Genome Cytosine Methylation May Affect Growth and Wood Property Traits in Populations of Populus tomentosa

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Phenotypic Data Collection
2.3. Genotyping by Methylation-Sensitive Amplified Polymorphism
2.4. Linear Correlation Analysis and Association Analysis
2.5. Candidate Gene Screening and Gene Expression
3. Results
3.1. Variation in Growth and Wood Characteristics
3.2. Linear Correlation between Phenotype and DNA Methylation Levels
3.3. MSAP Markers Associated with Phenotypic Traits within the Populations
3.4. Sequencing and Functional Prediction by Homology Alignment
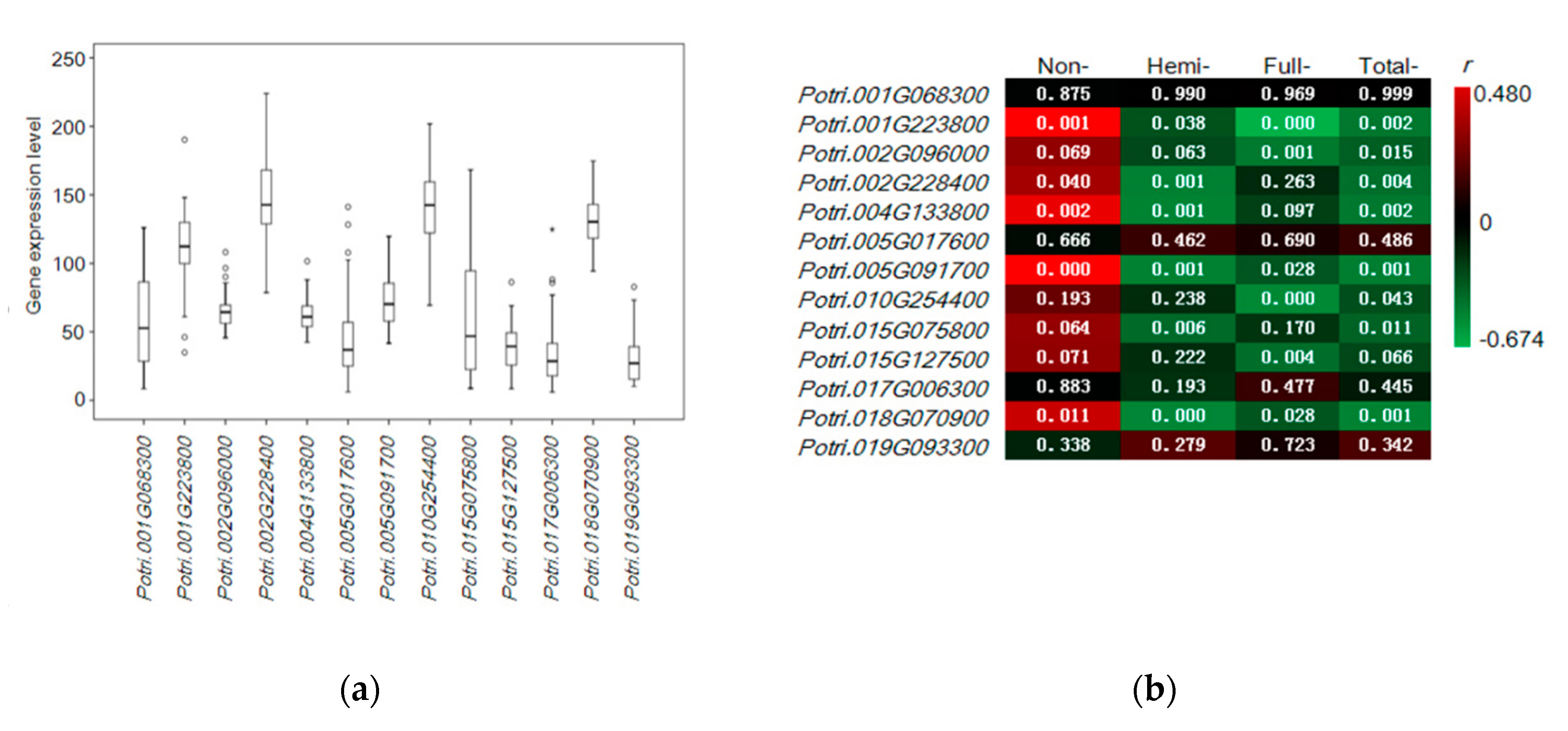
3.5. Quantitative Expression of Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Sequencing Data
References
- Labra, M.; Ghiani, A.; Citterio, S.; Sgorbati, S.; Sala, F.; Vannini, C.; Vannini, C.; Ruffini-Castiglione, M.; Bracale, M. Analysis of cytosine methylation pattern in response to water deficit in pea root tips. Plant Biol. 2002, 4, 694–699. [Google Scholar] [CrossRef] [Green Version]
- Nicotra, A.B.; Atkin, O.K.; Bonser, S.P.; Davidson, A.M.; Finnegan, E.J.; Mathesius, U.; Poot, P.; Purugganan, M.D.; Richards, C.L.; Valladares, F.; et al. Plant phenotypic plasticity in a changing climate. Trends Plant Sci. 2010, 15, 684–692. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [Green Version]
- Richards, E.J. Natural epigenetic variation in plant species: A view from the field. Curr. Opin. Plant Biol. 2011, 14, 204–209. [Google Scholar] [CrossRef]
- Teixeira, F.K.; Colot, V. Repeat elements and the Arabidopsis DNA methylation landscape. Heredity 2010, 105, 14–23. [Google Scholar] [CrossRef]
- Herrera, C.M.; Bazaga, P. Epigenetic differentiation and relationship to adaptive genetic divergence in discrete populations of the violet Viola cazorlensis. New Phytol. 2010, 187, 867–876. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Preis, J.J.; Morgan, H.; Whitelaw, E. The marks, mechanisms and memory of epigenetic statesin mammals. Biochem. J. 2001, 356, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Blewitt, M.E.; Druker, R.; Preis, J.I.; Whitelaw, E. Metastable epialleles in mammals. Trends Genet. 2002, 18, 348–351. [Google Scholar] [CrossRef]
- Foerster, A.M.; Dinh, H.Q.; Sedman, L.; Wohlrab, B.; Scheid, O.M. Genetic rearrangements can modify chromatin features at epialleles. PLoS Genet. 2011, 7, e1002331. [Google Scholar] [CrossRef] [Green Version]
- Vaughn, M.W.; Tanurdžic, M.; Lippman, Z.; Jiang, H.; Carrasquillo, R.; Rabinowicz, P.D.; Dedhia, N.; McCombie, W.R.; Agier, N.; Bulski, A.; et al. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol. 2007, 5, e174. [Google Scholar] [CrossRef] [PubMed]
