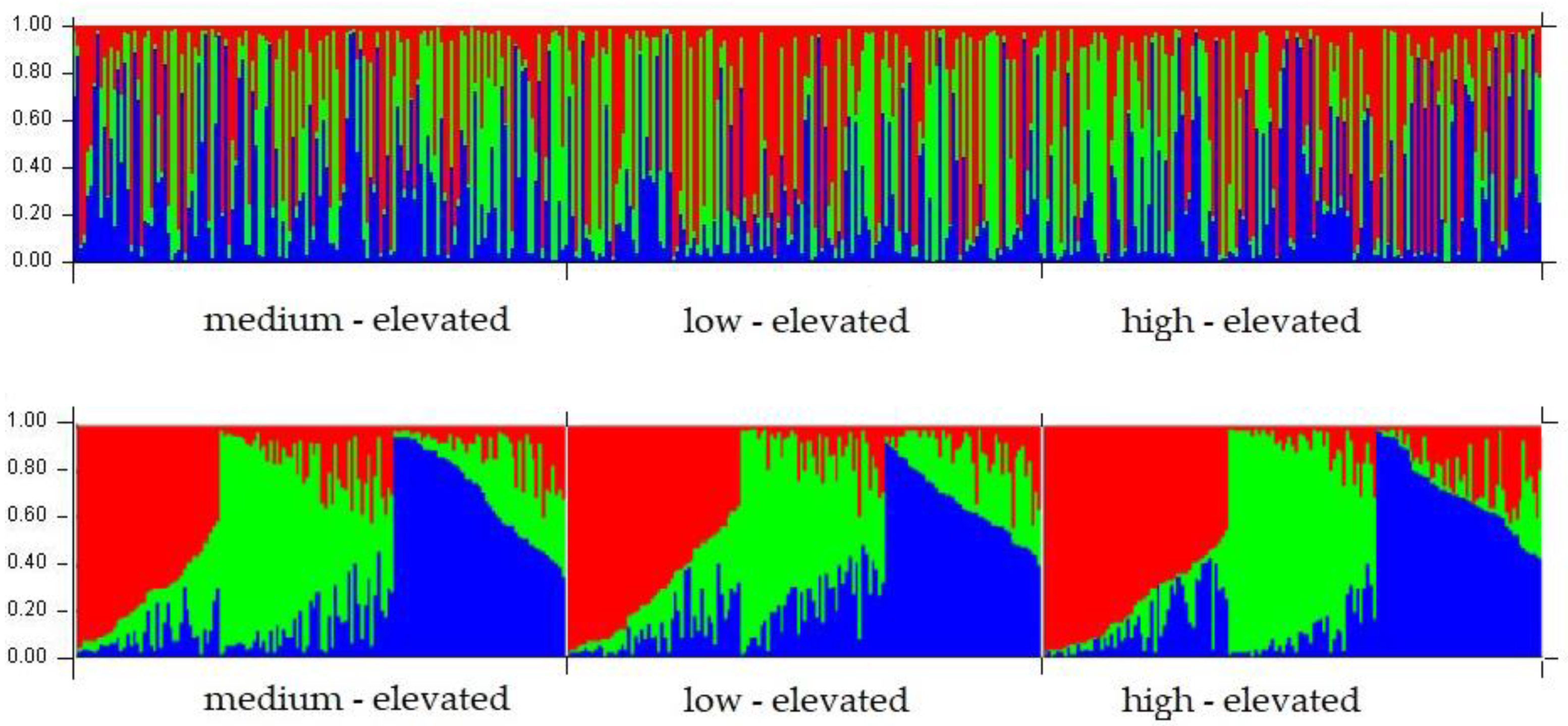{kind=link}
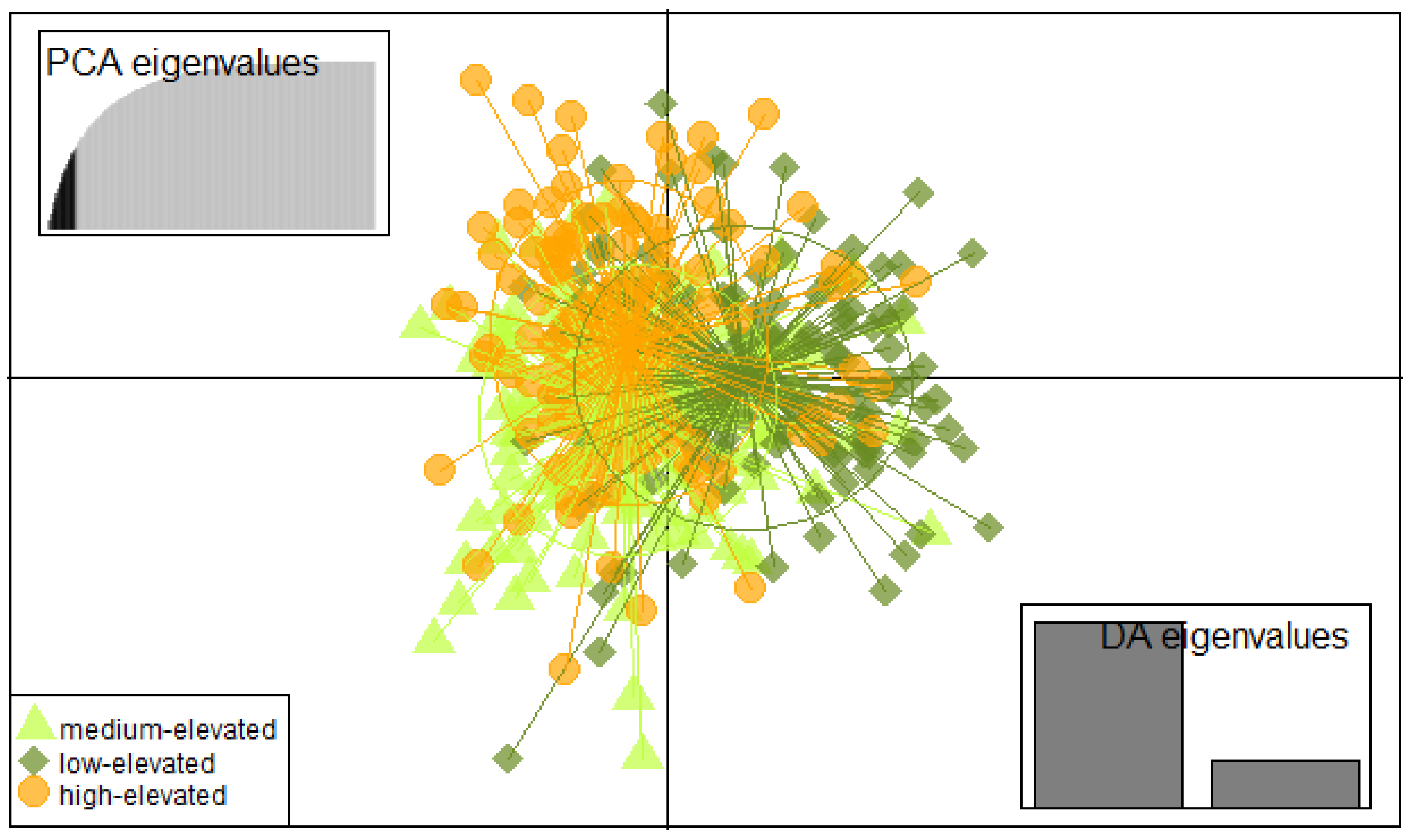{kind=link}
| Ecotypic Form | Elevation | N | E, m a.s.l. | T, °C | P, mm |
|---|---|---|---|---|---|
| acuminata | Low | 149 | 190–400 | 8.2 | 752 |
| europaea | Medium | 148 | 650–850 | 5.9 | 821 |
| obovata | High | 141 | 1100–1250 | 4.9 | 946 |
| SSR Locus | EMBL Accesion Number | Primer Sequences | Repeats | Allele Size Range | Fluorescent Dye | Source Reference | ||
|---|---|---|---|---|---|---|---|---|
| F | R | |||||||
| Multiplex 1 | paGB3 | AJ133748 | AGTGATTAAACTCCTGACCAC | CACTGAATACACCCATTATCC | (AT)11 | 112–130 | PET | [52] |
| WS0022.B15 * | CN480899 | TTTGTAGGTGCTGCAGAGATG | TGGCTTTTTATTCCAGCAAGA | (AG)12 | 174–214 | NED | [47] | |
| SpAGD1 | G31824 | GTCAACCAACTTGTAAAGCCA | ACTTGTTTGGCATTTTCCC | (AG)25 | 122–182 | NED | [48] | |
| SpAGC1 | G31822 | TTCACCTTAGCCGAGAACC | CACTGGAGATCTTCGTTCTGA | (TC)5TT(TC)10 | 73–123 | VIC | [48] | |
| SpAG2 | G31821 | GCTCTTCACGTGTACTTGATC | TTCGAAGATCCTCCAAGATAC | (TC)16 | 87–115 | NED | [48] | |
| EATC1E03 | AJ296702 | CCOCTTATTCCTAAOGTCAAA | TACCAGTGGTGACAACGATG | (CAT)4 CGT(CAT)8 | 121–143 | 6-FAM | [51] | |
| WS00716.F13 * | CN480905 | TCAAGTAATGGACAAACGATACA | TTTCCAATAGAATGGTGGATTT | (GA)10 | 206–252 | 6-FAM | [47] | |