- Angers, B.; Castonguay, E.; Massicotte, R. Environmentally induced phenotypes and DNA methylation: How to deal with unpredictable conditions until the next generation and after. Mol. Ecol. 2010, 19, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, R.; Sasaki, T.; Kudoh, H.; Taylor, J.M.; Kakutani, T.; Dennis, E.S. Epigenetic variation in the FWA gene within the genus Arabidopsis. Plant J. 2011, 66, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shiu, S.; Cal, A.; Borevitz, J.O. Global analysis of genetic, epigenetic and transcriptional polymorphisms in Arabidopsis thaliana using whole genome tiling arrays. PLoS Genet. 2008, 4, e1000032. [Google Scholar] [CrossRef]
- Long, Y.; Xia, W.; Li, R.; Wang, J.; Shao, M.; Feng, J.; King, G.J.; Meng, J. Epigenetic QTL mapping in Brassica napus. Genetics 2011, 189, 1093–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foust, C.; Preite, V.; Schrey, A.W.; Alvarez, M.; Robertson, M.; Verhoeven, K.J.F.; Richards, C.L. Genetic and epigenetic differences associated with environmental gradients in replicate populations of two salt marsh perennials. Mol. Ecol. 2016, 25, 1639–1652. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Sun, L.; Cheng, T.; Pan, H.; Wang, J.; Zhang, Q. Epigenetic variance, performing cooperative structure with genetics, is associated with leaf shape traits in widely distributed populations of ornamental tree Prunus mume. Front. Plant Sci. 2018, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Cervera, M.T.; Ruiz-Garcia, L.; Martinez-Zapater, J.M. Analysis of DNA methylation in Arabidopsis thaliana based on methylation-sensitive AFLP markers. Mol. Genet. Genom. 2002, 268, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Lisch, D. Epigenetic regulation of transposable elements in plants. Ann. Rev. Plant Biol. 2009, 60, 43–66. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, T.; Shin, J.; Uzawa, R.; Silva, P.; Cohen, S.; Bauer, M.J.; Hashimoto, M.; Kirkbride, R.C.; Harada, J.J.; Zilberman, D.; et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc. Natl. Acad. Sci. USA 2011, 108, 1755–1762. [Google Scholar] [CrossRef] [Green Version]
- Uthup, T.K.; Ravindran, M.; Bini, K.; Thakurdas, S. Divergent DNA methylation patterns associated with abiotic stress in Hevea brasiliensis. Mol. Plant 2011, 4, 996–1013. [Google Scholar]
- Diez, C.M.; Roessler, K.; Gaut, B.S. Epigenetics and plant genome evolution. Curr. Opin. Plant Biol. 2014, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rico, L.; Ogaya, R.; Barbeta, A.; Pnuelas, J. Changes in DNA methylation fingerprint of Quercus ilex trees in response to experimental field drought simulating projected climate change. Plant Biol. 2014, 16, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Tao, X.; Wang, Y.; Ma, D.; Li, D.; Yang, H.; Ma, X. Analysis of DNA methylation of perennial ryegrass under drought using the methylation-sensitive amplification polymorphism (MSAP) technique. Mol. Genet. Genom. 2014, 289, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Kakutani, T.; Munakata, K.; Richards, E.; Hirohiko, H. Meiotically and mitotically stable inheritance of DNA hypomethylation induced by ddm1 mutation of Arabidopsis thaliana. Genetics 1999, 151, 831–838. [Google Scholar] [PubMed]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Kashkush, K.; Khasdan, V. Large-scale survey of cytosine methylation of retrotransposons and the impact of read out transcription from long terminal repeatson expression of adjacent rice genes. Genetics 2007, 177, 1975–1985. [Google Scholar] [CrossRef] [Green Version]