| Multiplex 2 | WS0073.H08 * | CN480903 | TGCTCTCTTATTCGGGCTTC | AAGAACAAGGCTTCCCAATG | (AT)14 | 199–217 | VIC | [47] |
| PA_28 * | GT887962 | GGCCGAAAGTGCTACTGCTA | TGCTCCAGAAGAACACTCACA | (TCG)n | 151–172 | VIC | [54] | |
| PAAC3 | AJ131104 | CGCTACCTCAGATTTCTCCA | AGATATTCCCTCACAAAGTTGG | (CA)16 | 240–320 | VIC | [49] | |
| WS0092.A19 * | CN480888 | TGTGGTTTTCTGCTTGGAAA | CCCATTTTGACTTTGAAATAAGC | (AC)9 | 213–237 | NED | [47] | |
| WS0023.B03 * | CN480900 | AGCAGCTGGGGTCAAAGTT | AAAGAAAGCATGCATATGACTCAG | (AT)10 | 163–211 | NED | [47] | |
| PA_33 * | GT884592 | GGTCGAGGAGGAGGAGGTAG | CACCGCTAGTGCAGTCTCTG | (CGG)n | 99–108 | 6-FAM | [54] | |
| WS0019.F22 * | CN480896 | AAGCGTTTCTCATTTTCTTGG | GGGCCCAGAACTAACAATGA | (AT)13 | 355–387 | 6-FAM | [47] | |
| Pa_56 * | GT884695 | ATCGTCTGCATTGCATTCAC | CTTCGTTCCTTCCTGATCCA | (AGGTG)n | 124–139 | PET | [54] | |
| PAAC23 | AJ131109 | TGTGGCCCCACTTACTAATATCAG | CGGGCATTGGTTTACAAGAGTTGC | (GT)14 | 181–315 | PET | [49] | |
| Locus | k | Ho | He | PIC | HW | F(Null) |
|---|---|---|---|---|---|---|
| paGB3 | 8 | 0.624 | 0.779 | 0.747 | *** | 0.1105 |
| WS0022.B15 † | 20 | 0.843 | 0.869 | 0.858 | NS | 0.0139 |
| SpAGD1 | 30 | 0.742 | 0.953 | 0.950 | *** | 0.1269 |