- Vining, K.J.; Pomraning, K.R.; Wilhelm, L.J.; Priest, H.D.; Pellegrini, M.; Mockler, T.C.; Freitag, M.; Strauss, S.H. Dynamic DNA cytosine methylation in the Populus trichocarpa genome: Tissue-level variation and relationship to gene expression. BMC Genom. 2012, 13, 27. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, S.E.; Meyerowitz, E.M. Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 1997, 277, 1100–1103. [Google Scholar] [CrossRef] [Green Version]
- Bender, J.; Fink, G.R. Epigenetic control of an endogenous gene family is revealed by a novel blue fluorescent mutant of Arabidopsis. Cell 1995, 83, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, T.; Miura, A.; Choi, Y.; Kinoshita, Y.; Cao, X.; Jacobsen, S.E.; Fischer, R.L.; Kakutani, T. One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science 2004, 303, 521–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, R.J.; He, Y.; Valdés-López, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G.; et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ci, D.; Song, Y.; Du, Q.; Tian, M.; Han, S.; Zhang, D. Variation in genomic methylation in natural populations of Populus simonii is associated with leaf shape and photosynthetic traits. J. Exp. Bot. 2016, 67, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z. Collection, conservation and utilization of plus tree resources of Populus tomentosa in China. J. Beijing For. Univ. 1992, 14, 1–25. [Google Scholar]
- Zhu, Z.; Zhang, Z. The status and advavces of genetic improvement of Populus tomentosa Carr. J. Beijing For. Univ. 1997, 6, 1–7. [Google Scholar]
- Zhang, D.; Zhang, Z.; Yang, K.; Li, B. Genetic mapping in (Populus tomentosa × Populus bolleana) and P. tomentosa Carr. Using AFLP markers. Theor. Appl. Genet. 2004, 108, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Pan, W.; Xu, B.; Li, B.; Zhang, D. Polymorphic simple sequence repeat (SSR) loci within cellulose synthase (PtoCesA) genes are associated with growth and wood properties in Populus tomentosa. New Phytol. 2013, 197, 763–776. [Google Scholar] [CrossRef]
- Ma, K.; Song, Y.; Jiang, X.; Zhang, Z.; Li, B.; Zhang, D. Photosynthetic response to genome methylation affects the growth of Chinese white poplar. Tree Genet. Genomes 2012, 8, 1407–1421. [Google Scholar] [CrossRef]
- Ma, K.; Song, Y.; Yang, X.; Zhang, Z.; Zhang, D. Variation in genomic methylation in natural populations of Chinese white poplar. PLoS ONE 2013, 8, e63977. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ci, D.; Li, T.; Li, P.; Song, Y.; Chen, J.; Quan, M.; Zhou, D.; Zhang, D. The role of DNA methylation in xylogenesis in different tissues of poplar. Front. Plant Sci. 2016, 7, 1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Wang, B.; Wei, Z.; Zhang, D.; Li, B. Genetic diversity and population structure of Chinese white poplar (Populus tomentosa) revealed by SSR markers. J. Hered. 2012, 103, 853–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Meng, X.; Cheng, J.; Zhao, L.; Wang, X. Study on estimating the volume of a single tree using normal form factor. Forest Res. Manag. 2005, 1, 39–41. [Google Scholar]