| SpAGC1 | 25 | 0.580 | 0.610 | 0.600 | NS | 0.0345 |
| SpAG2 | 17 | 0.857 | 0.892 | 0.881 | NS | 0.0182 |
| EATC1E03 | 8 | 0.276 | 0.38 | 0.351 | *** | 0.1599 |
| WS00716.F13 † | 23 | 0.874 | 0.934 | 0.929 | ND | 0.0335 |
| WS0073.H08 † | 7 | 0.660 | 0.670 | 0.608 | NS | 0.0068 |
| PA_28 † | 7 | 0.766 | 0.687 | 0.637 | *** | −0.0663 |
| PAAC3 | 36 | 0.457 | 0.879 | 0.868 | *** | 0.3145 |
| WS0092.A19 † | 13 | 0.722 | 0.794 | 0.767 | * | 0.0513 |
| WS0023.B03 † | 25 | 0.378 | 0.859 | 0.846 | *** | 0.3904 |
| PA_33 † | 4 | 0.088 | 0.101 | 0.098 | ND | 0.0737 |
| WS0019.F22 † | 16 | 0.355 | 0.629 | 0.577 | *** | 0.2785 |
| Pa_56 † | 4 | 0.679 | 0.637 | 0.567 | NS | −0.0346 |
| PAAC23 | 36 | 0.457 | 0.879 | 0.868 | *** | 0.3145 |
| Mean | 17.437 | 0.585 | 0.722 | 0.697 | ||
| SD | 10.541 | 0.222 | 0.218 | 0.223 | ||
| SE | 2.974 | 0.062 | 0.061 | 0.063 |
| Populations. | N | Na | Ne | I | Ho | He | Fis | p-Value | |
|---|---|---|---|---|---|---|---|---|---|
| Low-elevation | Mean | 143 | 13.813 | 6.249 | 1.803 | 0.609 | 0.715 | 0.137 | 0.000 |
| SE | 2.199 | 1.258 | 0.200 | 0.050 | 0.055 | 0.039 | |||
| Medium-elevation | Mean | 150 | 15.625 | 5.984 | 1.749 | 0.599 | 0.701 | 0.128 | 0.006 |
| SE | 2.115 | 1.321 | 0.201 | 0.058 | 0.056 | 0.053 | |||
| High-elevation | Mean | 143 | 13.125 | 5.594 | 1.730 | 0.596 | 0.698 | 0.175 | 0.000 |
| SE | 1.805 | 1.258 | 0.188 | 0.070 | 0.056 | 0.072 | |||
| All individuals | 436 | 14.211 | 5.943 | 1.760 | 0.601 | 0.705 | 0.146 | 0.002 | |
| SE | 2.041 | 1.279 | 0.197 | 0.059 | 0.055 | 0.054 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bínová, Z.; Korecký, J.; Dvořák, J.; Bílý, J.; Zádrapová, D.; Jansa, V.; Lstibůrek, M. Genetic Structure of Norway Spruce Ecotypes Studied by SSR Markers. Forests 2020, 11, 110. https://doi.org/10.3390/f11010110
Bínová Z, Korecký J, Dvořák J, Bílý J, Zádrapová D, Jansa V, Lstibůrek M. Genetic Structure of Norway Spruce Ecotypes Studied by SSR Markers. Forests. 2020; 11(1):110. https://doi.org/10.3390/f11010110
Chicago/Turabian StyleBínová, Zuzana, Jiří Korecký, Jakub Dvořák, Jan Bílý, Dagmar Zádrapová, Václav Jansa, and Milan Lstibůrek. 2020. "Genetic Structure of Norway Spruce Ecotypes Studied by SSR Markers" Forests 11, no. 1: 110. https://doi.org/10.3390/f11010110
APA StyleBínová, Z., Korecký, J., Dvořák, J., Bílý, J., Zádrapová, D., Jansa, V., & Lstibůrek, M. (2020). Genetic Structure of Norway Spruce Ecotypes Studied by SSR Markers. Forests, 11(1), 110. https://doi.org/10.3390/f11010110