- Andersson, S.; Serimaa, R.; Torkkeli, M.; Paakkari, T.; Saranpaa, P.; Pesonen, E. Microfibril angle of Norway spruce [Piceaabies (L.) Karst.] compression wood: Comparison of measuring techniques. J. Wood Sci. 2000, 46, 343–349. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Zhao, R.; Mccord, J.; Rials, T.G.; Wang, S. Anatomical characteristics, microfibril angle and micromechanical properties of cottonwood (Populus deltoides) and its hybrids. Biomass Bioenergy 2016, 93, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Hart, J.F.; De-Araujo, F.; Thomas, B.R.; Mansfield, S.D. Wood quality and growth characterization across intra- and inter-specific hybrid aspen clones. Forests 2013, 4, 786–807. [Google Scholar] [CrossRef] [Green Version]
- Porth, I.; Klápště, J.; Skyba, O.; Lai, B.S.K.; Geraldes, A.; Muchero, W.; Tuskan, G.A.; Douglas, C.J.; El-Kassaby, Y.A.; Mansfield, S.D. Populus trichocarpa cell wall chemistry and ultrastructure trait variation, genetic control and genetic correlations. New Phytol. 2013, 197, 777–790. [Google Scholar] [CrossRef]
- Schimleck, L.R.; Kube, P.D.; Raymond, C.A. Genetic improvement of kraft pulp yield in Eucalyptus nitens using cellulose content determined by near infrared spectroscopy. Can. J. Forest Res. 2004, 34, 2363–2370. [Google Scholar] [CrossRef]
- Aleong, J.; Howard, D. Extensions of the Duncan’s Multiple Range Test for unbalanced data. J. Appl. Stat. 1985, 12, 83–90. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucl. Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [Green Version]
- Tixier, M.H.; Sourdille, P.; Röder, M.S.; Leroy, P.; Bernard, M. Detection of wheat microsatellites using a non-radioactive silver-nitrate staining method. J. Genet. Breed. 1997, 51, 175–177. [Google Scholar]
- Song, Y.; Ma, K.; Bo, W.; Zhang, Z.; Zhang, D. Sex-specific DNA methylation and gene expression in andromonoecious poplar. Plant Cell Rep. 2012, 31, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Song, Y.; Huang, Z.; Lin, L.; Zhang, Z.; Zhang, D. The low fertility of Chinese white poplar: Dynamic changes in anatomical structure, endogenous hormone concentrations, and key gene expression in the reproduction of a naturally occurring hybrid. Plant Cell Rep. 2013, 32, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Du, Q.; Xu, B.; Zhang, Z.; Li, B. The actin multigene family in Populus: Organization, expression and phylogenetic analysis. Mol. Genet. Genom. 2010, 284, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Zhao, S.; Wang, L.; Dong, W.; Liu, L.; He, Y. Structure of the Arabidopsis thaliana NADPH-cytochrome P450 reductase 2 (ATR2) provides insight into its function. FEBS J. 2017, 284, 754–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundin, L.; Vanholme, R.; Geerinck, J.; Goeminne, G.; Hofer, R.; Kim, H.; Ralph, J.; Boerjan, W. Mutation of the inducible ARABIDOPSIS THALIANA CYTOCHROME P450 REDUCTASE2 alters lignin composition and improves saccharification. Plant Physiol. 2014, 166, 1956–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.E.; Hemmingsen, S.M. Arabidopsis thaliana type I and II chaperonins. Cell Stress Chaperon 2001, 6, 190–200. [Google Scholar] [CrossRef]
- Peng, L.; Fukao, Y.; Myouga, F.; Motohashi, R.; Shinozaki, K.; Shikanai, T. A chaperonin subunit with unique structures is essential for folding of a specific substrate. PLoS Biol. 2011, 9, e1001040. [Google Scholar] [CrossRef]
- Tiwari, L.D.; Grover, A. Cpn60β4 protein regulates growth and developmental cycling and has bearing on flowering time in Arabidopsis thaliana plants. Plant Sci. 2019, 286, 78–88. [Google Scholar] [CrossRef]
- Micheli, F. Pectin methylesterases: Cell wall enzymes with important roles in plant physiology. Trends Plant Sci. 2001, 6, 414–419. [Google Scholar] [CrossRef]
- Wolf, S.I.; Mouille, G.; Pelloux, J. Homogalacturonan methyl-esterification and plant development. Mol. Plant 2009, 2, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Hongo, S.; Sato, K.; Yokoyama, R.; Nishitani, K. Demethylesterification of the primary wall by PECTIN METHYLESTERASE35 provides mechanical support to the Arabidopsis stem. Plant Cell 2012, 24, 2624–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siedlecka, A.; Wiklund, S.; Peronne, M.; Micheli, F.; Lesniewska, J.; Sethson, I.; Edlund, U.; Richard, L.; Sundberg, B.; Mellerowicz, E.J. Pectin methyl esterase inhibits intrusive and symplastic cell growth in developing wood cells of Populus. Plant Physiol. 2007, 146, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, M.D.; Delannoy, M.; Navarre, C.; Boutry, M. NtSCP1 from tobacco is an extracellular serine carboxypeptidase III that has an impact on cell elongation. Plant Physiol. 2012, 158, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, F.; Gonzalez, M.D.; Cejudo, F.J. A germination-related gene encoding a serine carboxypeptidase is expressed during the differentiation of the vascular tissue in wheat grains and seedlings. Planta 2002, 215, 727–734. [Google Scholar] [CrossRef]
- Escamillatrevino, L.L.; Chen, W.; Card, M.L.; Shih, M.; Cheng, C.; Poulton, J.E. Arabidopsis thalianaβ-glucosidases BGLU45 and BGLU46 hydrolyse monolignolglucosides. Phytochemistry 2006, 67, 1651–1660. [Google Scholar] [CrossRef]
- Chapelle, A.; Morreel, K.; Vanholme, R.; Le-Bris, P.; Morin, H.; Lapierre, C.; Boerjan, W.; Jouanin, L.; Demont-Caulet, N. Impact of the absence of stem-specific β-glucosidases on lignin and monolignols. Plant Physiol. 2012, 160, 1204–1217. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Wang, S.; Zhang, B.; Shang-guan, K.; Shi, Y.; Zhang, D.; Liu, X.; Wu, K.; Xu, Z.; Fu, X.; et al. A gibberellin-mediated DELLA-NAC signaling cascade regulates cellulose synthesis in rice. Plant Cell 2015, 27, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wang, X.; Lu, W.; Liu, R.; Tian, Q.; Sun, Y.; Luo, K. A poplar R2R3-MYB transcription factor, PtrMYB152, is involved in regulation of lignin biosynthesis during secondary cell wall formation. Plant Cell Tiss. Org. 2014, 119, 553–563. [Google Scholar] [CrossRef]
- Guillaumie, S.; Mzid, R.; Méchin, V.; Léon, C.; Hichri, I.; Destrac-Irvine, A.; Trossat-Magnin, C.; Delrot, S.; Lauvergeat, V. The grapevine transcription factor WRKY2 influences the lignin pathway and xylem development in tobacco. Plant Mol. Biol. 2010, 72, 215–234. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, X.; Yang, F.; Fan, D.; Jiang, Y.; Luo, K. PtrWRKY19, a novel WRKY transcription factor, contributes to the regulation of pith secondary wall formation in Populus trichocarpa. Sci. Rep. 2016, 6, 18643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büttner, M.; Truernit, E.; Baier, K.; Scholz-starke, J.; Sontheim, M.; Lauterbach, C.; Huss, V.A.R.; Sauer, N. AtSTP3, a green leaf-specific, low affinity monosaccharide-H+symporter of Arabidopsis thaliana. Plant Cell Environ. 2000, 23, 175–184. [Google Scholar] [CrossRef]
- Schneidereit, A.; Scholzstarke, J.; Buttner, M. Functional characterization and expression analyses of the glucose-specific AtSTP9 monosaccharide transporter in pollen of Arabidopsis. Plant Physiol. 2003, 133, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Scholz-Starke, J.; Büttner, M.; Sauer, N. AtSTP6, a new, pollen-specific H+-monosaccharide symporter from Arabidopsis. Plant Physiol. 2003, 131, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Schofield, R.A.; Bi, Y.; Kant, S.; Rothstein, S.J. Over-expression of STP13, a hexose transporter, improves plant growth and nitrogen use in Arabidopsis thaliana seedlings. Plant Cell Environ. 2009, 32, 271–285. [Google Scholar] [CrossRef]
- Liu, Q.; Dang, H.; Chen, Z.; Wu, J.; Chen, Y.; Chen, S.; Luo, L. Genome-wide identification, expression, and functional analysis of the sugar transporter gene family in cassava (Manihotesculenta). Int. J. Mol. Sci. 2018, 19, 987. [Google Scholar] [CrossRef] [Green Version]
- Alejandro, S.; Lee, Y.; Tohge, T.; Sudre, D.; Osorio, S.; Park, J.; Bovet, L.; Lee, Y.; Geldner, N.; Fernie, A.R.; et al. AtABCG29 is a monolignol transporter involved in lignin biosynthesis. Curr. Biol. 2012, 22, 1207–1212. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Watanabe, A.; Tamura, M.; Tsutsumi, Y. The gene expression analysis of Arabidopsis thaliana ABC transporters by real-time PCR for screening monolignol-transporter candidates. J. Wood Sci. 2018, 64, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, M.; Schuetz, M.; Lin, B.S.P.; Chanis, C.; Hamberger, B.; Western, T.L.; Ehlting, J.; Samuels, A.L. ABC transporters coordinately expressed during lignification of Arabidopsis stems include a set of ABCBs associated with auxin transport. J. Exp. Bot. 2011, 62, 2063–2077. [Google Scholar] [CrossRef] [Green Version]
- Hauben, M.; Haesendonckx, B.; Standaert, E.; Kelen, K.V.D.; Azmi, A.; Akpo, H.; Breusegem, F.V.; Guisez, Y.; Bots, M.; Lambert, B.; et al. Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc. Natl. Acad. Sci. USA 2009, 106, 20109–20114. [Google Scholar] [CrossRef] [Green Version]
- Tsaftaris, A.S.; Kafka, M.; Polidoros, A.; Tani, W. Epigenetic Changes in Maize DNA and Heterosis. In The Genetics and Exploitation of Heterosis in Crops; CIMMYT: Mexico City, Mexico, 1997; pp. 112–113. [Google Scholar]
- Gourcilleau, D.; Bogeat-Triboulot, M.; Thiec, D.L.; Lafon-Placette, C.; Delaunay, A.; El-Soud, W.A.; Brignolas, F.; Maury, S. DNA methylation and histone acetylation: Genotypic variations in hybrid poplars, impact of water deficit and relationships with productivity. Ann. For. Sci. 2010, 67, 208. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| L (%) | HC (%) | α- (%) | FL (mm) | FW (µm) | MFA (°) | DBH (cm) | H (m) | V (m3) | Non- (%) | CNG (%) | CG (%) | Uninformative (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HC (%) | −0.229 ** | ||||||||||||
| α- (%) | −0.248 ** | 0.611 ** | |||||||||||
| FL (mm) | −0.000 | −0.012 | −0.034 | ||||||||||
| FW (µm) | −0.004 | −0.017 | −0.041 | 0.206 ** | |||||||||
| MFA (°) | −0.008 | 0.043 | 0.037 | −0.383 ** | −0.093 | ||||||||
| DBH (cm) | 0.159 ** | −0.120 * | −0.181 ** | 0.280 ** | 0.157 ** | −0.114 * | |||||||
| H (m) | 0.083 | −0.001 | −0.095 | 0.246 ** | −0.001 | −0.143 ** | 0.634 ** | ||||||
| V (m3) | 0.134 ** | −0.080 | −0.161 ** | 0.273 ** | 0.095 | −0.085 | 0.945 ** | 0.728 ** | |||||
| Non- (%) | 0.010 | −0.011 | −0.049 | −0.039 | 0.037 | 0.015 | 0.010 | −0.109 * | −0.049 | ||||
| CNG (%) | 0.035 | 0.028 | 0.032 | 0.016 | −0.002 | 0.041 | −0.064 | 0.002 | −0.021 | −0.741 ** | |||
| CG (%) | −0.039 | 0.011 | 0.017 | 0.056 | 0.071 | −0.005 | 0.077 | 0.122 * | 0.096 | −0.413 ** | 0.356 ** | ||
| CG/CNG (%) | −0.032 | −0.017 | 0.032 | 0.014 | −0.088 | −0.060 | 0.009 | 0.097 | 0.044 | −0.518 ** | −0.103 * | −0.254 ** | |
| Total (%) | 0.013 | 0.026 | 0.032 | 0.035 | 0.027 | 0.030 | −0.019 | 0.051 | 0.022 | −0.748 ** | 0.931 ** | 0.671 ** | −0.181 ** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, K.; Song, Y.; Ci, D.; Zhou, D.; Tian, M.; Zhang, D. Genome Cytosine Methylation May Affect Growth and Wood Property Traits in Populations of Populus tomentosa. Forests 2020, 11, 828. https://doi.org/10.3390/f11080828
Ma K, Song Y, Ci D, Zhou D, Tian M, Zhang D. Genome Cytosine Methylation May Affect Growth and Wood Property Traits in Populations of Populus tomentosa. Forests. 2020; 11(8):828. https://doi.org/10.3390/f11080828
Chicago/Turabian StyleMa, Kaifeng, Yuepeng Song, Dong Ci, Daling Zhou, Min Tian, and Deqiang Zhang. 2020. "Genome Cytosine Methylation May Affect Growth and Wood Property Traits in Populations of Populus tomentosa" Forests 11, no. 8: 828. https://doi.org/10.3390/f11080828
APA StyleMa, K., Song, Y., Ci, D., Zhou, D., Tian, M., & Zhang, D. (2020). Genome Cytosine Methylation May Affect Growth and Wood Property Traits in Populations of Populus tomentosa. Forests, 11(8), 828. https://doi.org/10.3390/f11080828